
ABSTRACT
Lymphoma-associated hemophagocytic syndrome (LAHS) is a rare and life-threatening clinical syndrome with rapidly deteriorating health and high mortality. We retrospectively analyzed clinical features and prognostic factors from 117 patients diagnosed with LAHS. The cumulative incidence rate of LAHS was 4.0% (117/2906). Patients were classified into B-cell LAHS (B-LAHS, n = 22) and T/natural killer (NK)-cell LAHS (T/NK-LAHS, n = 95) groups. Patients with T/NK-LAHS were younger and had lower neutrophil counts and fibrinogen values, higher LDH and transaminase levels, and were more likely to develop hemophagocytic syndrome (HPS) during the clinical course than those with B-LAHS. The median survival time for the entire cohort was 57 days, and for the T/NK-LAHS and B-LAHS groups, it was 52 and 154 days, respectively, after the diagnosis of LAHS. Patients with B-LAHS had superior 1-year OS (p = 0.003, 36.4% versus 14.5%) compared with those with T/NK-LAHS. Prognostic factor analysis revealed that elevated LDH levels (LDH > 1000 IU/L) (p = 0.004), T/NK-cell lineage (p < 0.001) and HPS onset at relapse (p = 0.001) were strongly associated with early death. For patients diagnosed with T/NK-LAHS, in addition to LDH levels and HPS onset status, high EBV-DNA copies (≥4,450 copies/mL) (p = 0.016) were also related to poor prognosis of T/NK-LAHS.
Introduction
Hemophagocytic syndrome (HPS) or hemophagocytic lymphohistiocytosis (HLH) is a life-threatening clinical syndrome characterized by sustained pathologic immune activation resulting in an uncontrolled hyperinflammatory response with rare morbidity and high mortality [Citation1]. HPS is divided into two different forms, including primary and secondary genres. Primary HPS principally originates from chromosomal or genetic abnormalities and occurs in pediatric patients, while secondary HPS is ordinarily caused by serious underlying disorders, including malignancies, infections, and autoimmune diseases and develops at any age, especially in adults. Malignancy is one of the most common triggers found in adults with secondary HPS, and lymphoma accounts for 67% in adults in the context of reported neoplasm-associated HPS [Citation2].
HPS can sometimes mask certain lymphomas that are difficult to diagnose, such as intravascular B cell lymphoma, T-cell histiocyte-rich large B-cell lymphoma, and angioimmunoblastic T-cell lymphoma. HPS is characterized by fever, hepatosplenomegaly, cytopenia, coagulopathy, multiple organ failure, and hemophagocytosis in bone marrow (BM) or other viscera [Citation3,Citation4]. According to previous reports, most cases of lymphoma-associated hemophagocytic syndrome (LAHS) are related to T cell or natural killer (NK)/T cell lymphoma, and LAHS secondary to B cell lymphoma is quite infrequent [Citation5–9]. The outcome of LAHS is quite poor, with a median survival time of 1–2 months [Citation10,Citation11,Citation12]. The first-line treatment for LAHS is still ill-defined due to its low rate of incidence, difficulty in diagnosis, fast progression, and poor physical condition of individuals at the time of diagnosis. Furthermore, few investigations in LAHS with a large sample size have been carried out because of the rarity and heterogeneity of this syndrome [Citation13]. Therefore, we performed this retrospective study, with the largest sample size to-date, for the purpose of describing the clinical, biological and treatment-related characteristics of patients diagnosed with LAHS. In addition, this study sought to evaluate the overall survival (OS) and the mortality-associated parameters within this specific group.
Patients and methods
Patients, diagnostic criteria and data collection
We retrospectively identified 2906 patients diagnosed with lymphoma in our hospital between August 2008 and July 2019. The diagnosis of lymphoma was confirmed by pathology and immunohistochemistry based on the WHO classification [Citation14].
In 1991, the Histiocyte Society put forward a set of 5 diagnostic criteria applied to the prospective HLH-1994 clinical trial [Citation15]. These criteria were revised for HLH-2004 [Citation16] and patients fulfilling at least five of the following eight criteria were included in the present study: (1)fever (temperature≥38.5°C); (2) splenomegaly; (3) cytopenia affecting two or three lines in the peripheral blood (hemoglobin < 90 g per liter(/L), platelets < 100 × 109/L, neutrophils < 1.0 × 109/L), (4) hypertriglyceridemia and/or hypofibrinogenemia (fasting triglycerides≥3.0 mmol/L, fibrinogen≤1.5 g/L); (5) hyperferritinemia (ferritin≥500 ng/mL); (6) soluble CD25 ≥ 2400 U/ml, (7) low or absent NK cell activity; and (8) hemophagocytosis in BM or other organs. Due to the limitations of the inspection methods, NK-cell activity was undetectable in our center. According to HLH-2004 criteria, patients who met 5 of 8 diagnostic criteria were included as LAHS.
Clinical and laboratory data from medical records were collected including age, gender, the Eastern Cooperative Oncology Group (ECOG) performance status (PS), lymphoma characteristics (B symptoms, Ann Arbor stage, organ involvement, BM invasion, extranodal involvement, lactate dehydrogenase (LDH), Epstein–Barr virus DNA (EBV-DNA) copies, the International Prognostic Index (IPI)), infection status, and diagnostic indicators associated with HPS including complete blood count, coagulation function, liver function, hemophagocytosis etc. Because of the testing conditions, when ferritin levels exceeded 2000ng/mL and soluble CD25 levels surpassed 7500 U/mL, no exact value was reported for a portion of patients. Consequently, these parameters were not compared between patients with different lymphoma subtypes. The stage of lymphoma was assessed through the Ann Arbor staging system, and systemic imaging, including computed tomography (CT) or magnetic resonance (MR) of the nasopharynx, neck, chest and abdomen or positron emission tomography/CT (PET/CT), was employed to evaluate the range of disease. Concurrent infection was defined by any infectious diseases during hospitalization for the LAHS.
Statistical analysis
In the descriptive analysis, continuous variables were presented as medians with interquartile ranges (IQRs). OS was defined as the time from HPS diagnosis to death due to any cause or the date of last follow-up. Survival functions were calculated by the Kaplan-Meier method, and differences were compared using the log-rank test. Univariate analysis was performed with the log-rank test to confirm the prognostic variables. We compared categorical characteristics between the B cell and T/NK cell lymphoma groups using the Mann–Whitney U or chi-squared test, where appropriate. Statistical significances were established if a p value< 0.05. All statistical analyses were carried out using SPSS software, version 26.0 (IBM SPSS).
Results
Patient characteristics
A total of 2906 patients were diagnosed with lymphoma between August 2008 and July 2019. A total of 117 patients (85 male, 32 female) met the HPS diagnostic criteria and had complete follow-up data, with an incidence of 4.0% (117/2906). The baseline clinical and biological characteristics of patients diagnosed with LAHS are presented in . The median age was 44 years (range, 13–72 years). According to the occurrence time of HPS as described by previous report [Citation17], 82 patients exhibited HPS at initial diagnosis of lymphoma, and 35 patients presented with HPS during the clinical course, especially during disease progression. The most common histological subtype among T/NK cell lymphomas was nasal type, extranodal natural killer (NK)/T cell lymphoma (ENKTL), which occurred in 83 of 95 patients (87.4%) and accounted for 70.9% of all LAHS. Among B cell lymphomas, the most frequent pathological subtype was diffuse large B cell lymphoma (DLBCL), with a proportion of 50% in B-LAHS. All patients presented with fever, cytopenia and elevated ferritin levels. Hemophagocytosis was identified in 56 (53.3%) of 105 patients based upon BM histology. 85 cases (72.6%) developed hepatosplenomegaly and 105 cases (89.7%) were classified into advanced stages (Ann Arbor III/IV). LDH values were elevated in 115 (98.3%) of 117 patients, with a median level of 732 IU/L (range, 207–4379 IU/L). All patents had ferritin≥500 ng/mL, and 94% (110/117) of patients had ferritin values of more than 2000ng/mL without exact concentrations. The data for soluble CD25 levels were available for 76 patients. Among the 76 patients, 3 patients had soluble CD25 level < 2400 U/mL, 27 patients had elevated levels of > 2400 U/mL with exact values and the highest value of 36685 U/mL, and 44 patients had elevated levels > 7500 U/mL without precise quantitative values. The detection of EBV-DNA was available for 86 patients. Among patients with data for peripheral blood EBV-DNA status, 10 (11.6%) had a negative EBV-DNA load, 76 (88.4%) had detectable viral titers and the highest value was 2.23 × 108 copies/mL, which occurred in ENKTL (data not shown).
Table 1. Characteristics of patients with LAHS.
Treatment
Treatment variables were decided by the pathogenesis of HPS and individual physician management. Among the 22 patients with B-cell lymphoma-associated HPS (B-LAHS), 11 patients were treated by HLH-1994 [Citation18] or HLH-2004 protocols [Citation16]. Six patients received anthracycline-based combination chemotherapy regimens combined with or without rituximab or other cytotoxic drugs such as etoposide, and one of these patients had autologous stem cell transplantation (auto-HSCT) after combination chemotherapy. Four patients did not receive chemotherapy and were put on supportive care because of poor performance status, and one patient accepted steroids, rituximab and cyclosporine A. Of the 95 patients diagnosed with T/NK cell lymphoma-associated HPS (T/NK-LAHS), 23 patients received HLH-1994 or HLH-2004 protocols, 43 acquired asparaginase-based combined chemotherapy, 23 accepted nonasparaginase-based combined chemotherapy, and four of these patients received allogeneic hematopoietic stem cell transplantation (allo-HSCT) after induction therapy. Because of poor performance, 4 cases underwent optimal supportive treatment, one received steroids and one was given chidamide.
Comparison of variables between patients with B-LAHS and T/NK-LAHS
Comparisons of characteristics between the patients with B-LAHS and T/NK-LAHS are shown in . The results revealed that patients with T/NK-LAHS were younger (P = 0.032) and were more likely to develop HPS during the clinical course (P = 0.018). Laboratory data showed that patients with T/NK-LAHS had lower neutrophil counts (P = 0.045) and fibrinogen values (P < 0.001) and higher LDH (P = 0.046) and transaminase levels (alanine aminotransferase and aspartate aminotransferase, P < 0.001 and P = 0.002 respectively). Patients with B-LAHS had lower hemoglobin (HB) values than those with T/NK-LAHS. No significant differences were observed in gender, ECOG PS, hepatosplenomegaly, tumor stage, BM involvement, IPI score, extranodal sites, concurrent infection etc.
Table 2. Comparisons between patients with B-LAHS and T/NK-LAHS.
Outcome and survival analysis
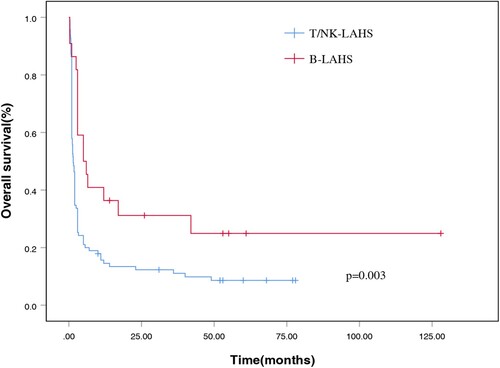
A total of 109 patients (93.2%) received a specific treatment for LAHS or primary tumor. The median time from HPS diagnosis to treatment was 9 days (range: 0–56 days). The median OS for the entire cohort was 57 days after the diagnosis of LAHS. For the T/NK-LAHS and B-LAHS groups, the median OS were 52 and 154 days, respectively. The 1-month survival rate was 67.5%, the 2-month survival rate was 42.7%, the 3-month survival rate was 35.0%, the 6-month survival rate was 25.6%, and the 1-year survival rate was 18.7% (). Patients with B-LAHS had superior OS (p = 0.003, with a 1-year OS of 36.4% versus 14.5%, ) compared with those with T/NK-LAHS.
Figure 1. Survival curves of 117 patients with lymphoma-associated hemophagocytic syndrome.
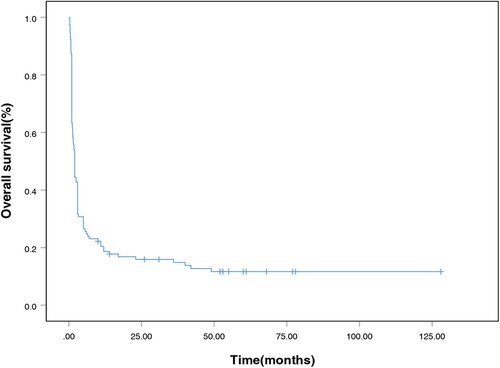
Figure 2. Overall survival curves for patients with B cell lymphoma-associated hemophagocytic syndrome and T/natural killer cell lymphoma-associated hemophagocytic syndrome.
Prognostic factors
When assessing the prognosis of LAHS, it is difficult to accurately determine whether patients died from HPS, lymphoma progression, or treatment-associated toxicity. Consequently, we adopted the concept of ‘early death’, defined as death within the first 120 days after the diagnosis of HPS as previously described [Citation19]. For LDH concentration and EBV-DNA titer, there is no standard cutoff value used to predict the prognosis of LAHS. We set the cutoff values of LDH and EBV-DNA to 1000 IU/L [Citation9,Citation20] and 4450 copies/mL [Citation21], respectively, according to previous reports. In the univariate analysis, elevated LDH level (LDH > 1000 IU/L) (p = 0.004), T/NK-cell lineage (p < 0.001) and HPS onset at relapse (p = 0.001) had the strongest association with early death, while other factors showed no significance for survival (). Because of limited factors, Cox multivariate analysis could not be conducted.
Table 3. Univariate analysis of the prognostic factors of LAHS.
In this study, T/NK-LAHS accounted for the majority of patients diagnosed with LAHS. Therefore, we further performed univariate analysis to evaluate the association between prognostic factors and survival by histological subtypes of T/NK-LAHS using the concept of ‘early death’ as well. Similar to the entire cohort, elevated LDH levels (LDH > 1000 IU/L) (p = 0.042) and HPS onset at relapse (p = 0.037) were strongly associated with early death (). In addition, high EBV-DNA copies (≥4,450 copies/mL) (p = 0.016) were also related to the prognosis of T/NK-LAHS ().
Table 4. Univariate analysis of the prognostic factors of T/NK-LAHS.
Discussion
LAHS is a disease characterized by defective cytotoxic cell control of an initial immune response leading to uncontrollable macrophage activation and hyperinflammation [Citation22]. Currently, data as reported in the literature is insufficient for predicting the incidence of LAHS [Citation8–13,Citation17,Citation19]. In the present study, LAHS occurred in 4.0% of all patients with lymphoma. Most cases of LAHS were associated with T/NK-cell lymphoma, and LAHS cases secondary to B-cell lymphoma were relatively rare, which was consistent with previous reports [Citation8,Citation10,Citation23]. Among patients diagnosed with LAHS, ENKTL was the most frequent pathological subtype and accounted for 70.9% of all cases, while DLBCL accounted for 9.4% in this cohort. Many previous studies have demonstrated that T/NK-LAHS has a worse prognosis and higher mortality rate than B-LAHS [Citation19, Citation24, Citation25]. Additionally, in our study, patients with T/NK-LAHS had worse OS (1-year OS of 14.5% versus 36.4%) than those with B-LAHS.
In the present study, patients had a variety of clinical manifestations. All patients presented with cytopenia, fever, and high ferritin levels. Hepatosplenomegaly was observed in 72.6% of cases. BM involvement and hemophagocytosis were present in more than half of the cases. The clinical characteristics of patients with B-LAHS and T/NK-LAHS were compared in this study. T/NK-LAHS individuals were younger than those with B-LAHS, which was consistent with previous reports [Citation10, Citation19]. Most of the 117 patients (70.1%) developed LAHS at the time of lymphoma diagnosis. Subgroup analysis found that B-cell lymphoma patients were more likely to develop HPS at lymphoma onset than T/NK-cell lymphoma patients. Laboratory findings, including cytopenia, usually beginning with thrombocytopenia evolving into severe pancytopenia, hypofibrinogenemia, elevated LDH concentration, and liver dysfunction, were more frequent in T/NK-LAHS than in B-LAHS.
We further investigated the prognostic factors associated with LAHS survival. Univariate analysis of prognostic factors demonstrated that early death after developing LAHS was associated with T/NK-cell lineage, elevated LDH level (LDH > 1000 IU/L), and HPS onset at relapse. To date, the cumulative incidence of LAHS has been higher in patients with T/NK cell lymphoma than in those with B-cell lymphoma [Citation8, Citation10, Citation19, Citation23]. We carried out subgroup analysis in T/NK-LAHS, and the results indicated that in addition to LDH level and HPS onset status, high EBV-DNA titers were also related to early mortality.
EBV is a widely spread γ-herpes virus that promotes the proliferation of infected lymphocytes by expressing growth-promoting latency genes and membrane proteins [Citation26]. EBV-DNA is a well-known indirect tumor burden index because ENKTL tumor cells are permanently infected with virus, and the diagnosis of ENKTL is based on the intensity of positive virus copies in in situ hybridization [Citation27]. EBV positivity has been thought to be significantly associated with the poor survival of ENKTL and DLBCL [Citation28,Citation29]. Viral infections are the common reason for secondary HLH, and EBV is the virus most frequently associated with HLH [Citation30]. In a global survey carried out in Japan, EBV-HLH accounted for approximately 30% of HLH, followed by other infection- or lymphoma-associated HLH [Citation30]. EBV-DNA is defined as positive if there are any detectable concentrations in plasma, however, EBV titers are typically high in patients with LAHS; thus, this definition of EBV positivity may not be suitable. We previously reported that an EBV-DNA titer detected in plasma at more than 4450 copies/mL was a predictor for developing NK/T-LAHS [Citation21]. Hence, we used 4450 copies/mL as the cutoff value. In the present study, in addition to LDH level and HPS onset status, an EBV-DNA titer more than 4450 copies/ml was also correlated with poor survival in T/NK-LAHS. The expression of programed death protein-1 (PD-1) on CD8 T cells increased after EBV infection [Citation31]. Nivolumab provides a potential cure for relapsed/refractory EBV-HLH, most likely by restoring a defective anti-EBV response [Citation32]. In this study, three patients with T/NK-LAHS underwent treatment with programed death-1 inhibitor after induction therapy. Of these, one survived for more than 3 years, one survived for more than 2 years, and one died within 3 months.
According to HLH-2004 diagnostic criteria, ferritin > 500 ng/mL and soluble CD25 > 2400 U/mL were used to diagnose HPS. However, in our study, all patients had ferritin > 500 ng/mL, and most patients (73/76) had soluble CD25 > 2400 U/mL in this study; thus, these cutoff values were unsuitable for prognostic analysis. A recent study proposed an optimized HLH inflammatory (OHI) index comprised of elevated levels of soluble CD25 (>3900 U/mL) and ferritin (>1000 ng/mL), showing diagnostic and prognostic value in HLH [Citation33]. However, elevated ferritin (>1000 ng/mL) and soluble CD25 (>3900 U/mL) levels did not have sufficient discriminatory power for predicting early-death in this study. This could be partially explained by the fact that most of the patients had ferritin > 1000 ng/mL (94.3%) and soluble CD25 > 3900 U/mL (86.8%), and the unbalanced distribution of patients resulted in a negative prognostic value for these two indicators.
The clinical characteristics of LAHS are caused by hypercytokinemia in terms of tumor necrosis factor-α, interferon-gamma, interleukin (IL)−10, and IL-18 discharged by highly activated lymphocytes and macrophages [Citation34]. In patients diagnosed with LAHS, etoposide-containing treatment should be taken into serious consideration [Citation35]. Etoposide can inhibit topoisomerase II, resulting in the breakage of double-stranded DNA [Citation36], and it can selectively consume activated T cells to inhibit the production of inflammatory cytokines and improve survival in HPS [Citation37]. A previous study showed that patients receiving etoposide-based therapeutic regimens had a lower rate of early mortality than those who did not accept etoposide-containing treatment [Citation13]. However, our study did not find that etoposide-based treatment had a significant influence on the prognosis of LAHS patients. This could be partially explained by the fact that most patients (82.9%) received etoposide-containing therapy, and the survival differences caused by etoposide treatment were difficult to distinguish.
Excessive cytokines can bind to a specific set of receptors and lead to activation of the downstream JAK-STAT dependent signaling pathway, promoting the transcription of abundant downstream proinflammatory genes. Blocking the JAK-STAT pathway by inhibiting the downstream signaling of HPS related cytokines may be more effective in alleviating HPS related immunopathology. Ruxolitinib inhibits both JAK1 and JAK2, which function downstream of interferon-gamma and several other cytokines, such as IL-2 IL-6, IL-10, and IL-12. Recently, relevant studies have confirmed the efficacy of ruxolitinib in HPS [Citation38, 39,40]. However, in this study, patient data was included in the relatively early years, and they did not accept ruxolitinib, so we could not further evaluate its efficacy in LAHS.
In fact, in addition to the malignancy itself, malignancy immunotherapy such as immune-checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR)-T cells, can also lead to HLH. Several reports have confirmed that nivolumab could result in HLH, which could be relieved with corticosteroids [Citation41,Citation42,Citation43]. CAR-T cell therapy is always associated with cytokine release syndrome (CRS), which is an acute toxicity resulting in hyperinflammation. The most serious form of CRS progresses to fulminant HLH. The diagnostic criteria for CAR-T associated HLH include ferritin > 10,000 ng/mL, and two of the following abnormalities: grade≥3 organ damage involving the liver, kidney, or lung or haemophagocytosis in the bone marrow or other organs [Citation44]. Anti-IL-6 therapy, corticosteroids, cytokine-directed therapy or etoposide could be considered for the management of CAR-T associated HPS [Citation44].
The limitations of the present study are primarily its single-institution, retrospective characteristics. Furthermore, the incidence rate of LAHS may be undervalued because NK-cell activity could not be detected, and the soluble CD25 levels were not tested for some patients. However, to our knowledge, this is the largest single center study for LAHS, a rare entity, conducted in an Asian country to date.
In general, LAHS is a rare disorder with rapid progression and poor prognosis, and there are currently no efficient treatment protocols. We compared clinical features between B-LAHS and T/NK-LAHS, and established prognostic factors for LAHS based on a large cohort of affected patients, which would be valuable for risk stratification.
Declarations
Ethics approval and consent to participate
All data in this study were approved by the Ethics Committee on Biomedical Research of our hospital and were in line with the Helsinki Declaration. This study is a retrospective study without informed consent.
Authors’ contributions
LQZ designed this study and revised the manuscript. NL gathered and analyzed the data, executed the research and finished the article. MJ, WCW and HJZ offered valuable suggestions. All authors read and approved the final manuscript and submitted version. All authors agreed to be responsible for their own contributions and to guarantee the veracity or integrity of any part of the research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69–86.
- Ramos-Casals M, Brito- Zerón P, López -Guillermo A, et al. Adult haemophagocytic syndrome. The Lancet. 2014;383(9927):1503–1516.
- Allory Y, Challine D, Haioun C, et al. Bone marrow involvement in lymphomas With hemophagocytic syndrome at presentation. Am J Surg Pathol 2001;25(7):865–874.
- Kumakura S. Hemophagocytic syndrome. Intern Med. 2005;44:278–280.
- Cheung MM, Chan JK, Lau WH, et al. Primary non-hodgkin’s lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol. 1998;16:70–77.
- Falini B, Pileri S, De Solas I, et al. Peripheral T-cell lymphoma associated with hemophagocytic syndrome [see comments]. Blood. 1990;75:434–444.
- Florena AM, Iannitto E, Quintini G, et al. Bone marrow biopsy in hemophagocytic syndrome. Virchows Arch. 2002;441:335–344.
- Shimazaki C, Inaba T, Shimura K, et al. B-cell lymphoma associated with haemophagocytic syndrome: a clinical, immunological and cytogenetic study. Br J Haematol 1999;104:672–679.
- Li F, Li P, Zhang R, et al. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): An analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Medical Oncology. 2014;31:902.
- Han AR, Lee HR, Park BB, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol 2007;86(7):493–498.
- Takahashi N, Miura I, Chubachi A, et al. A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol 2001;74:303–308.
- Li N, Zhang L, Liu J, et al. A clinical study of 21 patients with hemophagocytic syndrome in 295 cases diagnosed with nasal type, extranodal nature killer/T cell lymphoma. Cancer Biol Ther 2017;18(4):252–256.
- Bigenwald C, Fardet L, Coppo P, et al. A comprehensive analysis of lymphoma-associated haemophagocytic syndrome in a large French multicentre cohort detects some clues to improve prognosis. Br J Haematol 2018;183(1):68–75.
- Sabattini E, Bacci F, Sagramoso C, et al. WHO classification of tumors of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102:83–87.
- Henter JI, Elinder G, Ost A. The FHL study group of the histiocyte society. diagnostic guidelines for hemophagocytic lymphohistiocytosis. Semin Oncol. 1991;18(1):29–33.
- Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131.
- Chubachi A, Miura AB. Lymphoma-associated hemophagocytic syndrome in adults: a review of the literature. Rinsho Ketsueki. 1994;35:837–845.
- Henter JI, Arico M, Egeler M, et al. HLH-94: A treatment protocol for hemophagocytic lymphohistiocytosis. Med Pediatr Oncol 1997;28:342–347.
- Sano H, Kobayashi R, Tanaka J, et al. ATF7IP as a novel PDGFRB fusion partner in acute lymphoblastic leukaemia in children. Br J Haematol 2014;165:836–841.
- Jia J, Song YQ, Lin NG, et al. Clinical features and survival of extranodal natural killer/T cell lymphoma with and without hemophagocytic syndrome. Ann Hematol 2016;95(12):2023–2031.
- Li N, Jiang M, Wu WC, et al. How to identify patients at high risk of developing nasal-type, extranodal nature killer/T-cell lymphoma-associated hemophagocytic syndrome. Front Oncol. 2021;11:704962.
- Weaver LK, Behrens EM. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol. 2014;26:562–569.
- Bhagwati NS, Oiseth SJ, Abebe LS, et al. Intravascular lymphoma associated with hemophagocytic syndrome: a rare but aggressive clinical entity. Ann Hematol 2004;83:247–250.
- Yu JT, Wang CY, Yang Y, et al. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol 2013;92(11):1529–1536.
- Bhatt NS, Oshrine B, An Talano J. Hemophagocytic lymphohistiocytosis in adults. Leuk Lymphoma. 2019;60(1):19–28.
- Lei KI, Chan LY, Chan WY, et al. Quantitative analysis of circulating cell-free epstein-barr virus (EBV) DNA levels in patients with EBV -associated lymphoid malignancies. Br J Haematol. 2000;111(1):239–246.
- Wang L, Wang H, Wang JH, et al. Post-treatment plasma EBV-DNA positivity predicts early relapse and poor prognosis for patients with extranodal NK/T cell lymphoma in the era of asparaginase. Oncotarget. 2015;6(30):30317–30326.
- Fei Q, Tian XK, Wu J, et al. Prognostic significance of epstein–barr virus DNA in NK/T-cell lymphoma: a meta-analysis. Onco Targets Ther. 2018;Volume 11:997–1004.
- Castillo JJ, Beltran BE, Miranda RN, et al. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2018 update on diagnosis, risk-stratification and management. Am J Hematol 2018;93(7):953–962.
- Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. 2018;18(2):91–104.
- Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol 2007;86:58–65.
- Liu PP, Pan XY, Chen C, et al. Nivolumab treatment of relapsed/refractory epstein-barr virus-associated hemophagocytic lymphohistiocytosis in adults. Blood. 2020;135(11):826–833.
- Zoref-Lorenz A, Murakami J, Hofstetter L, et al. An improved index for diagnosis and mortality prediction in malignancy associated hemophagocytic lymphohistiocytosis. Blood. 2021;139(7):1098–1110.
- Larroche C, Mouthon L. Pathogenesis of hemophagocytic syndrome (HPS). Autoimmun Rev. 2004;3:69–75.
- Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125:2908–2914.
- Johnson TS, Terrell CE, Millen SH, et al. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. The Journal of Immunology. 2014;192:84–91.
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematol Am Soc Hematol Educ Program. 2009;1:127–131.
- Broglie L, Pommert L, Rao S, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Advances. 2017;1(19):1533–1536.
- Wang J, Wang Y, Wu L, et al. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica. 2020;105(5):e210–e212.
- Slostad J, Hoversten P, Haddox CL, et al. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a single patient experience. Am J Hematol 2018;93(2):e47–e49.
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150–153.
- Malissen N, Lacotte J, Du-Thanh A, et al. Macrophage activation syndrome: a new complication of checkpoint inhibitors. Eur J Cancer. 2017;77:88–89.
- Takeshita M, Anai S, Mishima S, et al. Coincidence of immunotherapyassociated hemophagocytic syndrome and rapid tumor regression. Ann Oncol. 2017;28(1):186–189.
- Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nature Reviews Clinical Oncology. 2018;15:47–62.