
ABSTRACT
Objectives
BCR-ABL1 and JAK2 V617F coexistence in myeloproliferative neoplasms has been described as concomitant or sequential events. Despite this, we present a unique case of chronic myeloid leukemia (CML) not referable to either of the known scenarios.
Methods
BCR-ABL1 molecular monitoring was performed by real-time quantitative PCR (RQ-PCR). At the time of molecular relapse, a targeted next-generation sequencing analysis with a customized panel of 26 genes commonly mutated in myeloid diseases was performed. To investigate the kinetics of the JAK2 variant and its association with the BCR-ABL1 rearrangement, RQ-PCR was performed at different time points during the patient’s follow-up.
Results
While negative at CML diagnosis, the JAK2 mutation was first detected 9 years later (VAF: 7.2%). The mutational burden of JAK2 remained stable in multiple determinations, with minor fluctuations independent of BCR-ABL1 kinetics. At the last available time point, the patient was in deep molecular response (MR4), the JAK2 mutational burden was 7%, and no clinical-laboratory findings of Ph-MPN were detectable.
Discussion
In the presented case, the JAK2variantoccurring during the course of the disease seems to stay in the shadows of CML, just as a bystander. The impact of this event (that may be considered suggestive of clonal hematopoiesis of indeterminate potential) on the disease outcome, even if seemingly irrelevant, has still to be explored.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders of the hematopoietic stem cells currently classified according to their clinical, phenotypical, and genomic features [Citation1]. The most frequent are chronic myeloid leukemia (CML), essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF). While ontologically they belong to the same group, these entities are historically divided, in clinical practice, according to the presence of the Philadelphia chromosome (Ph)/BCR-ABL1 fusion transcript. On the one hand, Ph+ CML; on the other, Ph-MPNs, comprising ET, PV and PMF.
These latter are frequently associated with the hotspot mutation (V617F) of JAK2 gene occurring in approximately∼90% of PV and ∼50% of ET and PMF [Citation2].
Historically, BCR-ABL1and JAK2 V617Fwere considered mutually exclusive driver genetic aberrations [Citation3]. Notwithstanding, there is evidence that both may co-exist in various ways (). The first scenario is a coexistence of the two alterations at the diagnosis. Patients usually present with either a typical CML or a Ph-MPN phenotype but in some circumstances, a ‘hybrid’ phenotype with overlapping features has been reported [Citation4] (Supplementary Table 1). The concomitance of mutations should remind us,on one hand, that BCR-ABL1 has been detected in healthy individuals [Citation5]; on the other hand, JAK2 V617F accounts for ∼3% of cases of clonal hematopoiesis of indeterminate potential(CHIP) [Citation6].
Figure 1. Different possible scenarios of BCR-ABL1 and JAK2 (V617F) co-occurrence in MPNs. (A) A CML patient can show the JAK2 mutation at diagnosis and present a ‘hybrid’ phenotype. (B) A CML patient can acquire the JAK2 mutation during follow-up, together with a phenotype change. (C) A JAK2 mutated Ph-MPN can acquire BCR-ABL1 during follow-up and change the phenotype. CML: chronic myeloid leukemia; Ph-MPN: Philadelphia-negative myeloproliferative neoplasm; PLT: platelets; HCT: hematocrit; WBC: white blood cells.
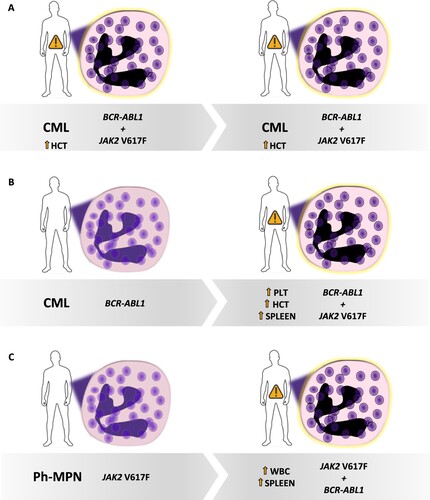
A second possibility is that of the sequential detection of mutations (Supplementary Table 1). In this context, patients presenting one of the two driver variants could develop the other, at some point. In both cases, a phenotype change generally suggests the emergence of a second mutation ().
This report presents a unique case of CML not referable to either of the scenarios mentioned above.
Case presentation
In 2007, a 49-year-old male was referred to our center for neutrophilic leukocytosis, unexplained weight loss and early satiety. His blood counts showed: white blood cells 77 × 109/L, hemoglobin 13.6 g/dL, platelets 380 × 109/L. A physical examination revealed splenomegaly. Peripheral blood (PB) smear showed the presence of myeloid precursor and ∼2% of blasts. Total RNA was extracted from bone marrow (BM) cells using the RNeasy Mini kit (Qiagen), and the presence of a b3a2 BCR-ABL1 fusion transcript was revealed by reverse-transcription PCR (RT–PCR) analysis. A BM aspiration and biopsy were consistent with a CML diagnosis. Conventional cytogenetic analysis of G-banded BM metaphase cells showed the following karyotype: 46,XY,t(9;22)(q34;q11.2)[20]. The Sokal risk score was intermediate (0,86). The patient was started on Imatinib Mesylate (IM) treatment at 400 mg/die. After 6 months, he reached a complete cytogenetic response. Molecular monitoring was initially performed by RT–PCR and then, starting from 2012, by real-time quantitative PCR (RQ-PCR) analysis using the BCR-ABL1Mbcr Kit (Ipsogen). The patient was regularly monitored in our outpatients clinic. In 2019, he expressed his will to stop IM and attempt treatment-free remission (TFR) [Citation7]. At that time, he was in deep molecular response (MR4.5), with undetectable disease according to the international scale (IS) lasting4 years.
After 3 months from IM withdrawal, the transcript increased to 1,8%IS, thus prompting therapy resumption. The patient immediately reached major molecular response (MR3 by IS). At present, he is in MR4 with undetectable disease.
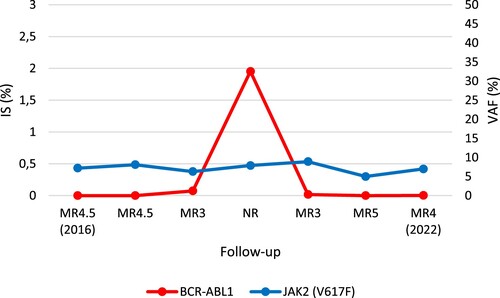
The rapid loss of MR, even if consistent with the available evidence [Citation7], prompt us to make a more in-depth genomic evaluation of the patient. On a PB sample at the time of the TFR failure, we performed targeted next-generation sequencing (NGS) analysis with a customized panel encompassing 26 genes involved in the pathogenesis of myeloid malignancies [Citation8]. A JAK2V617F mutation with a variant allele frequency (VAF) of 4.6% was detected. To trace the kinetics of the JAK2 variant and to investigate its relationship with the BCR-ABL1 rearrangement, RQ-PCR with JAK2MutaQuant Kit (Ipsogen) was performed on PB genomic DNA at different time points during the patient’s follow-up. While negative at diagnosis, the JAK2mutation was first detected 9 years later (VAF: 7.2%). At that time, the patient was 58 year-old, under treatment with IM, and in MR4.5 with undetectable disease. His blood counts were normal, and no clinical-laboratory signs of Ph-MPNwere present. Even after failure of the TFR and IM retreatment, the mutational burden of JAK2 V617F remained stable in multiple determinations, with minor fluctuations (min. 4.6% – max. 8.9%, mean: 6.9%), independent of BCR-ABL1kinetics (). At the last available time point, the patient, still in treatment with IM, was still in MR4 (as described above), the JAK2mutational burden was7%, yet no clinical-laboratory findings of Ph-MPN were detectable.
Figure 2. BCR-ABL1 and JAK2 (V617F) kinetics during the CML follow-up. JAK2 V617F remained stable with minor fluctuations independent of BCR-ABL1 kinetics. The evaluations shown refer to the 2016–2022 time range. IS: international scale; MR: molecular response; NR: non responder; VAF: variant allele frequency.
Discussion
A growing body of evidence has changed the paradigm of the relationship between BCR-ABL1and JAK2 in MPNs. Historically considered as driver mutant genes and mutually exclusive in the context of CML and Ph-MPN, respectively, their coexistence has now been established both in well-defined and in overlapping clinical contexts [Citation3,Citation4] (). In the context of CML, this phenomenon is not limited to JAK2but also comprisesCALRand MPL mutations, typically occurring in Ph-MPNs [Citation9].
Moreover, various studies have revealed that both the BCR-ABL1 and JAK2 variants may occur in the same clone or in independent ones, thus suggesting different pathological routes (Supplementary Table 1).
Notwithstanding, a common feature in those ‘double mutant’ scenarios is the presence of overlapping CML/Ph-MPN clinical-laboratory signs (). This is even more accentuated in the sequential detection of mutations, suggesting that those signs may indicate a more complex genomic architecture [Citation10,Citation11]. Furthermore, various studies have reported a certain interplay between BCR-ABL1 and JAK2 V617F, such that the latter burden may increase or decrease upon CML treatment with tyrosine kinase inhibitors (TKI) [Citation12]
Meanwhile, physicians are becoming ever more aware of CHIP in clinical practice; consistently, the biological weight of mutations should prompt considerations of the diagnostic and prognostic significance of CHIP in clinical decisions making. In this regard, both BCR-ABL1 and JAK2 V617F have been detected in otherwise healthy individuals [Citation5,Citation6]. Moreover, there is evidence demonstrating that a somatic JAK2 V617F mutation may be acquired in utero, eventually leading to overt disease within a time frame of about 30 years [Citation13].
Against this background, the case reported here illustrates a peculiar situation, never before described, in which we were able to demonstrate the occurrence ofJAK2 V617F in CML, with no overlapping clinical-laboratory features. JAK2 V617F was detectable nine years after the CML diagnosis, when the patient had reached the age of 58 years. The CHIP prevalence in individuals aged 50–60 years is expected to be roughly around 5% [Citation14]. Moreover, the JAK2 mutation burden remained essentially stable during the 5 years of follow-up, regardless of the BCR-ABL1kinetics () and in the absence of Ph-MPN signs.
The onset of the JAK2mutation in a CML patient in MR4.5 (nine years from diagnosis) with normal blood counts, the absence of any phenotype change and the independence of the JAK2 mutational burden from theBCR-ABL1 kinetics, are all suggestive of CHIP. The impact of this event on the disease outcome, even if seemingly irrelevant, has still to be explored. Notably, evidence suggests that the presence of the JAK2 mutation in the CML context could have a detrimental effect on the response to TKI therapy [Citation15,Citation16]; otherwise, no data are available regarding its impact on TFR. Could the presence of CH feed a pro-inflammatory state, eventually altering the equilibrium of TFR? In light of these data, the late appearance of the JAK2 gene mutation could be interpreted as a consequence of previous prolonged stimulation of the hematopoietic system operated by the CML. This phenomenon has already been demonstrated in other situations of hematopoietic proliferative stress exerted by various pathologies [Citation17,Citation18].
Conclusion
Based on our hypothesis, the presence of CHIP in CML patients could represent an indicator of the ‘biological age’ of the disease: in patients with an early CML diagnosis, the hematopoietic stress will be short-lived, and therefore the patient will not develop CHIP over time (or at least, not because of the CML). On the contrary, in a CML patient with a late diagnosis, the disease will be associated with an older ‘biological age’, due to a more extended period of hematopoietic proliferative stress, and there will be a greater risk of developing CHIP.
Further studies are needed to better characterize the epidemiological, biological, and clinical aspects of these cases. In our future practice, the patient's follow-up will hopefully reveal the role of JAK2: will it unleash its ‘driving force’, or will it stay in the shadows of CML, just as a bystander?
Etichs
The local ethics committee approved the study. Informed consent was obtained from the patient before study inclusion, in accordance with the Declaration of Helsinki. His record/information was anonymized and de-identified before analysis. Consent for publication was obtained before his enrollment in the present study.
Author contributions
Conceptualization: FT, CC, FA. Data curation: FT, CC, AZ, EP. Investigation: AZ, EP, LA, LA, NC, GT, CFM, IR, ARR, MRC, GS, PM. Writing - review & editing: FT, CC, FA. Supervision: FA. All authors approved the final version of manuscript.
Supplemental Table
Download MS Word (94.8 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405.
- Greenfield G, McMullin MF, Mills K. Molecular pathogenesis of the myeloproliferative neoplasms. J Hematol Oncol. 2021;14:103–121.
- Jelinek J, Oki Y, Gharibyan V, et al. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome–negative CML, and megakaryocytic leukemia. Blood. 2005;106:3370–3373. Available from: http://ashpublications.org/blood/article-pdf/106/10/3370/1636628/zh802205003370.pdf.
- Pieri L, Spolverini A, Scappini B, et al. Concomitant occurrence of BCR-ABL and JAK2V617F mutation. Blood. 2011;118:3445–3446. Available from: http://ashpublications.org/blood/article-pdf/118/12/3445/1340381/zh803811003445.pdf.
- Ismail SI, Naffa RG, Yousef AMF, et al. Incidence of bcr–abl fusion transcripts in healthy individuals. Mol Med Rep. 2014;9:1271–1276. Available from: https://pubmed.ncbi.nlm.nih.gov/24535287/.
- Sano S, Walsh K. Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis. J Cardiovasc Aging. 2021;1:5–8.
- Sharf G, Marin C, Bradley JA, et al. Treatment-free remission in chronic myeloid leukemia: the patient perspective and areas of unmet needs. Leukemia. 2020;34:2102–2112. Available from: https://www.nature.com/articles/s41375-020-0867-0.
- Cumbo C, Tota G, de Grassi A, et al. RUNX1 gene alterations characterized by allelic preference in adult acute myeloid leukemia. Leuk Lymphoma. 2021 May 24:1–5. doi:10.1080/10428194.2021.1929960.
- Langabeer SE. CALR-mutated myeloproliferative neoplasm. EXCLI J. 2020;19:86–88. doi:10.17179/excli2019-2063.
- Lorenzo M, Grille S, Stevenazzi M. Emergence of BCR-ABL1 chronic myeloid leukemia in a JAK2-V617F polycythemia vera. J Hematol. 2020;9:23–29.
- Bornhäuser M, Mohr B, Oelschlaegel U, et al. Concurrent JAK2(V617F) mutation and BCR-ABL translocation within committed myeloid progenitors in myelofibrosis. Leukemia. 2007;21:1824–1826. Available from: https://pubmed.ncbi.nlm.nih.gov/17476275/.
- Ali EAH, Al-Akiki S, Yassin MA. A case report of BCR-ABL-JAK2-positive chronic myeloid leukemia with complete hematological and major molecular response to dasatinib. Case Rep Oncol. 2021;14:690–694. Available from: https://www.karger.com/Article/FullText/514632.
- Williams N, Lee J, Mitchell E, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature. 2022;602:162–168.
- McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–1245.
- Fava C, Cambrin GR, Ferrero D, et al. Coexistence of a JAK2 mutated clone may cause hematologic resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Clin Lymphoma Myeloma. 2009;9:E41.
- Frikha R, Turki F, Kassar O, et al. Co-existence of BCR-ABL and JAK2V617F mutation in resistant chronic myeloid leukemia in the imatinib era: Is there a correlation? J Oncol Pharm Pract. 2021;27:1784–1789. doi:10.1177/1078155221991646?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0pubmed.
- Heyde A, Rohde D, McAlpine CS, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. 2021;184:1348–1361. e22.
- Dawoud AAZ, Tapper WJ, Cross NCP. Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia. 2020;34:2660–2672. Available from: https://pubmed.ncbi.nlm.nih.gov/32518416/.