
ABSTRACT
Objectives
Here we report two rare α-globin chain variants in two unrelated families: Hb Val de Marne [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C] and Hb Dongguan [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G]. Notably, HBA2: c.400A > C is an unreported new variant in the third exon of the α2 gene, and simple heterozygous unstable Hb Dongguan haematological characteristics are proposed for the first time.
Methods
Hb analysis was performed by using capillary electrophoresis (CE). Twenty-three common mutations were detected using a suspension array system. Mutations were identified by DNA sequencing.
Results
The CE results showed an abnormal peak with incomplete separation from Hb A at zone 8 in two members of Family 1. DNA sequencing confirmed the presence of Hb Val de Marne [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C]. Five members of Family 2 exhibited an abnormal peak at zone 11, and DNA sequencing confirmed the presence of Hb Dongguan [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G].
Conclusions
The discovery of HBA2: C.400A > C expands the existing spectrum of α-globin variants. The carriers of simple heterozygous Hb Dongguan generally do not have obvious clinical symptoms. The information in this study will help clinicians understand the screening, molecular diagnosis and clinical significance of Hb variants.
Introduction
Haemoglobinopathy is a common single-gene disease. Haemoglobin (Hb) variants are mainly composed of missense mutations (single amino acid substitutions) in the globin chain, resulting in abnormal or ‘varied’ Hb tetramers, affecting the molecular conformation and function of Hb, such as destabilizing Hb and altering Hb-O2 affinity. Nevertheless, most variants are stable and clinically silent [Citation1,Citation2]. To date, 1414 naturally occurring human Hb variants have been recorded in the HbVar database (http://globin.bx.psu.edu/hbvar); among them, 103 kinds of Hb with high oxygen affinity and 155 unstable Hbs. Guangdong Province, located in southern China, have been identified with a high incidence of thalassemia. The identification and diagnosis of new or unstable mutations are of great significance for laboratory research and clinical genetic counselling. In recent years, an increasing number of unstable variants and variants with the same protein change but different α1- or α2-globin gene mutations have been reported [Citation3–5]. Here, we present two rare α-globin chain variants. The first is the previously undescribed point mutation Hb Val de Marne [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C]. The second variant is the unstable variant Hb Dongguan [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G]. Simple heterozygous haematological characteristics and Hb analytical profiles based on capillary electrophoresis (CE) of this variant are described in this paper.
Materials and methods
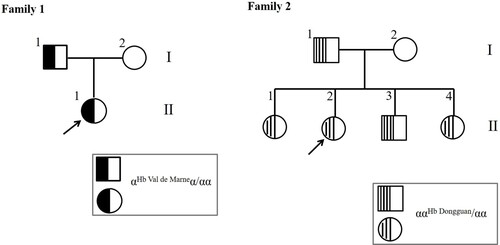
Family 1 and Family 2 were from Guangzhou, Guangdong Province. The proband in Family 1 (, Family 1; II-1) was a 35-year-old female who came to our hospital for genetic counselling due to fertility problems and then underwent thalassemia screening. The proband in Family 2 (, Family 2; II-2) was a 28-year-old woman who came to our hospital for routine thalassemia screening in the maternity insurance program. These two families were unrelated. All studies were approved by the Ethics Committee of Guangdong Women and Children Hospital.
Figure 1. Pedigrees of the families participating in this study.
Peripheral blood samples were collected using EDTA as an anticoagulant. Red blood cell indices were analysed using a Sysmex XN5000 automated haematology analyser (Sysmex Corporation, Kobe, Japan). Hb analysis (separation and quantitation of Hb sub-types) was performed using an automated capillary 2 electrophoresis system (Sebia, Lisses, France). All human materials were collected after informed consent was obtained from both families.
Genomic DNA was isolated from peripheral blood leukocytes using the Lab-Aid 820 automation system (Xiamen Zeesan Biotech Company, Fujian, China). To exclude the presence of twenty-three common mutations in individuals from southern China, including 3 deletions and 3 non-deletion mutations associated with α-thalassemia and 17 point mutations associated with β-thalassemia, molecular analysis was carried out in our laboratory using a suspension array system as previously described [Citation6].
Specific PCR amplification for α1-, α2-, and β-globin was performed as previously described [Citation7], and the genes were subsequently sequenced by Sangon Biotech (Shanghai, China) Co., Ltd. The sequences were compared to National Centre for Biotechnology Information reference sequences (HBA1: NC_000016.10: 176680-177522; HBA2: NC_000016.10: 172876-173710; HBB: NC_000011.10: 5225464-5227071) using Sequence Scanner software (Applied Biosystems). Polymorphism Phenotyping v2 (PolyPhen-2) was used to predict the possible impact of amino acid substitution on the structure and function of Hb.
Results
Routine haematology studies were carried out for all patients in the study, and the results are shown in . None of the members of either family had any abnormal haematology data, except II2 in Family 2 (microcytic hypochromic anaemia). Hb studies using CE revealed abnormal peaks for carriers of Hb Val de Marne or Hb Dongguan.
Table 1. Hematological and molecular data from families in this study.
Family 1: Hb Val de Marne [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C]
Hb analysis of the proband showed an obvious anomalous peak with incomplete separation from the Hb A peak, which together accounted for 96.5% of the total; the Hb A2 peak accounted for 2.8%, and the Hb A2-variant peak accounted for 0.7% (a). Her father was also a carrier with 96.9% Hb A + Hb variant, 2.5% Hb A2, and 0.6% Hb A2-variant.
Figure 2. Capillary electrophoresis results of the probands in the two families. a. Hb Val de Marne is characterized by incomplete separation of an abnormal peak from the Hb A peak; b: Hb Dongguan is characterized by an anomalous peak at zone 11.

Initial molecular screening of the proband and her parents determined that they were negative for α-thalassemia and β-thalassemia by the suspension array system. DNA sequencing of the α-globin genes revealed that both I1 and II1 were heterozygous for one previously undescribed single nucleotide substitution in which adenine (A) had mutated to cytosine (C) in exon 3 of HBA2 (HBA2: c.400A > C), replacing serine with arginine at position 133 of the α2-globin chain (a).
Figure 3. Sequencing result of the α-globin genes. a. Reverse sequencing result of the HBA2 gene in the proband. The arrow indicates the A > C heterozygous mutation substitution at codon 133 [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C]. b. Forward sequencing result of the HBA1 gene in the proband. The arrow indicates the A > C heterozygous mutation substitution at codon 52 [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G].
![Figure 3. Sequencing result of the α-globin genes. a. Reverse sequencing result of the HBA2 gene in the proband. The arrow indicates the A > C heterozygous mutation substitution at codon 133 [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C]. b. Forward sequencing result of the HBA1 gene in the proband. The arrow indicates the A > C heterozygous mutation substitution at codon 52 [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G].](/cms/asset/26af45d4-9a71-486f-bdd8-719e5cbcc294/yhem_a_2109324_f0003_oc.jpg)
Family 2: Hb Dongguan [α52(E6) Ser > Cys (TCT > TGT); HBA1: c.158C > G]
As shown in b, Hb analysis of the proband using a CE system identified, in addition to Hb A, Hb F and HbA2 + Hb A2-variant, an anomalous peak at zone 11. The quantitative Hb assessment of her parents and affected siblings is summarized in . Her father and her siblings were all carriers of Hb Dongguan with a normal red blood cell index, except for her sister (II1), who had typical microcytic hypochromic anaemia and a high red blood cell distribution width (RDW) of 19.6%. It is suspected that this individual had iron deficiency anaemia. This finding indicates that the heterozygotes for Hb Dongguan were generally asymptomatic, but the carriers had abnormal peaks ranging from 12% to 16% in zone 11 using CE. We used the suspension array system to screen for routine thalassemia with negative results.
Table 2. Hematological and molecular data from families in this study and related references.
DNA sequencing of the α-globin genes revealed that all members except the mother had a substitution at codon 52 of the α1-globin gene (TCT > TGT), resulting in replacement of serine with cysteine, and this mutation had been previously reported as the unstable Hb Dongguan variant (b).
Furthermore, we used PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) to predict the potential effects of amino acid substitution on protein function. Hb Val de Marne [HBA2: c.400A > C] was predicted to be ‘POSSIBLY DAMAGING’ with scores of 0.851 and 0.508 in HumDiv and HumVar, respectively. Hb Dongguan [HBA1: c.158C > G] was predicted to be ‘PROBABLY DAMAGING’, with scores of 1.000 and 0.990 in HumDiv and HumVar, respectively.
Discussion
Many Hb variants caused by missense mutations confer no physical abnormalities or clinical problems, making these variants easy to ignore. However, as laboratory personnel or clinicians, we must gain insight into how these substitutions interact with other positions on the gene and whether these mutations lead to alterations in oxygen affinity, changes in binding stability between globin chains, structural defects with thalassemia effects, or changes in physical behaviour. This acknowledgement has led an increasing number of laboratories to focus on identifying mutations and determining their clinical significance.
The mutation causing amino acid changes at codon 133 [α133(H16) Ser > Arg] corresponds to that found in Hb Val de Marne [Citation4,Citation8–11], which was reported to present normal haematologic parameters, with 16% abnormal Hb [Citation8]. Additionally, the Hb stability of Hb Val de Marne was normal [Citation8, Citation9]. However, the oxygen affinity was increased, while the haem-haem interaction was slightly reduced and the chloride ion binding and Bohr effect remained normal. The reason may be that serine (a133(H16)) is the residue at the C-terminus of the α-globin chain, which is in contact with the haem pocket (Phe a98(C5)) and other internal residues (Lys a99(G6), Ser a102(G9) and Ser a138 (H21)), and this replacement of Ser by Arg may help oxygen or water interact with the haem iron. Regarding the molecular characteristics of Hb Val de Marne, Edmond Shiu-Kwan Ma et al. [Citation4] first revealed an AGC > AGA mutation at codon 133 of the α2 gene in a Chinese family. Based on amino acid analysis, replacing serine at 133 of the codon of the α2 gene with arginine requires only a single mutation in the codon, which can be A→C (our report), C→G, or C→A (2004).
In our study, the proband of Family 1 is the first with a base A→C change at codon 133 of the α2 gene, and HBA2: c.400A > C is an unreported new single nucleotide substitution in the third exon of the α2 gene. The haematologic parameters of the carriers are consistent with those described in the literature. These normal parameters can easily lead to underestimation of this mutation in patients. We identified an obvious abnormal peak by CE, which was not completely separated from the Hb A peak. This property has never been described. CE appears to have a good ability to discriminate this variant in laboratory analysis.
Regarding the second variant in Family 2 that we reported, Hb Dongguan was first described in a 67-year-old Chinese male, who was compound heterozygous for Hb Dongguan and – –SEA α-thal deletion with an anomalous peak accounting for 37.8% of the total Hb and presented typical microcytic hypochromic anaemia [Citation12]. However, the previous study did not provide a haematological phenotype of the simple heterozygous Hb Dongguan. This limitation has been addressed in the current report. Our description of the haematological and molecular features of individuals carrying only the Hb Dongguan variant could facilitate laboratory detection and clinical understanding of this variant. Moreover, it has been reported that the isopropanol test is strongly positive [Citation12], indicating that Hb Dongguan is unstable in nature. Although the heterozygous state of the two variants in this study did not lead to obvious symptoms of anaemia, these variants did cause some functional changes, such as affinity changes and the generation of unstable Hb according to the literature [Citation12] and protein function prediction.
Conclusions
In this study, we characterized a novel α-globin gene mutation Hb Val de Marne [α133(H16) Ser > Arg (AGC > CGC); HBA2: c.400A > C] and performed haematological and molecular characterization. This variant was detected for the first time was localized by using CE. In addition, this study presents the haematological and molecular characterization of individuals carrying only the unstable variant Hb Dongguan.
Although the two Hb variants in our study are unlikely to produce significant haemolytic processes or anaemia in the simple heterozygous state, understanding the haematologic characteristics and Hb composition of these variants and obtaining a clear diagnosis of the type of variant are necessary for laboratory research and clinical genetic counselling. Moreover, it cannot be ignored that these asymptomatic variants may produce more relevant haematologic phenotypes if they are associated with α 0-thal defects, the clinical significance of which remains to be discovered by studying more cases in the future.
Declarations
Availability of data and material
All the data in this study were available in the figures and tables in the manuscript.
Author contributions
Cuize Yao and Danqing Qin performed the assays, analyzed data and wrote the manuscript; Jicheng Wang, Xiuqin Bao and Jie Liang collected the samples and performed the confirmed assays. Li Du designed the study and revised the article. All authors reviewed, edited, and approved the version to be submitted.
Ethics approval
This study was approved by the Ethics Committee of Guangdong Women and Children Hospital and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent to participate and for publication
The patients agreed to participate and be publicized in this study and signed the informed consent.
Acknowledgement
We thank the patients for their willingness to participate in this study. Financial support from the Science and Technology Program of Guangzhou, China (grant number 202002030390) is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem. 2000 Aug;46(8 Pt 2):1284–90. PMID:10926923.
- Thom CS, Dickson CF, Gell DA, et al. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harbor Perspect Med. 2013;3:a011858.
- Chan NC, Chow K-H, Leung RF, et al. First Report of Hb Kent [β37 (C3) Trp→ Cys (TG G> TG C) HBB: c. 114G> C] in a Chinese Family. Hemoglobin. 2017;41:283–285.
- Ma ESK, Chan AYY, Lee ACW. Molecular Characterization of Hb Val de Marne [α133 (H16) Ser→ Arg; AG C→ AG A;(α2)] in a Chinese Family. Hemoglobin. 2004;28:213–216.
- Wajcman H, Traeger-Synodinos J, Papassotiriou I, et al. Unstable and thalassemic α chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin. 2008;32:327–349.
- Yin A, Zhang L, Luo M, et al. Development of bead-based suspension array technology for the diagnosis of thalassemia. Am J Hematol. 2014 Dec;89(12):1158–1159. doi: 10.1002/ajh.23830. PMID: 25118110.
- Qin D, Du L, Wang J, et al. Compound heterozygosity for hemoglobin variant Hb-Broomhill and the Southeast Asian α-thalassemia deletion does not worsen outcome: a case report of two unrelated patients. J Int Med Res. 2020;48:0300060520967825.
- Wajcman H, Kister J, M'rad A, et al. Hb Val de Marne [α133 (H16) Ser→ Arg]: a new hemoglobin variant with moderate increase in oxygen affinity. Hemoglobin. 1993;17:407–417.
- Owen M, Hendy J. Hb Footscray or α133 (H16) Ser→ Arg: a new hemoglobin variant. Hemoglobin. 1994;18:19–27.
- Akbari MT, Hamid M. (2012). Identification of α-globin chain variants: A report from Iran.
- Okumura J, Shimauti E, Silva D, et al. Hemoglobin (Hb) Val de Marne (Hb Footscray) in Brazil: the first case report. Genetics and Molecular Research: GMR. 14 Jul 2016;vol. 15,2; doi:10.4238/gmr.15028294
- Chen W-D, Ren Y-X, Wang Y-J, et al. Compound Heterozygosity for an Unstable Novel Hemoglobin Variant, Hb Dongguan [α52 (E1) Ser→ Cys (TC T> TGT); HBA1: c. 158C> G], and the––SEA (Southeast Asian) α-Thalassemia Deletion. Hemoglobin. 2019;43:286–288.