
ABSTRACT
Objective
The mechanism of immunomodulatory drugs (IMiDs) resistance to multiple myeloma (MM) cells has been gradually demonstrated by recently studies, and some potential novel strategies have been confirmed to have antimyeloma activity and be associated with IMiD activity in MM.
Methods
This article searched the Pubmed library, reviewed some recently studies related to IMiD resistance to MM cells and summarized some potent agents to improve IMiD resistance to MM cells.
Results
Studies have confirmed that cereblon is a primary direct protein target of IMiDs. IRF4 not only is affected by the IKZF protein but also can directly inhibit the expression of BMF and BIM, thereby promoting the survival of MM cells. Additionally, the expression of IRF4 and MYC also plays an important role in three important signaling pathways (Wnt, STAT3 and MAPK/ERK) related to IMiD resistance. Notably, MYC, a downstream factor of IRF4, may be upregulated by BRD4, and upregulation of MYC promotes cell proliferation in MM and disease progression. Recently, some novel therapeutic agents targeting BRD4, a histone modification-related ‘reader’ of epigenetic marks, or other important factors (e.g. TAK1) in relevant signaling pathways have been developed and they may provide new options for relapse/refractory MM therapy, such as BET inhibitors, CBP/EP300 inhibitors, dual-target BET-CBP/EP300 inhibitors, TAK1 inhibitors, and they may provide new options for relapsed/refractory MM therapy.
Conclusions
Accumulated studies have revealed that some key factors associated with the mechanism of IMiD resistance to MM cells. Some agents represent promising new therapeutics of MM to regulate the IRF4/MYC axis by inhibiting BRD4 expression or signaling pathway activation.
Multiple myeloma (MM) is the second most common hematological malignancy and is characterized by malignant proliferation and tissue invasion of monoclonal plasma cells in bone marrow; in addition, MM cells retain the biological characteristics of antibody production and secretion[Citation1–3]. Moreover, 50-60% of MM cases are hyperdiploid MM, including genetic abnormalities such as Ras mutation, TP53 mutation and IgH gene translocation[Citation3–6]. According to the International Staging System (ISS) and the revised ISS criteria, MM patients with high-risk stratification usually have worse survival. Recent studies have suggested that MM with double or triple hits may have the worst prognosis, and the criteria are any two or three of the following diagnostic parameters: IgH gene translocation (t(4; 14), t(14; 16) or t(14; 20)), 17p deletion, 1q gain and TP53 mutation[Citation5, Citation6]. Furthermore, minimal residual disease (MRD), as an independent prognostic marker, is significantly correlated with progression-free survival (PFS) and overall survival (OS) in MM patients[Citation7–12]. The persistence of MRD may be the primary cause of recurrence in MM patients since the residual cells may be drug resistant[Citation13]. Moreover, in the bone marrow microenvironment (BMME), interaction between stromal cells and MM cells involving cytokines, receptors and adhesion molecules promotes the proliferation, angiogenesis and drug resistance of MM cells[Citation14].
Although there are antimyeloma agents, such as traditional chemotherapeutic drugs, proteasome inhibitors, monoclonal antibodies (anti-CD38 and anti-CS1), and B-cell maturation antigen (BCMA) or CD19 chimeric antigen receptor (CAR)-T-cell therapy[Citation15–18], immunomodulatory drugs (IMiDs) are one of the most common basic drugs used to treat MM for several reasons: i) they have strong antimyeloma activity, ii) they are readily available to patients, iii) they are conveniently orally administrated, and iv) they have definitive antimyeloma effects. IMiDs include thalidomide and its derivatives, and fourth-generation have been developed. First-generation agents include thalidomide, which can effectively inhibit tumor necrosis factor α (TNF-α), accelerate abnormal monoclonal plasma cell apoptosis, prevent angiogenesis and immune regulation, and regulate the BMME[Citation19–21]. Second-generation agents include lenalidomide, which inhibits TNF-α 50000 times more than does thalidomide, and its ability to affect costimulatory T cells is stronger than that of thalidomide. Lenalidomide can inhibit regulatory T-cell activity and exert a stronger role in immune regulation[Citation22]. Third-generation agents include pomalidomide, which has an ability to inhibit tumor cell proliferation in relapsed/refractory MM (RRMM) that is 10 times stronger than that of lenalidomide and 100 times stronger than that of thalidomide[Citation23–25]. Fourth-generation agents include iberdomide (CC-220) and CC-92480, more exactly called cereblon (CRBN) E3 ligase modulators (CELMoDs), which have stronger antimyeloma activity, even in MM cells that are resistant to lenalidomide or pomalidomide or have CRBN dysregulation[Citation26, Citation27]. Ye et al. reported that iberdomide have different dose-dependent immunomodulatory effects on B cells and T cells in a first human clinical trial in healthy people[Citation28].
However, since MM inevitably develops resistance to IMiDs, it is a challenge for clinicians to make decisions about RRMM treatment, and this issue has aroused extensive research interest. Hence, an increasing number of studies are exploring the potential mechanism of MM resistance to IMiDs. Some target molecules, such as CRBN, upstream or downstream molecules of CRBN, substrates of CRBN, and some adhesion molecules in the BMME, have been confirmed to play a key role in the mechanism of drug resistance. This review focuses on the mechanism of MM resistance to IMiDs and describes some promising drugs to resensitize MM cells to IMiDs and/or increase or synergistically enhance the sensitivity of MM cells to IMiDs. For example, CREB binding protein (CBP)/adenovirus E1A-related transcriptional coactivating protein (EP300) inhibitors, TGF-β activated kinase-1 (TAK1) inhibitors, protein translation inhibitors, and proteolysis-targeting chimeras (PROTACs) have been identified as possible agents in MM.
Antimyeloma mechanism of IMiDs
Studies have confirmed that CRBN, a substrate receptor of cullin-RING ligase 4 (CRL4), is a primary direct target of IMiDs, and CRL4CRBN directly binds to the E3 ubiquitin ligase complex (A). Ikaros zinc finger (IKZF) protein, a substrate of CRBN, is ubiquitinated and then degraded by the 26S proteasome to inhibit the interferon regulatory factor 4 (IRF4)/MYC axis, resulting in inhibition of MM cell proliferation (A)[Citation29–33]. In addition to CRBN expression, numerous factors are also related to IMiD sensitivity, such as CRBN alterations (point mutations, copy loss/structural variations and specific variant transcripts), upstream factors of CRBN, substrates of CRBN and downstream factors of CRBN. MM cells with high CRBN expression are sensitive to IMiDs and have a longer survival time, while MM cells with downregulated or absent CRBN expression are highly resistant to IMiDs[Citation34–37]. Of note, UBE2G1, an E2 enzyme associated with ubiquitin, plays a key role in regulating the destruction of neomorphic substrates of CRBN[Citation38].
Figure 1. (A) Schematic diagram of crosstalk among key factors after IMiDs act on MM cells. (B) Schematic diagram of signaling pathways involved in IMiD resistance. Abbr. IMiDs, immunomodulatory drugs; MM, multiple myeloma; CRBN, cereblon; GS, glutamine synthetase; ZFP91, zinc-finger 91; IRF4, interferon regulatory factor 4; MMSET, multiple myeloma SET region; C/EBPβ, CCAAT/enhancer binding protein β; PC4, positive coactivator 4; HO-1, heme oxygenase-1; MAPK, mitogen-activated protein kinase; ERK, extracellular signal regulated kinase; Mcl-1, myeloid leukemia 1.
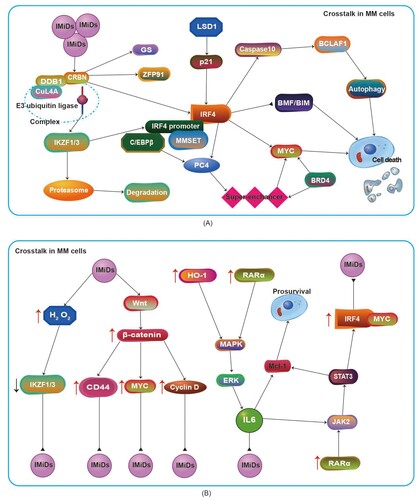
Cul4A, an upstream factor of CRBN, serves as the skeleton of the E3 ubiquitin complex (A), and downregulation of CuL4A expression can lead to IMiD resistance. DDB1, another upstream factor of CRBN, is very similar to Cul4A[Citation39]. Recent studies also found that Cul4A expression was correlated with an improvement in PFS, while DDB1 expression was negatively correlated with OS[Citation40].
Substrates of CRBN: IKZF, glutamine synthetase and zinc-finger protein 91
The IKZF transcription factor family plays a crucial role in lymphocyte development[Citation41]. The homologous proteins of IKZF, including Ikaros, Helios, Aiolos, Eos, and Pegasus (encoded, respectively by the IKZF1-5 genes), functions as transcription suppressors and activators in T-cell and B-cell differentiation and mature cell function[Citation41]. The IKZF protein exists in the CRBN complex and contains thalidomide binding groups[Citation29]. The transcription factor MEIS2, which plays an important role in human development, competitively binds to the same site of CRBN as thalidomide, and thalidomide enhances the interaction between CRBN and IKZF[Citation29]. IKZF, as the key substrate of CRBN (A), is not only related to sensitivity to IMiDs but also significantly related to OS in MM. Overexpression of IKZF protein is related to sensitivity to IMiDs and significantly prolongs OS in MM; in contrast, IKZF loss causes resistance to IMiDs (B) and shortens OS in MM [Citation42, Citation43]. Compared with lenalidomide and pomalidomide, iberdomide has a stronger ability to degrade IKZF1/3[Citation27].
Glutamine synthetase (GS), another important substrate of CRBN (A), plays a key role in metabolism and can act as the endogenous substrate of CRBN-Cul4A[Citation44, Citation45]. P97, a protein homologous to ATPase, can promote many cellular processes, including ubiquitination-dependent protein degradation, endoplasmic reticulum-related degradation and autophagy[Citation46]. The P97 is a key protein in IMiD binding with CRBN to ubiquitinate GS and degrade it via the proteasome, which can promote the degradation of IKZF1/3, substrates recruited by CRBN; this phenomenon has been verified by p97 inhibitors[Citation46]. USP15 is a key regulator of the CUL4A-CRBN-p97 pathway that controls the stability of GS, IKZF1/3, casein kinase 1 α (CK1α), and bromodomain-containing protein 4 (BRD4)[Citation47]. USP15 antagonizes ubiquitination of the Cul4A-CRBN target protein to prevent its degradation[Citation47]. USP15 is highly expressed in IMiD-resistant cells, and downregulation of USP15 can result in sensitivity to IMiDs[Citation47].
In addition, zinc finger protein 91 (ZFP91) contains a ZnF gene sequence related to IKZF1/3[Citation48]. It can also act as a substrate of CRBN-CRL4 for IMiDs (A) and play an important role in the process of IMiDs binding to CRBN, and IMiDs can promote the degradation of ZFP91[Citation48].
Moreover, IKZF is an inhibitor of the IL-2 promoter; IKZF degradation relieves inhibition of the IL-2 transcriptional promoter and stimulates T cells to secrete a large amount of IL-2, so IMiDs play an immunomodulatory role[Citation49, Citation50]. Notably, IMiDs can also activate natural killer (NK) cells to exert immunomodulatory effects on MM cells via both zeta-chain-associated protein kinase-70 and CRBN-dependent pathways[Citation51].
Downstream molecules of CRBN: the IRF4/MYC axis
IRF4 is a member of the interferon regulatory family that is located downstream of CRBN and expressed in bone marrow plasma cells of MM patients, and IRF4 serves as a key B-cell fate determinant and survival factor of MM cells[Citation33, Citation52–54]. Abnormal IRF4 activation plays an important role in the pathogenesis and progression of MM. IRF4 expression is regulated by IKZF1/3, CCAAT/enhancer binding protein β (C/EBPβ) and multiple myeloma SET region (MMSET)[Citation43, Citation49, Citation55, Citation56]. IKZF1/3 are transcriptional targets of IRF4, and IKZF protein degradation leads to the downregulation of IRF4 expression[Citation49, Citation57]. C/EBPβ is an IRF4 promoter binding protein, and its expression is regulated by the eIF4Eβ expression level[Citation55]. Downregulation of C/EBPβ leads to inhibition of IRF4 transcription and expression[Citation55]. MMSET, located upstream of IRF4, binds to the IRF4 promoter region, and loss of MMSET can downregulate IRF4 expression[Citation56]. Additionally, IRF4 also induces the expression of chromatin protein positive coactivator 4 (PC4) through a super enhancer (A). Knockdown of PC4 can downregulate IKZF1 expression, resulting in MM cell resistance to IMiDs[Citation58]. Knockdown or downregulation of IRF4 promotes MM cell apoptosis and sensitivity to IMiDs. In contrast, IRF4 overexpression promotes the survival of MM cells and resistance to IMiDs. IRF4 expression is related not only to IMiD resistance but also to ISS stage[Citation59]. Compared with ISS-I and ISS-II disease, ISS-III disease shows significantly upregulated IRF4 expression, indicating that IRF4 has a certain correlation with the prognosis of MM[Citation59].
Recent studies revealed that IRF4 can directly inhibit the expression of the proapoptotic molecules BMF (a Bcl-2 modifier) and BIM (encoded by Bcl-2L11) and promote the survival of MM cells[Citation52], impling that IRF4 overexpression boosts resistance to IMiDs (A). Notably, IRF4 downregulation might indirectly promote the death of MM cells via autophagy. IRF4 affects caspase10, which can regulate basal autophagy to avoid MM cell death, and its related protein CFLIPL in myeloma to produce BCLAF1, block the autophagy-dependent cell death pathway and promote the survival of MM cells (A)[Citation60]. IRF4 downregulation leads to downregulation of caspase 10, which disrupts the balance regulation of basic autophagy by caspase 10, enhances autophagy and promotes the death of MM cells (A)[Citation61].
The transcription factor MYC is usually expressed at low levels in mature plasma cells and can inhibit tumor occurrence, promote apoptosis, regulate CD47 and PD-L1 expression in the immune microenvironment and prevent tumor cell immune escape[Citation62]. Affer et al. revealed that IRF4 can directly bind to the MYC promoter region and upregulate MYC expression; IRF4 and MYC form an autoregulatory loop and transactivate each other. MYC rearrangement or regulation by IRF4 and superenhancers also results in dysregulation of MYC expression (A)[Citation63]. MYC overexpression, accompanied by the high expression of Bcl-2, which promotes the proliferation of MM and disease progression, induces resistance to IMiDs[Citation62]. Moreover, ARID2, a pomalidomide-induced neosubstrate of CRL4CRBN, is involved in transcriptional regulation of the pomalidomide target gene MYC. Pomalidomide is more effective than lenalidomide in degrading ARID2 and is capable of inhibiting MYC expression in and proliferation of lenalidomide-resistant cell lines[Citation64]. Importantly, inhibiting the activity of c-MYC can induce MM cell death, suggesting that c-MYC can serve as a potential therapeutic target[Citation65].
Mechanism of IMiD-resistance
IMiDs are an important strategy for treating MM, but patients with RRMM often have resistance to IMiDs, and this issue has attracted extensive attention from researchers. However, the mechanism of IMiD resistance in MM is complicated, and parts of the resistance mechanism have been gradually demonstrated by studies. Several studies have shown that IMiD resistance in MM is associated with downregulation or absence of CRBN expression[Citation34, Citation35, Citation37, Citation66]. IMiD resistance in MM is also related to some downstream molecules of CRBN, such as MYC and IRF4, which are associated with epigenetic modifications in MM. Moreover, IMiD resistance in MM is associated with some signaling pathways in MM. Of note, some novel therapeutic strategies related to epigenetics or relevant signaling pathways were found to have antimyeloma activity and were associated with IMiD activity. As such, this review clearifies the relationship between factors associated with epigenetics or relevant signaling pathways and the mechanism of IMiD resistance in MM.
Epigenetic factors related to IMiD resistance
Histone modification is an important epigenetic mechanism, and the dysregulation of epigenetic mechanisms leads to oncogene overexpression and tumor suppressor gene silencing[Citation67, Citation68]. CBP/EP300 is a histone acetyltransferase that is widely expressed in organisms and has acetyltransferase activity. The bromodomain (BRD) of CBP/EP300 can recognize acetylated lysine residues. The BRD is the ‘reader’ of lysine acetylation that is responsible for the transduction of signals carried by acetylated lysine residues and the translation of signals into normal or abnormal phenotypes[Citation69, Citation70].
TP53 gene mutation is an important factor in the double- or triple-hit theory of MM[Citation5, Citation6]. Interestingly, when DNA is damaged, CBP/EP300 can enhance TP53-dependent cell cycle arrest and transcriptional activation of DNA damage genes, thereby inhibiting tumor progression[Citation71]. Specifically, the TP53 gene is also related to MM drug resistance[Citation72, Citation73]. TP53-related protein kinase (TP53RK), which mediates TP53 activity, can serve as a therapeutic target for MM patients with a poor prognosis[Citation73]. IMiDs downregulate p21 expression in wild-type TP53 MM.1S cells and trigger apoptosis. A subsequent study found that IMiDs can bind to and inhibit TP53RK[Citation73]. Genetic or pharmacological inhibition of TP53RK can trigger MM cell apoptosis via TP53-MYC axis-dependent or TP53-MYC axis-independent pathways[Citation73].
Furthermore, IMiDs may regulate the activity of the histone demethylase LSD1, upregulate the cyclin-dependent kinase inhibitor p21waf−1, block MM cells in the G1 phase, and play an antimyeloma role (A)[Citation74, Citation75]. LSD1 is involved in the demethylation of histone H3K9[Citation74]. When LSD1 mutation leads to LSD1 silencing, the expression of p21waf−1 decreases, and resistance to IMiDs follows[Citation74]. Knockdown of CRBN can prevent the upregulation of p21 mediated by immune regulation and alleviate the inhibition of IRF4 (A)[Citation31]. In addition, a more recent study was the first to demonstrate that aberrant CRBN DNA methylation is associated with IMiD resistance in MM cell lines, but further studies are needed to confirm its efficacy[Citation76].
The bromodomain and extraterminal (BET) family includes BRDT, BRD2, BRD3, and BRD4[Citation77], and BRD4 is an important epigenetic and transcriptional regulator of embryogenesis and tumor development. Studies have revealed that super-enhancers in MM cells can show enrichment of BRD4 and mediator complexes, which is significantly related to the MYC oncogene in MM biology[Citation77]. More precisely, BRD4 was found to be associated with the IgH enhancer driving MYC gene expression (A)[Citation77], and c-MYC transcription driven by BRD4 can accelerate tumor cell proliferation and disease development in MM[Citation78]. Importantly, activation of c-MYC is crucial to increase myeloma cell proliferation[Citation79], and c-MYC is upregulated in MM cells with IMiD resistance (B). Recently, Zhu et al revealed that downregulation of BRD4 can result in obvious downregulation of c-MYC transcription and G1 phase arrest in MM cells[Citation80], which suggests that BRD4 is associated with IMiD resistance in MM.
Signaling pathways related to IMiD resistance
Activation of three signaling pathways (the Wnt/β-catenin, JAK2/STAT3 and mitogen activated protein kinase (MAPK)/extracellular signal regulated kinase (ERK) signaling pathways) has been confirmed to be involved in the mechanism of IMiD resistance (B)[Citation81–85].
The Wnt pathway is a key signaling pathway regulating the growth, development and death of organisms, and β-catenin is a key regulator of this pathway that can bind the axin-CK1α-APC-GSK3β complex and then be phosphorylated[Citation82]. β-Catenin expression is related to sensitivity to IMiDs, and the β-catenin downregulation induces sensitivity to IMiDs[Citation81]. After IMiDs bind CRBN, once the regulatory protein Wnt binds to the Frizzled receptor, the axin-CK1α-APC-GSK3β complex is degraded and the E3 ubiquitin ligase is inhibited. As a result, phosphorylated β-catenin cannot be ubiquitinated, so massive accumulation of β-catenin promotes the accumulation of downstream cyclinD and c-MYC, which reduces sensitivity to IMiDs and even induces drug resistance (B)[Citation82]. Moreover, CD44, a downstream transcription target of the Wnt/β-catenin signaling pathway and an adhesion molecule existing in the BMME, is related to sensitivity to IMiDs and to the prognosis and progression of MM[Citation86, Citation87]. CD44 downregulation induces sensitivity to IMiDs in MM; in contrast, upregulation of CD44 induces resistance to IMiDs in MM (B)[Citation87].
Furthermore, the mechanism of IMiD resistance mediated by Wnt/β-catenin is related to oxidative stress[Citation88, Citation89]. In MM cells with high CRBN expression, lenalidomide can increase the intracellular H2O2 level, cause dimerization of the immunoglobulin light chain, significantly increase endoplasmic reticulum stress and induce cytotoxicity[Citation89]. Moreover, H2O2-mediated oxidative stress promotes the degradation of IKZF1/3 to confer resistance to IMiDs in MM (B)[Citation89].
The JAK2/STAT3 and MAPK/ERK signaling pathways play an important role in regulating cell growth, differentiation and survival. The IL-6 receptor family can help regulate B-cell differentiation and plasma cell production. IL-6 can activate the STAT3 pathway, promote the upregulation of the expression of members of the IRF4/MYC axis, promote the survival of MM cells and reduce sensitivity to IMiDs (B)[Citation33]. IMiDs can also enhance the autocrine activity of IL-6, which may be because IMiDs inherently enhance the drug resistance of MM cells. Barrera et al. found that upregulation of heme oxygenase-1 (HO-1) was mediated by bortezomib and that HO-1 could regulate the production of IL-6 via the p38MAPK pathway[Citation90]. HO-1 and IL-6 play an important role in the growth and proliferation MM cells and in the inhibition of MM cell apoptosis, and high expression of HO-1 makes MM cells resistant to IMiDs (B)[Citation33, Citation91].
Novel strategies for MM treatment
Recently, some studies have found that some potential novel agents can resensitize MM cells to IMiDs, or increase the sensitivity of MM cells to IMiDs, or synergistically enhance the antimyeloma activity of IMiDs, and these agents are summarized in .
Table 1. Novel strategies for MM treatment associated with IMiDs. Abbr. CBP/EP300, CREB binding protein/adenovirus E1A-related transcriptional coactivating protein; IRF4, interferon regulatory factor 4; HDAC, histone deacetylase; TAK1, TGF-β activated kinase-1; Mcl-1, myeloid leukemia 1; ATO, arsenic trioxide; ATRA, all-trans retinoic acid; BET, bromodomain and extraterminal; PROTAC, proteolysis-targeting chimera.
Epigenetic therapy
Accumulating studies have shown that epigenetic therapy plays a potentially important role in the treatment of MM. SGC-CBP30, a selective EP300/CBP inhibitor, targets the IRF4 super enhancer and MYC regulatory region in MM cells[Citation54]. In the presence of CRBN, SGC-CBP30 can directly target CBP/EP300 in the regulatory region of the IRF4/MYC axis, downregulate IRF4 and MYC expression, and indirectly inhibit IL-6 expression and STAT3 pathway activation to make MM cells sensitive to lenalidomide ()[Citation33].
Increasing numbers of studies have confirmed that BRD4 inhibitors display good antimyeloma activity and reverse IMiD resistance[Citation32, Citation54, Citation92, Citation93]. For example, CPI203, a BET inhibitor, selectively inhibits BRD4 and inhibits MYC transcription, thereby downregulating the expression of IRF4, MYC, and IKZF1[Citation32]. The antimyeloma activity of CPI203 is unrelated to sensitivity to lenalidomide, but CPI203 can cooperate with lenalidomide to induce and block the MYC and IKZF1 signaling pathways and enhance the efficacy of lenalidomide plus dexamethasone ()[Citation32]. Moreover, other BET inhibitors (), such as JQ1, OTX015, and CPI0610, inhibit myeloma cell proliferation[Citation77, Citation92, Citation94–96]. Specifically, strategies inhibiting BRD4 may result in drug resistance due to recovery of the tumor-related genes or the accumulation of BRD4, which has been found in acute myeloid leukemia (AML) and breast cancer[Citation97, Citation98] but has not been reported in MM.
CBP/EP300 is a highly homologous transcriptional coactivator containing a BRD; it is often mutated in human cancer, affecting histone acetylation[Citation67]. Recent studies demonstrated that NEO1132 and NEO2734, which simultaneously inhibit CEP/EP300 and BET, can induce apoptosis in vitro and effectively reduce the survival rate of AML cells, improving the effectiveness of chemotherapy against AML in vivo ()[Citation99]. Importantly, the efficacy of both agents has also been confirmed in MM[Citation100]. The sensitivity to dual inhibitors does not depend on specific molecular subsets in MM but on the expression level of the c-MYC protein[Citation100]. The dual inhibitors NEO1132 and NEO2734 can effectively stall MM cells in G1 phase in vitro and reduce the protein levels of c-MYC and IRF4, showing significant antimyeloma activity[Citation100]. These agents are as effective as JQ1 and more effective than other single inhibitors[Citation100].
In addition, some novel selective histone deacetylase (HDAC) inhibitors have also shown antimyeloma activity. For example, A452, a selective HDAC6 inhibitor, plus IMiDs synergistically inhibited cell growth, reduced MM cell viability and increased the level of apoptosis (). Moreover, increased cell death was associated with inactivation of Akt and ERK1/2. Of note, A452 combined with IMiDs induced synergistic MM cell cytotoxicity without changing the expression of CRBN, which synergistically downregulated IKZF1/3, c-MYC and IRF4[Citation101]. BG45, a selective HDAC3 inhibitor, inhibits HO-1 expression by inhibiting the activation of the JAK2/STAT3 signaling pathway to promote MM cell apoptosis and inhibit MM cell proliferation[Citation83]. BG45 plus lenalidomide can also enhance sensitivity to IMiDs in MM ()[Citation83]. However, not all HDAC inhibitors can treat MM patients in clinical practice. For example, in phase 2 clinical trial, although romidepsin induced apoptosis by decreasing the expression of Bcl-2 family proteins (Bcl-XL and Mcl-1) in cell lines and induced G1 cell cycle arrest (by promoting the expression of p21 and p53), as a single agent, it had poor efficacy in refractory MM[Citation102].
Signaling pathway inhibition
Selumetinib, a selective and noncompetitive inhibitor of MAPK1/2, plus IMiDs as treatment for IMiD-resistant MM cells showed effective inhibition of the MAPK/ERK signaling pathway and resensitized MM cells to IMiDs ()[Citation103]. Unfortunately, it is unclear whether selumetinib as a single agent can treat RRMM. A phase II clinical trial showed that the efficacy of single-agent selumetinib was poor in RRMM patients, and grade 3 and above adverse events occured[Citation104]. Interestingly, selumetinib plus an HDAC inhibitor can effectively kill Ras/Raf-mutated drug-resistant MM cells[Citation4].
Morales et al. showed that arsenic trioxide (ATO) could induce the upregulation of three proapoptotic BH3 proteins (Noxa, BMF and puma) and the downregulation of two antiapoptotic proteins (Mcl-1 and Bcl-XL) (B)[Citation105]. Moreover, ATO can enhance the sensitivity of MM cells to IMiDs by upregulating the expression level of CRBN ()[Citation106]. Nevertheless, ATO may activate the p38MAPK pathway, resulting in ATO resistance to myeloma, while inhibition of p38MAPK may overcome ATO resistance to myeloma[Citation107]. These results seem contradictory and imply that ATO is not an ideal choice in the treatment of MM.
TAK1, a member of the MAPK kinase family, plays a key role in the growth and development of B cells, and it is continuously overexpressed and phosphorylated in MM cells, affecting factors including NF-κB, p38MAPK/ERK, and JNK[Citation108, Citation109]. Recent studies demonstrated that a TAK1 inhibitor (LLZ1640-2) could inhibit the activation of the NF-κB, p38MAPK/ERK, and STAT3 signaling pathways to reduce the expression of key mediators of MM growth and survival, including MYC, Mcl-1, and IRF4, accompanied by a significant reduction in the angiogenesis factor VEGF in MM cells; LLZ1640-2 decreases the adhesion between MM cells and bone marrow stromal cells (BMSCs) [Citation85, Citation109]. Moreover, TAK1 inhibition can abolish IL-6 and TNF-α-induced signaling in MM cell lines[Citation109]. Although the relationship between TAK1 inhibitors and IMiD resistance is unclear, several factors, such as IRF4, MYC, and Mcl-1, that are downregulated by TAK1 inhibitors are closely associated with IMiD resistance (Figure 2B); thus TAK1 inhibitors may improve IMiD efficacy by decreasing IMiD resistance-associated factor expression in MM, but this idea needs be confirmed by further studies. Notably, LLZ1640-2 may play a critical role in bone destruction in MM and may inhibit tumor cell proliferation and prevent bone destruction and loss ()[Citation84, Citation85, Citation109].
All-trans retinoic acid (ATRA) can downregulate β-Catenin and cell surface total CD44 expression, inhibit Wnt/β-catenin cascade-mediated cell proliferation and migration, reduce the expression of antiapoptotic proteins and the adhesion of drug-resistant cells, promote the expression of Fas antigen, promote cell apoptosis, and improve the sensitivity of MM cells to IMiDs[Citation87, Citation110]. Chen et al. confirmed that ATRA could inhibit the proliferation of MM cells by upregulating p21waf−1 expression, which was not dependent on downregulation of the IL-6 receptor[Citation111]. However, Liu et al. revealed that ATRA induced chemoresistance of MM cell via a noncanonical signaling in vitro[Citation112]. Notably, the expression of retinoic acid receptor α (RARα) is related to disease progression in MM. Wang et al. confirmed that ATRA is effective in MM patients with high expression of RARα (B, )[Citation113].
Protein translation inhibitors
Several studies have revealed that homoharringtonine and polyethylene glycol homoharringtonine liposomes can induce apoptosis and growth arrest of MM cells[Citation114–116]. Omacetaxine is a semisynthetic homoharringtonine that can bind to the ribosomal A-site cleft and block the initial extension of protein synthesis. Omacetaxine can inhibit protein translation by downregulating antiapoptotic oncoproteins of the Bcl-2 family, such as myeloid leukemia 1 (Mcl-1) and c-MYC, and it has been approved by the FDA for treating patients with chronic myeloid leukemia resistant or intolerant to two or more tyrosine kinase inhibitors[Citation117–121]. However, the level of the key survival-promoting factor Mcl-1 is directly related to the progression of MM, and c-MYC is usually highly expressed in IMiD-resistant MM cells[Citation122]. Walker et al. reported that omacetaxine resensitized MM cells in RRMM patients with IMiD resistance to IMiDs by affecting the IRF4/c-MYC axis, blocking the process of protein translation and exerting strong antimyeloma activity, indicating that omacetaxine is a promising antimyeloma agent ()[Citation2].
PROTACs
PROTACs, small, dual-functional molecular conjugates selectively target proteins (e.g. BRD4, and Mcl-1) and E3 ubiquitin ligases in the CRBN-binding region (e.g. IMiDs), thereby inducing the ubiquitination and degradation of proteins by the proteasome ()[Citation122–124].
ARV825, a BET PROTAC, interacts with the E3 ligase CRBN-binding region of pomalidomide and the BET-binding region of OTX015[Citation124, Citation125]. After the target is ubiquitinated, the PROTAC and E3 ligases can be separated and reused, and the induced PROTAC-mediated protein degradation is more effective than that induced by directly inhibitory drugs[Citation126, Citation127]. A preclinical model showed that PROTACs targeting BRD4 and other BET family members for ubiquitination and proteasome degradation could block cells in the G0/G1 phase, downregulate CDK4/6 levels, upregulate p21 levels, induce apoptosis, and significantly reduce the viability of MM cells in a time- and concentration-dependent manner[Citation80]. Of note, PROTACs can also overcome resistance to bortezomib, dexamethasone, lenalidomide and pomalidomide, and the activity of PROTACs is maintained in TP53-wild-type or TP53-deficient myeloma cells[Citation80].
Mcl-1, a prosurvival factor, is upregulated by IL-6 in a STAT3-dependent manner and overexpressed in MM[Citation128]. Of note, Mcl-1 can act as a crucial therapeutic target of MM via protein–protein interactions (PPIs) and is thus involved in diverse biological processes[Citation122]. Administration of a PROTAC selectively targeting Mcl-1 with A-121477 (an Mcl-1 inhibitor) and binding with an E3 ligase in the CRBN-binding region promoted the degradation of the antiapoptotic protein Mcl-1 via PPIs[Citation122], and this may also be a potent therapeutic strategy.
Overall, IMiDs, as basic agents for treating MM, have direct antimyeloma activity by targeting CRBN. However, accumulated studies have revealed that some factors associated with CRBN, such as downregulation or loss of CRBN expression, downregulation of IKZF protein, high MYC expression and high CD44 expression in BMME, affect sensitivity to IMiDs in MM. Importantly, IRF4 is not only affected by IKZF but also can directly inhibit the expression of BMF and BIM, thereby promoting cell survival of MM. Additionally, the expression of IRF4 and MYC also plays an important role in three important signaling pathways (Wnt, STAT3 and MAPK/ERK) related to IMiD resistance. Fortunately, other studies have discovered that some novel agents may improve sensitivity to IMiDs in MM by targeting these factors. For example, high expression of MYC promotes the proliferation of MM cells and disease progression, which may be caused by the driving effect of BRD4 (a histone modification ‘reader’). These are very important discoveries with potential for ameliorating resistance to IMiDs in MM, and agents such as BET inhibitors, CBP/EP300 inhibitors, dual-target BET-CBP/EP300 inhibitors, TAK1 inhibitors, protein translation inhibitors and PROTACs are promising. These agents represent potential new therapeutics to regulate the IRF4/MYC axis by inhibiting BRD4 expression or signaling pathway activation. Although these new strategies are effective in MM cells, research is still in early stage and more studies are needed to verify the effect of these strategies in clinical patients with MM.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Van De Donk N, Pawlyn C, Yong KL. Multiple myeloma. The Lancet. 2021;397(10272):410–427.
- Walker ZJ, Idler BM, Davis LN, et al. Exploiting protein translation dependence in multiple myeloma with omacetaxine-based therapy. Clin Cancer Res. 2021;27(3):819–830.
- Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3:17046.
- Ramakrishnan VG, Miller KC, Macon EP, et al. Histone deacetylase inhibition in combination with MEK or Bcl-2 inhibition in multiple myeloma. Haematologica. 2019;104(10):2061–2074.
- Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: A meta-analysis of the molecular profiles of 1905 trial patients. Leukemia. 2018;32(1):102–110.
- Walker BA, Mavrommatis K, Wardell CP, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159–170.
- Paiva B, Van Dongen JJ, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125(20):3059–3068.
- Flores-Montero J, Sanoja-Flores L, Paiva B, et al. Orfao A. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia. 2017;31(10):2094–2103.
- Lahuerta JJ, Paiva B, Vidriales MB, et al. Depth of response in multiple myeloma: A pooled analysis of three PETHEMA/GEM clinical trials. J Clin Oncol. 2017;35(25):2900–2910.
- Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of minimal residual disease With superior survival outcomes in patients With multiple myeloma. JAMA Oncol. 2017;3(1):28–35.
- Rawstron AC, Gregory WM, De Tute RM, et al. Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood. 2015;125(12):1932–1935.
- Paiva B, Gutiérrez NC, Rosiñol L, et al. High-Risk Cytogenetics and Persistent Minimal Residual Disease by Multiparameter Flow Cytometry Predict Unsustained Complete Response After Autologous Stem Cell Transplantation in Multiple Myeloma. Blood. 2012;119(3):687–691.
- Zuo X, Liu D. Progress in the application of minimal residual disease detection in multiple myeloma. J Hematop. 2021;14(2):97–107.
- Pagnucco G, Cardinale G, Gervasi F. Targeting multiple myeloma cells and their bone marrow microenvironment. Ann N Y Acad Sci. 2004;1028:390–399.
- Munshi NC, Anderson LD, Jr., Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716.
- Yan Z, Cao J, Cheng H, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019;6(10):e521–e529.
- Nerreter T, Letschert S, Gotz R, et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat Commun. 2019;10(1):3137.
- Yan L, Qu S, Shang J, et al. Sequential CD19 and BCMA-specific CAR T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer Med. 2021;10(2):563–574.
- Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–1571.
- D'amato RJ, Loughnan MS, Flynn E, et al. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91(9):4082–4085.
- Ati ZE, Lamia R, Cherif J, et al. Thalidomide-induced bronchiolitis obliterans organizing pneumonia in a patient with multiple myeloma. Saudi J Kidney Dis Transpl. 2019;30(4):974–977.
- Braga WM, Atanackovic D, Colleoni GW. The role of regulatory T cells and TH17 cells in multiple myeloma. Clin Dev Immunol. 2012;2012:293479.
- Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide–refractory multiple myeloma: intergroupe francophone du myélome 2009-02. Blood. 2013;121(11):1968–1975.
- Lacy MQ, Hayman SR, Gertz MA, et al. Pomalidomide (CC4047) Plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol. 2009;27(30):5008–5014.
- Lacy MQ, Allred JB, Gertz MA, et al. Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: comparison of 2 dosing strategies in dual-refractory disease. Blood. 2011;118(11):2970–2975.
- Bjorklund CC, Kang J, Amatangelo M, et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated crbn. Leukemia. 2020;34(4):1197–1201.
- Matyskiela ME, Zhang W, Man HW, et al. A cereblon modulator (CC-220) with improved degradation of ikaros and aiolos. J Med Chem. 2018;61(2):535–542.
- Ye Y, Gaudy A, Schafer P, et al. First-in-human, single- and multiple-ascending-dose studies in healthy subjects to assess pharmacokinetics, pharmacodynamics, and safety/tolerability of iberdomide, a novel cereblon E3 ligase modulator. Clin Pharmacol Drug Dev. 2021;10(5):471–485.
- Fischer ES, Bohm K, Lydeard JR, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53.
- Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350.
- Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326–2335.
- Diaz T, Rodriguez V, Lozano E, et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity inin vitro and in vivo models of multiple myeloma by blockade of ikaros and MYC signaling. Haematologica. 2017;102(10):1776–1784.
- Zhu YX, Shi CX, Bruins LA, et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019;9(2):19.
- Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–4779.
- Kortum KM, Mai EK, Hanafiah NH, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128(9):1226–1233.
- Broyl A, Kuiper R, Van Duin M, et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood. 2013;121(4):624–627.
- Gooding S, Ansari-Pour N, Towfic F, et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood. 2021;137(2):232–237.
- Lu G, Weng S, Matyskiela M, et al. UBE2G1 governs the destruction of cereblon neomorphic substrates. Elife. 2018;7(e40958.
- Angers S, Li T, Yi X, et al. Molecular architecture and assembly of the DBB1-CuL4A ubiquitin ligase machinery. Nature. 2006;443(7111):590–593.
- Barankiewicz J, Szumera-Cieckiewicz A, Salomon-Perzynski A, et al. The CRBN, The CRBN, CUL4A and DDB1 expression predicts the response to immunomodulatory drugs and survival of multiple myeloma patients. J Clin Med. 2021;10(12):2683.
- Heizmann B, Kastner P, Chan S. The ikaros family in lymphocyte development. Curr Opin Immunol. 2018;51:14–23.
- Zhu YX, Braggio E, Shi CX, et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood. 2014;124(4):536–545.
- Kronke J, Kuchenbauer F, Kull M, et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: A study of the German myeloma study group (DSMM). Leukemia. 2017;31(6):1363–1367.
- Nguyen TV, Lee JE, Sweredoski MJ, et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol Cell. 2016;61(6):809–820.
- Hensley CT, Wasti AT, Deberardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678–3684.
- Nguyen TV, Li J, Lu CJ, et al. P97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. 2017;114(14):3565–3571.
- Nguyen TV. USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. 2021;118(40):e2111391118.
- An J, Ponthier CM, Sack R, et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase. Nat Commun. 2017;8(15398).
- Krönke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014: 343(6168): 301-305.
- Quintana FJ, Jin H, Burns EJ, et al. Aiolos promotes TH17 differentiation by directly silencing IL2 expression. Nat Immunol 2012;13(8):770–777.
- Hideshima T, Ogiya D, Liu J, et al. Immunomodulatory drugs activate NK cells via both Zap-70 and cereblon-dependent pathways. Leukemia. 2021;35(1):177–188.
- Fedele PL, Liao Y, Gong JN, et al. The transcription factor IRF4 represses proapoptotic BMF and BIM to licence multiple myeloma survival. Leukemia. 2021;35(7):2114–2118.
- Low MSY, Brodie EJ, Fedele PL, et al. IRF4 activity is required in established plasma cells to regulate gene transcription and mitochondrial homeostasis. Cell Rep. 2019;29(9):2634–2645.e5 e2635.
- Conery AR, Centore RC, Neiss A, et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. Elife. 2016;5(e19432.
- Li S, Pal R, Monaghan SA, et al. IMiD immunomodulatory compounds block C/EBP{beta} translation through eIF4E down-regulation resulting in inhibition of MM. Blood. 2011;117(19):5157–5165.
- Xie Z, Bi C, Chooi JY, et al. MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia. 2015;29(12):2347–2354.
- Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science. 2014;343(6168):305–309.
- Ochiai K, Yamaoka M, Swaminathan A, et al. Chromatin protein PC4 orchestrates B cell differentiation by collaborating with ikaros and IRF4. Cell Rep. 2020;33(12):108517.
- Bai H, Wu S, Wang R, et al. Bone marrow IRF4 level in multiple myeloma: An indicator of peripheral blood TH17 and disease. Oncotarget. 2017;8(49):85392–85400.
- Lamy L, Ngo VN, Emre NC, et al. Control of Autophagic Cell Death by Caspase-10 in multiple myeloma. Cancer Cell. 2013;23(4):435–449.
- Morelli E, Leone E, Cantafio ME, et al. Selective targeting of IRF4 by synthetic microRNA-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia. 2015;29(11):2173–2183.
- Shaffer AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–231.
- Affer M, Chesi M, Chen WG, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28(8):1725–1735.
- Yamamoto J, Suwa T, Murase Y, et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat Chem Biol. 2020;16(11):1208–1217.
- Kuehl WM, Bergsagel PL. MYC addiction: A potential therapeutic target in MM. Blood. 2012;120(12):2351–2352.
- Gandhi AK, Mendy D, Waldman M, et al. Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Br J Haematol. 2014;164(2):233–244.
- Spriano F, Gaudio E, Cascione L, et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4(17):4124–4135.
- Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463.
- Ntranos A, Casaccia P. Bromodomains: translating the words of lysine acetylation into myelin injury and repair. Neurosci Lett. 2016;625:4–10.
- Perez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics. 2017;12(5):323–339.
- Grossman SR. P300/CBP/p53 interaction and regulation of the p53 response. Eur J Biochem. 2001;268(10):2773–2778.
- Munawar U, Roth M, Barrio S, et al. Assessment of TP53 lesions for p53 system functionality and drug resistance in multiple myeloma using an isogenic cell line model. Sci Rep. 2019;9(1):18062.
- Hideshima T, Cottini F, Nozawa Y, et al. P53-related protein kinase confers poor prognosis and represents a novel therapeutic target in multiple myeloma. Blood. 2017;129(10):1308–1319.
- Escoubet-Lozach L, Lin IL, Jensen-Pergakes K, et al. Pomalidomide and lenalidomide induce p21WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res 2009;69(18):7347–7356.
- Hideshima T, Chauhan D, Shima Y, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96(9):2943–2950.
- Haertle L, Barrio S, Munawar U, et al. Cereblon enhancer methylation and IMiD resistance in multiple myeloma. Blood. 2021;138(18):1721–1726.
- Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334.
- Doroshow DB, Eder JP, Lorusso PM. BET inhibitors: A novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787.
- Holien T, Vatsveen TK, Hella H, et al. Addiction to c-MYC in multiple myeloma. Blood. 2012;120(12):2450–2453.
- Zhang X, Lee HC, Shirazi F, et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia. 2018;32(10):2224–2239.
- Bjorklund CC, Ma W, Wang ZQ, et al. Evidence of a role for activation of Wnt/β-catenin signaling in the resistance of plasma cells to lenalidomide. J Biol Chem. 2011;286(13):11009–11020.
- Butrym A, Rybka J, Lacina P, et al. Polymorphisms within beta-catenin encoding gene affect multiple myeloma development and treatment. Leuk Res. 2015;39(12):1462–1466.
- Tang S, Cheng B, Zhe N, et al. Histone deacetylase inhibitor BG45-mediated HO-1 expression induces apoptosis of multiple myeloma cells by the JAK2/STAT3 pathway. Anticancer Drugs. 2018;29(1):61–74.
- Tenshin H, Teramachi J, Oda A, et al. TAK1 inhibition subverts the osteoclastogenic action of TRAIL while potentiating its antimyeloma effects. Blood Adv. 2017;1(24):2124–2137.
- Haland E, Moen IN, Veidal E, et al. TAK1-inhibitors are cytotoxic for multiple myeloma cells alone and in combination with melphalan. Oncotarget. 2021;12(21):2158–2168.
- Paiva B, Corchete LA, Vidriales MB, et al. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: a new model to understand chemoresistance. Blood. 2016;127(15):1896–1906.
- Bjorklund CC, Baladandayuthapani V, Lin HY, et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: therapeutic implications. Leukemia. 2014;28(2):373–383.
- Yin L, Tagde A, Gali R, et al. MUC1-C is a target in lenalidomide resistant multiple myeloma. Br J Haematol. 2017;178(6):914–926.
- Sebastian S, Zhu YX, Braggio E, et al. Multiple myeloma cells’ capacity to decompose H2O2 determines lenalidomide sensitivity. Blood. 2017;129(8):991–1007.
- Barrera LN, Rushworth SA, Bowles KM, et al. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle. 2012;11(12):2248–2252.
- Wu W, Ma D, Wang P, et al. Potential crosstalk of the interleukin-6-heme oxygenase-1-dependent mechanism involved in resistance to lenalidomide in multiple myeloma cells. FEBS J. 2016;283(5):834–849.
- Siu KT, Ramachandran J, Yee AJ, et al. Preclinical Activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia. 2017;31(8):1760–1769.
- Abruzzese MP, Bilotta MT, Fionda C, et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of cMYC-IRF4-miR-125b interplay. J Hematol Oncol. 2016;9(1):134.
- Amorim S, Stathis A, Gleeson M, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196–e204.
- Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917.
- Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073.
- Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417.
- Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547.
- Van Gils N, Martiañez Canales T, Vermue E, et al. The novel oral BET-CBP/p300 dual inhibitor NEO2734 is highly effective in eradicating acute myeloid leukemia blasts and stem/progenitor cells. Hemasphere. 2021;5(8):e610.
- Ryan KR, Giles F, Morgan GJ. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur J Haematol. 2021;106(1):90–99.
- Won HR, Lee DH, Yeon SK, et al. HDAC6-selective inhibitor synergistically enhances the anticancer activity of immunomodulatory drugs in multiple myeloma. Int J Oncol. 2019;55(2):499–512.
- Niesvizky R, Ely S, Mark T, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117(2):336–342.
- Ocio EM, Fernandez-Lazaro D, San-Segundo L, et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia. 2015;29(3):705–714.
- Holkova B, Zingone A, Kmieciak M, et al. A phase II trial of AZD6244 (selumetinib. ARRY; 142886), an oral MEK1/2 inhibitor, in relapsed/refractory multiple myeloma. Clin Cancer Res. 2016;22(5):1067–1075.
- Morales AA, Gutman D, Lee KP, et al. BH3-only proteins noxa, Bmf, and Bim are necessary for arsenic trioxide-induced cell death in myeloma. Blood. 2008;111(10):5152–5162.
- Jian Y, Gao W, Geng C, et al. Arsenic trioxide potentiates sensitivity of multiple myeloma cells to lenalidomide by upregulating cereblon expression levels. Oncol Lett. 2017;14(3):3243–3248.
- Wen J, Cheng HY, Feng Y, et al. P38 MAPK inhibition enhancing ATO-induced cytotoxicity against multiple myeloma cells. Br J Haematol. 2008;140(2):169–180.
- Buglio D, Palakurthi S, Byth K, et al. Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood. 2012;120(2):347–355.
- Teramachi J, Tenshin H, Hiasa M, et al. TAK1 is a pivotal therapeutic target for tumor progression and bone destruction in myeloma. Haematologica. 2021;106(5):1401–1413.
- Navas TA, Nguyen AN, Hideshima T, et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L). Mcl; 1, and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia. 2006;20(6):1017–1027.
- Chen Y-H, Lavelle D, Desimone J, et al. Growth inhibition of a human myeloma cell line by All-transRetinoic acid Is Not mediated through downregulation of interleukin-6 receptors but through upregulation of p21WAF1. Blood. 1999;94(1):251–259.
- Liu Z, Li T, Jiang K, et al. Induction of chemoresistance by all-trans retinoic acid via a noncanonical signaling in multiple myeloma cells. PLoS One. 2014;9(1):e85571.
- Wang S, Tricot G, Shi L, et al. RARα2 expression is associated with disease progression and plays a crucial role in efficacy of ATRA treatment in myeloma. Blood. 2009;114(3):600–607.
- Lou YJ, Qian WB, Jin J. Homoharringtonine induces apoptosis and growth arrest in human myeloma cells. Leuk Lymphoma. 2007;48(7):1400–1406.
- Li M, Shi F, Fei X, et al. PEGylated long-circulating liposomes deliver homoharringtonine to suppress multiple myeloma cancer stem cells. Exp Biol Med (Maywood). 2017;242(9):996–1004.
- Li M, Fei X, Shi F, et al. Homoharringtonine delivered by high proportion PEG of long- circulating liposomes inhibits RPMI8226 multiple myeloma cells in vitro and in vivo. Am J Transl Res. 2016;8(3):1355–1368.
- Damlaj M, Lipton JH, Assouline SE. A safety evaluation of omacetaxine mepesuccinate for the treatment of chronic myeloid leukemia. Expert Opin Drug Saf. 2016;15(9):1279–1286.
- Gurel G, Blaha G, Moore PB, et al. Determines the species specificity of the a-site cleft antibiotics: The structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2504;2009:389(1): 146-156.
- Tang R, Faussat AM, Majdak P, et al. Semisynthetic homoharringtonine induces apoptosis via inhibition of protein synthesis and triggers rapid myeloid cell leukemia-1 down-regulation in myeloid leukemia cells. Mol Cancer Ther. 2006;5(3):723–731.
- Allan EK, Holyoake TL, Craig AR, et al. Omacetaxine may have a role in chronic myeloid leukaemia eradication through downregulation of Mcl-1 and induction of apoptosis in stem/progenitor cells. Leukemia. 2011;25(6):985–994.
- Chen R, Guo L, Chen Y, et al. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011;117(1):156–164.
- Papatzimas JW, Gorobets E, Maity R, et al. From inhibition to degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (Mcl1). J Med Chem. 2019;62(11):5522–5540.
- Buckley DL, Crews CM. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53(9):2312–2330.
- Sun B, Fiskus W, Qian Y, et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 2018;32(2):343–352.
- Lim SL, Damnernsawad A, Shyamsunder P, et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: efficacy, biomarker and drug combinations. Haematologica. 2019;104(6):1209–1220.
- Roy MJ, Winkler S, Hughes SJ, et al. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem Biol 2019;14(3):361–368.
- Bondeson DP, Mares A, Smith IE, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11(8):611–617.
- Gupta VA, Matulis SM, Conage-Pough JE, et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood. 2017;129(14):1969–1979.