
ABSTRACT
Background:
The V617F mutation of Janus-associated kinase 2 (JAK2) is common in myeloproliferative neoplasms (MPN). JAK2 V617F mutation can be detected in patients with de novo acute myeloid leukemia (AML), but de novo acute promyelocytic leukemia (APL) with JAK2 V617F mutation is rare.
Case presentation:
We report a case of APL with both the t(15;17) translocation as well as the JAK2 V617F mutation that transformed into MPN (PV/ET).
Conclusions:
A de novo APL patient presented initially with JAK2 V617F. After ATRA and ATO dual induction and chemotherapy consolidation, the patient achieved complete remission (CR) with undetectable PML/RARα. However, the JAK2 V617F remained positive, and the patient developed MPN (PV/ET) 22 months later, which responded well to interferon therapy.
AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; ATO, arsenic trioxide; BM, bone marrow; CR, complete remission; ET, essential thrombocythemia; Hb, hemoglobin; JAK2, Janus-associated kinase 2; MPN, myeloproliferative neoplasms; PLT, platelets; PMF, primary myelofibrosis; PML/RARα; PV, polycythemia vera; WBC, white blood cells.
Introduction
JAK2, a nonreceptor tyrosine kinase, plays a fundamental role in the signal transduction of hematopoiesis [Citation1]. The activating V617F mutation of JAK2 tyrosine kinase is a hot spot mutation and is well-studied in BCR/ABL-negative chronic myeloproliferative neoplasms (MPN), which inlcude polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) [Citation2]. However, the JAK2 V617F mutation is also common in acute myeloid leukemia (AML) that follows an MPN, a condition that is often termed as secondary AML or blast-phase MPN (AML-MPN). The JAK2 V617F mutation can occur, although rarely, in AML without an antecedent MPN, which is described as de novo AML. JAK2 V617F mutation is more common in patients with erythroid or megakaryoblastic AML. De novo acute promyelocytic leukemia (APL) with JAK2 V617F mutation is extremely rare [Citation3–7]. Here, we describe the clinicopathologic findings of a de novo APL case with JAK2 V617F that transformed into MPN after 22 months of APL treatment. Our report provides insights into underlying pathogenesis and offers potential therapeutic targets that might be useful in treating this type of patient.
Case presentation
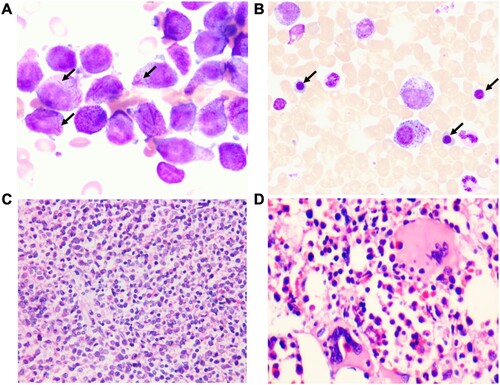
A 44-year-old female was initially admitted in February 2020 for recurrent skin and gingival mucosal bleeding. Her complete blood count revealed a white blood cell (WBC) count of 23.5 × 109/L (normal values: 4 ∼ 10 × 109/L), RBC 7.94 × 1012/L (normal values: 3.5 ∼ 5 × 109/L), hemoglobin (Hb) 210 g/L (normal values: 110 ∼ 150 g/L), platelet (PLT) 10 × 109/L (normal values: 100 ∼ 300 × 109/L). She did not have splenomegaly on admission. Bone marrow (BM) aspiration revealed accumulation of abnormal promyelocytic blasts ((A)). BM biopsy showed hypercellularity and diffuse proliferation of immature myeloid cells. Erythroid precursors were markedly decreased and megakaryocytes are occasionally seen. No reticulin fibrosis was detected ((C)). Flow cytometry of peripheral mononuclear cells showed that the abnormal cell population mainly expressed CD13, CD33, CD64, CD117, and MPO. Molecular studies were positive for PML/RARα (bcr3 type). Cytogenetics revealed t(15;17)(q24;q21) in 20 metaphases. A diagnosis of high-risk APL was made.
Figure 1. Morphologic features of BM sample from the patient. (A) BM aspiration smear on admission in February 2020 revealed abnormal accumulations of immature cells containing multiple Auer rods in the cytoplasm (arrow) (100×). (B) BM aspiration smear in November 2020 after APL CR revealed erythroid cells were significantly increased, mainly composed of middle and late juvenile erythrocytes. (arrow) (100×). (C) BM biopsy on admission in February 2020 showing hypercellularity of immature atypical myeloid cells (H&E-stained image, 40×). (D) BM biopsy in November 2020 after APL CR. H&E-stained sections show active hyperplasia of nucleated myeloid and erythroid cells. The morphology of those cells was generally normal (40×).
The patient was immediately treated with oral ATRA (20 mg/m2/day) and intravenous arsenic trioxide (ATO, 0.16 mg/kg/day) (). Daunorubicin and cytarabine were used as a cytoreductive therapy. However, after three days of treatment, her WBC count began to increase rapidly, reaching as high as 36.5 × 109/L on day 7. A diagnosis of differentiation syndrome was made, which was successfully managed with treatments of dexamethasone and other supportive measures. She became PCR negative for PML/RARα after achievement of CR at two months with the completion of dual induction chemotherapy. The patient was then put on consolidation chemotherapy with daunorubicin/harringtonine and cytarabine for a total of three cycles. She continued ATRA and ATO treatment throughout consolidation.
Figure 2. Clinical course of the patient. White blood counts (WBC), hemoglobin (Hb) and platelet (PLT) counts during the treatment. DS, differentiation syndrome; DA, daunorubicin + cytarabine; HA, harringtonine + cytarabine;
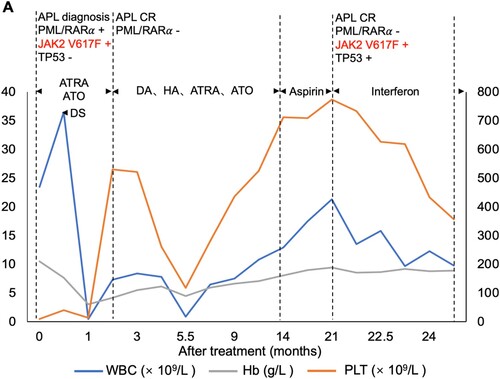
Surprisingly, at the 19th month after APL CR, the patient had a rapid increase in WBC, Hb, and PLT (WBC 21.3 × 109/L, RBC 6.65 × 1012/L, Hb 189 g/L, PLT 774 × 109/L). BM aspiration smear at this point revealed increased mature neutrophils. Erythrocytes were mainly composed of acidophilic normoblasts. The numbers of megakaryocytes and platelets were significantly increased. BM biopsy showed active hyperplasia of nucleated, myeloid, and erythroid cells. No reticulin fibrosis was seen. Molecular studies were negative for the PML/RARα, but positive for mutations of JAK2 V617F and TP53 P278R. Cytogenetics revealed a normal karyotype.
To determine whether the patient has the JAK2 V617F mutation at the initial diagnosis of APL, RNA was extracted from paraffin sections of bone marrow biopsy samples at the time of initial diagnosis, and then cDNA was synthesized for gene mutation analysis. The results showed the patient had the JAK2 V617F mutation at the initial diagnosis of APL. Since there were no dysplastic megakaryocytes or reticulin fibrosis, based on clinical and laboratory findings, the patient was diagnosed as PV/ET. She was given Interferon 5,000,000 IU once every two days. At the time of preparation of this report, the patient’s condition has been considerably improved.
Discussion
The main clinical manifestations of APL are leukopenia and severe bleeding, which are extremely dangerous with high mortality [Citation8]. However, the discoveries of ATRA and ATO dual induction have turned the highly fatal form of leukemia into a curable disease. Currently, ∼90% of newly diagnosed APL patients achieve CR, and over 70% of patients are curable after ATRA and ATO dual induction therapy followed by subsequent consolidation chemotherapies [Citation9]. APL accounts for ∼10% of de novo adult AML. APL is typically characterized by the fusion oncogene PML/RARα, resulted from the unique chromosomal translocation t(15;17), which initiates APL by blocking differentiation and increasing self-renewal of leukemic progenitor cells [Citation10, Citation11]. Other than the leukemogenic t(15;17), APL may have chromosomal and genetic abnormalities. However, the association of APL with the JAK2 V617F as reported in this study is very rare.
JAK2 encodes Janus kinase 2, belonging to a nonreceptor tyrosine kinase (JAK family), which plays a fundamental role in the signal transduction of hematopoiesis-related cytokines and growth factors such as erythropoietin and thrombopoietin. JAK2 V617F is an activation mutation in BCR/ABL-negative MPN [Citation12], which is present in 95% of patients with PV, and in 50% of patients with ET and PMF [Citation13]. As a result, JAK2 V617F constitutively activates thrombopoietin and erythropoietin signaling pathways, thereby contributing to the pathogenesis of ET and PV [Citation14]. The JAK2 inhibitors and long-acting interferon have significantly improved the prognosis of MPN patients [Citation15–18]. Interferon is recommended for the first-line treatment of some special MPN groups, such as younger patients, especially women with high-risk PV and those who need cytoreductive therapy [Citation16, Citation19]. Interestingly, our patient developed severe differentiation syndrome after ATRA and ATO treatment. It is speculated that the clinical presentation with severe differentiation syndrome may be the result of JAK2 V617F mutation, which enhances the inflammatory downstream signals leading to excessive inflammatory response [Citation7].
Transformation of MPN, especially PV and PMF, into AML (AML-MPN) is well-studied, with an incidence of about 15%. Among the AML subtypes transformed from MPN, AML with t (8; 21) (q22; q22) translocation is more common. Patients with AML-MPN were more common with splenomegaly, complex karyotype, MPN-like megakaryocyte abnormalities, and higher JAK2 V617F mutation frequency. The prognosis of AML-MPN is poor, which may be related to complex gene variation. Studies have shown that JAK2 V617F is an independent indicator of poor prognosis, and the recurrence rate of t (8; 21) (q22; q22) AML patients with this mutation is high [Citation20].
JAK2 V617F mutation in de novo AML are rare [Citation6, Citation7]. However, it is more common in patients with erythroid or megakaryoblastic AML [Citation3–5, Citation21, Citation22]. JAK2 V617F mutation in de novo APL are extremely rare. Mamorska-Dyga et al. reported a young male with concurrent primary APL and JAK2 V617F-positive MPN. When the patient reached molecular remission after double induction and consolidation therapies, JAK2 V617F mutation persisted and PLT gradually increased to 1500 × 109/L, accompanied by typical PMF manifestations such as megakaryocyte dysplasia and bone marrow reticular fibrosis. It is speculated that the dominant APL clone at the initial diagnosis masked the phenotype of MPN. The MPN clone became dominant after the APL clone was suppressed [Citation6]. We found that the TP53 P278R mutation was present when the patient progressed to MPN. However, this mutation was not detected at the APL presentation via RNA extraction form original paraffin samples. The TP53 P278R mutation has been detected in AML, CLL/SLL, MDS, lymphoplasmacytic lymphoma, and solid tumors. This mutation is located in the DNA binding domain of tp4s3 protein and is a loss-of-function mutation. The TP53 P278R mutation is a significant adverse prognostic factor in MDS and secondary AML [Citation23].
In summary, we report a case of APL with JAK2 V617F mutation transformed into chronic MPN. The patient initially had no MPN phenotypes such as splenomegaly, megakaryocyte proliferation, and bone marrow reticular fibrosis, indicating no previous history of MPN. Therefore, the patient initially had a typical de novo APL with JAK2 V617F mutation. The MPN clone with JAK2 V617F mutation progressed into MPN 22 months after APL achieved CR.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Marneth AE, Mullally A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med. 2020;10(2):a034876.
- Skov V. Next generation sequencing in MPNs. lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers (Basel). 2020;12(8), article no. 2194.
- Lee JW, Kim YG, Soung YH, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25(9):1434–1436.
- Swaminathan S, Madkaikar M, Ghosh K, et al. Novel immunophenotypic and morphologic presentation in acute myeloid leukemia (AML) with JAK2 V617F mutation. Eur J Haematol. 2010;84(2):180–182.
- Vicente C, Vázquez I, Marcotegui N, et al. JAK2-V617F activating mutation in acute myeloid leukemia: prognostic impact and association with other molecular markers. Leukemia. 2007;21(11):2386–2390.
- Mamorska-Dyga A, Wu J, Khattar P, et al. Acute promyelocytic leukemia co-existing with JAK2 V617F positive myeloproliferative neoplasm: a case report. Stem Cell Investig. 2016;3:8.
- Braun TP, Maxson JE, Agarwal A, et al. Acute promyelocytic leukemia with JAK2 V617F and severe differentiation syndrome. Leuk Res Rep. 2015;4(1):8–11.
- Salhotra A, Mei M. Acute promyelocytic leukemia: update on risk stratification and treatment practices. Cancer Treat Res. 2021;181:45–55.
- Maimaitiyiming Y, Zhu HH, Yang C, et al. Biotransformation of arsenic trioxide by AS3MT favors eradication of acute promyelocytic leukemia: revealing the hidden facts. Drug Metab Rev. 2020;52(3):425–437.
- Soriani S, Mura C, Panico AR, et al. Rapid detection of t(15;17)(q24;q21) in acute promyelocytic leukaemia by microwave-assisted fluorescence in situ hybridization. Hematol Oncol. 2017;35(1):94–100.
- Maimaitiyiming Y, Wang QQ, Yang C, et al. Hyperthermia selectively destabilizes oncogenic fusion proteins. Blood Cancer Discov. 2021;2(4):388–401.
- Leonard JP, Martin P, Roboz GJ. Practical implications of the 2016 revision of the world health organization classification of lymphoid and myeloid neoplasms and acute leukemia. J Clin Oncol. 2017;35(23):2708–2715.
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148.
- Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15.
- Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208.
- Loscocco GG, Vannucchi AM. Role of JAK inhibitors in myeloproliferative neoplasms: current point of view and perspectives. Int J Hematol. 2022;115(5):626–644.
- Bose P, Verstovsek S, Cortes JE, et al. A phase 1/2 study of ruxolitinib and decitabine in patients with post-myeloproliferative neoplasm acute myeloid leukemia. Leukemia. 2020;34(9):2489–2492.
- How J, Hobbs G. Use of interferon alfa in the treatment of myeloproliferative neoplasms: perspectives and review of the literature. Cancers (Basel). 2020;12(7), article no. 1954.
- Aynardi J, Manur R, Hess PR, et al. JAK2 V617F-positive acute myeloid leukaemia (AML): a comparison between de novo AML and secondary AML transformed from an underlying myeloproliferative neoplasm. A study from the bone marrow pathology group. Br J Haematol. 2018;182(1):78–85.
- Jelinek J, Oki Y, Gharibyan V, et al. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005;106(10):3370–3373.
- Aivalioti MM, Bartholdy BA, Pradhan K, et al. PU.1-dependent enhancer inhibition separates Tet2-deficient hematopoiesis from malignant transformation. Blood Cancer Discov. 2022;3(5):444–467.
- Sallman DA, McLemore AF, Aldrich AL, et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood. 2020;136(24):2812–2823.