
ABSTRACT
A 37-year-old Chinese man complained of recurrent dyspnea, with low pulse oximetry (SpO2) and normal partial pressure of oxygen (PaO2) by blood gas analysis. The patient’s P50 was elevated at 47.3 mmHg, while hemoglobin fraction by capillary electrophoresis revealed an abnormal HbD quantified at 43.2%. Further DNA analysis by whole exome sequencing (WES) identified heterozygosity for a mutation in exon 2 at codon 38 (HBB: c.112T>A, p.Trp38Arg), which decodes to a substitution of the amino acid tryptophan (Try) by arginine (Arg), known as Hb Rothschild. The patient’s mother and daughter were also diagnosed to have low SpO2 readings and his daughter was confirmed to carry Hb Rothschild. This is the first known familial case of Hb Rothschild in China. As Hb Rothschild may be underdiagnosed in Asian descent, better understanding in this area would help avoid unnecessary cardiorespiratory interventions.
Introduction
Hemoglobin (Hb) is a heterotetramer composed of two α-subunits and two β-subunits (an α1β1 dimer and an α2β2 dimer in HbA). According to Monod, Wyman and Changeaux (MWC) model, there are two conformationally stable states of Hb quaternary structure. The relaxed, high oxygen affinity state is known as the R state, and the tense, low oxygen affinity state, is known as the T state. The pathophysiologic mechanism of altered oxygen affinity often involves mutations in critical regions of the globin chain that affect stabilization or destabilization of the T and R states. With regard to oxygen delivery, high-affinity variants deliver less oxygen to tissue than normal Hb A, while low oxygen affinity variants provide more oxygen to the tissues.
Up to now, more than 70 low oxygen affinity variants of Hbs, including Hb Rothschild, Chico, and Bassett, have been reported, and chronically low oxygen saturation (SpO2) has been documented in patients with these variants. However, low pulse oximetry with respiratory symptoms is mainly presented in diseases involving the pulmonary or cardiac systems. Certain inherited variant Hbs are quite rare causes and pulmonologist, cardiologist and emergency care physicians ought to be aware of. Here, we present the first report of a Chinese family with low pulse oximetry caused by Hb Rothschild (HBB: c.112T>A) as a rare addition to the hematological literature.
Case report
A 37-year-old Chinese man complained of recurrent dyspnea for 2 years. The symptom occurred intermittently in the awake state and was improved after rest. There was no cold, fever, cough, sputum, chest tightness and chest pain at onset, night sweats, or weight loss. There was no history of tobacco use or exposure to noxious stimuli. No special medical history was reported.
On physical examination, the patient had normal blood pressure and heart rate. The SpO2 measured by pulse oximetry was 80% on room air, but increased to 96% after oxygen therapy (FiO2 33%). No cyanosis, pallor, jaundice or plethora was found. There was no polish or scarring on his nails that could interfere with pulse oximetry readings. Cardiac, respiratory, abdominal exams and muscle strength were normal.
His compete blood count revealed a hemoglobin of 14.7 g/dL and basic chemistry panel was unremarkable. As the likeliest causes of dyspnea are diseases involving the pulmonary or cardiac systems, a computed tomography (CT) scan of the chest was performed the same day, which showed almost clear lung fields except for some strips and calcifications. Pulmonary function tests (PFTs) showed FEV1 3.74 L (predicted value 3.53 L), FVC 4.48 L (predicted value 4.2 L), FEV1/FVC ratio 83.3% (normal ≥ 70%), carbon monoxide diffusing capacity (DLCO) 32.79 mL/min/mmHg (predicted value 29.44 mL/min/mmHg) which was 111.4% of the predicted value (normal ≥ 80% predicted). Assessment of airway responsiveness with methacholine provocation was negative. Electrocardiography demonstrated normal sinus rhythm. Echocardiogram suggested normal cardiac structure and function. No evidence of pulmonary hypertension or anatomic shunting was revealed. Ventilation and perfusion scan indicated a low likelihood of pulmonary embolus. Fiberoptic bronchoscopy showed no abnormality in all levels of bronchus. Cultures of bronchial alveolar lavage fluid (BALF) and sputum were negative.
Unexplained dyspnea may also be associated with neuropsychiatric disorders. Our psychological assessment results indicated that the patient may have no or minimal negative emotions (depression and/or anxiety) or related mental health problems.
Arterial blood gas (ABG) was performed when dyspnea occurred, and the results were: pH 7.39 (normal 7.35–7.45), PaO2 94.2 mmHg (normal 80–100 mmHg), SpO2 87.1% (normal 95–98%), PaCO2 43.6 mmHg (normal 35–45 mmHg) and bicarbonate 25 mmol/L (normal 22–26 mmol/L). Notably, there was a mismatch between the patient’s SpO2 and PaO2. Thus, ABG was repeated, with similar results to the previous. However, his carboxyhemoglobin and deoxyhemoglobin were increased to 2.2% (normal 0.5–1.5%) and 12.5% (normal1.4–4.9%), respectively. Methemoglobin (0.6%, normal 0.2–0.8%) was undetectable. Toxicological screening test including methylamphetamine, methylenedioxymethamphetamine, ketamine, morphine and heroin were negative. Therefore, an altered oxygen affinity variant hemoglobin (Hb) was suspected.
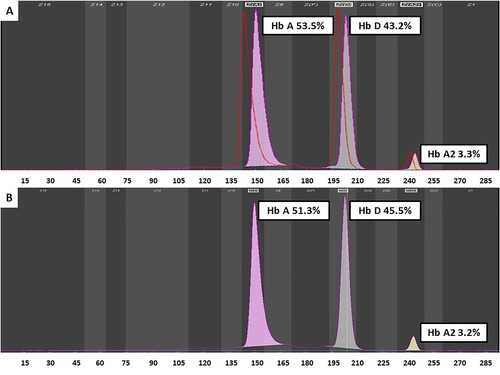
The patient’s P50 was elevated at 47.3 mmHg (normal 24–27 mmHg), consistent with a low oxygen affinity variant. Hemoglobin fraction by capillary electrophoresis revealed Hb A 53.5% (normal 94.5–97.3%), Hb A2 3.3% (normal 2.5–3.5%), Hb F 0% (normal 0–2.5%). However, an abnormal HbD quantified at 43.2% was found. These results were suggestive of a β-globin chain variant [Citation1].
Altered oxygen affinity variant Hbs are caused by rare mutations in globin genes. A family history of hypoxemia may strongly indicate an underlying genetic disorder. Upon further questioning, the patient reported that some of his family members also had low SpO2 readings (). Since only screening test for thalassemia that covered mutations in hot spots or whole exome sequencing (WES) was available, WES was undertaken using the next generation sequence (NGS) method, and the results were verified by Sanger sequence method. DNA analysis identified heterozygosity for a mutation in exon 2 at codon 38 (HBB: c.112T>A, p.Trp38Arg), which decodes to a substitution of the amino acid tryptophan (Try) by arginine (Arg), known as Hb Rothschild (). His 3-year-old daughter revealed the same Try to Arg amino acid change by Sanger sequence method, with the abnormal HbD quantified at 45.5% (). However, no such mutation was found in the Hb gene in the patient’s brother.
Figure 1. Pedigree of the patient’s family with low SpO2 readings.
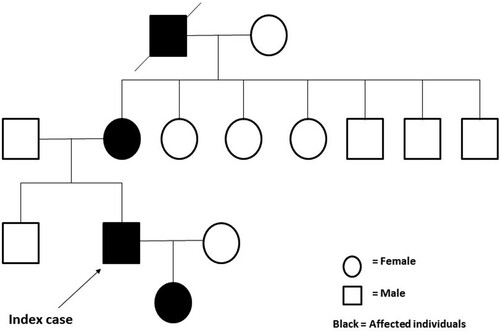
Figure 2. Sanger sequence identified heterozygosity for a mutation of the β-hemoglobin chain in exon 2 at codon 38 (HBB: c.112T>A, p.Trp38Arg).
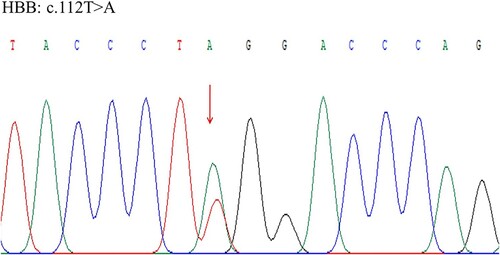
Figure 3. The electropherogram of the patient (A) and his daughter (B) derived from capillary electrophoresis.
Discussion
Pulse oximetry is a common non-invasive method for measuring oxygen saturation in clinical practice. However, this case highlights the limitations of pulse oximetry. ABG samples using modern blood gas analyzers can overcome these limitations to provide more reliable results. Typically, low oxygen affinity Hb variants have low SpO2 readings accompanied by low SaO2, with no evidence of cardiac or respiratory disease. P50 is one of the common metrics to quantify Hb oxygen affinity. When an altered oxygen affinity variant is suspected, P50 testing should be performed. Nevertheless, in some unusual Hb variants (eg, Hb Titusville and Hb Bonn), altered absorption spectra results in falsely low SpO2 and normal SaO2 [Citation2,Citation3], and P50 should be interpreted carefully since it relies on optical properties of Hb at a 560 nm wavelength.
Diagnosis of such a hemoglobinopathy requires a strong suspicion of the disease, which relies on relevant clinical background and experience. The family history of this patient initially missed has critical significance, suggesting the possibility of a genetic disorder. In fact, hemoglobinopathies are common inherited disorders which affect around 3–7% of world population and increasing cases are being discovered [Citation4]. Most hemoglobinopathies are asymptomatic, which may be the reason why the affected family members of our patient did not consult doctors. To complete the family study, DNA analysis was performed and revealed the same missense mutation in HBB gene of both the patient and his daughter, which leads to amino acid change from Try to Arg at position 38 of the β-hemoglobin chain. Tryptophan at this position is reported as an important contact point at the α1β2 and α2β1 interfaces. Due to the destabilizing effect of the substitution of tryptophan by arginine, Hb Rothschild has a tendency to dissociate into αβ dimers with lower oxygen affinity than the intact tetramer [Citation5]. One explanation for the asymptomatic disposition of Hb Rothschild subjects is that deoxygenation may increase the tetramer stability and the oxygen affinity of T-state Hb Rothschild [Citation6,Citation7]. Hb Rothschild is an uncommon hemoglobin abnormality found mainly in Caucasians [Citation8]. To the best of our knowledge, this is the first report of Hb Rothschild in a Chinese family. However, atypical symptoms may result in underdiagnosis of Hb Rothschild. Considering the short-time consumption and low cost, Sanger sequence method rather than WES is preferred to confirm the diagnosis if Sanger sequencing for the whole Hb chain is available. For physicians (especially not in this field), this case also highlights the importance of rational diagnostic flow in such diseases to avoid unnecessary tests.
For most subjects with low oxygen affinity Hbs, no specific treatment is required and supportive care is indicated. Our patient received oxygen therapy until discharge with improvement of his symptoms and oxygen saturation. He is now followed up in outpatient. Previous studies have reported some long-term pulmonary complications of low oxygen affinity β-chain hemoglobinopathies, including pulmonary hypertension caused by chronic hypoxia, hemolysis-related alveolar fibrosis, and pulmonary dysfunction [Citation9,Citation10]. The contributory effects of Hb Rothschild on pulmonary diseases still needs long-term follow-up and further evaluation. Also, little is known about the phenotype if this variant is combined to another HBB or HBA variant. Better understanding in this area and screening patients with suspected family history during genetic counseling would help avoid unnecessary cardiorespiratory interventions. Among those for whose diagnosis remains elusive and unexplainable, specialty referral (e.g. pulmonologist, cardiologist, hematologist or multidisciplinary discussion) may help identify a potentially treatable underlying cause.
Statement of ethics
This is a non-interventional study. Written informed consents were obtained from the patients for clinical diagnosis and research purposes.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Keren DF, Shalhoub R, Gulbranson R, et al. Expression of hemoglobin variant migration by capillary electrophoresis relative to hemoglobin A2 improves precision. Am J Clin Pathol. 2012;137(4):660–664.
- Verhovsek M, Henderson MP, Cox G, et al. Unexpectedly low pulse oximetry measurements associated with variant hemoglobins: a systematic review. Am J Hematol. 2010;85(11):882–885.
- Zur B, Bagci S, Ludwig M, et al. Oxygen saturation in pulse oximetry in hemoglobin anomalies. Klin Padiatr. 2012;224(4):259–265.
- Ghosh K, Ghosh K, Agrawal R, et al. Recent advances in screening and diagnosis of hemoglobinopathy. Expert Rev Hematol. 2020;13(1):13–21.
- Alli NA, Wessels P, Rampersad N, et al. Detection of Hb Rothschild HBB: c.[112T>A or 112T>C], through high index of suspicion on abnormal pulse oximetry. Hemoglobin. 2017;41(2):137–139.
- Sharma VS, Newton GL, Ranney HM, et al. Hemoglobin Rothschild (beta 37(C3)Trp replaced by Arg): a high/low affinity hemoglobin mutant. J Mol Biol. 1980;144(3):267–280.
- Perutz MF, Muirhead H, Cox JM, et al. Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: the atomic model. Nature. 1968;219(5150):131–139.
- Bruns CM, Thet LA, Woodson RD, et al. Hemoglobinopathy case finding by pulse oximetry. Am J Hematol. 2003;74(2):142–143.
- Wu Y, Ramani GV, Gai Q, et al. Rare hemoglobinopathy presenting as progressive dyspnea. Am J Hematol. 2010;85(5):355–357.
- Wille RT, Krishnan K, Cooney KA, et al. Familial association of primary pulmonary hypertension and a new low-oxygen affinity beta-chain hemoglobinopathy, Hb Washtenaw. Chest. 1996;109(3):848–850.