
ABSTRACT
Objectives
To explore the application of third-generation sequencing (TGS) for genetic diagnosis and prenatal genetic screening of thalassemia genes.
Methods
Two groups of subjects were enrolled in this study. The first group included 176 subjects with positive hematological phenotypes for thalassemia. Thalassemia-associated genes were detected simultaneously in each sample using both the PacBio TGS platform based on single-molecule real-time (SMRT) technology and the conventional PCR-reverse dot blot (PCR-RDB). Sanger sequencing was used for validation when results were discordant between the two methods. The second group included 53 couples with at least one partner having a positive thalassemia hematological phenotype, and they were screened for homotypic thalassemia variants by TGS, and the risk of pregnancies with babies presenting with severe thalassemia, was assessed.
Results
Of the 176 subjects, 175 had concordant genotypes between the two methods, including 63 normal subjects and 112 α- and/or β-thalassemia gene carriers, with a concordance rate of 99.43%. TGS detected a rare β-thalassemia gene variant −50 (G > A) that was not detected by conventional PCR-RDB. TGS identified seven of the 53 couples as homotypic thalassemia gene carriers, five of whom were at risk of pregnancies with severe thalassemia.
Conclusion
TGS could effectively detect common and rare thalassemia variants with high accuracy and efficiency. This approach would be suitable for prenatal thalassemia genetic screening in areas with high incidence of thalassemia.
Introduction
Thalassemia is a heterogeneous group of hereditary hemolytic disorders caused by the absence or insufficient synthesis of α-globin or β-globin and is one of the most common monogenic genetic diseases in the world[Citation1]. It is estimated that approximately 1-5% of the world's population are carriers of mutations within the thalassemia genes[Citation2]. It is mainly distributed over the Mediterranean coast, North Africa, the Middle East, the Indian mainland, Southeast Asia and Southern China [Citation1, Citation3]. Guizhou is a multi-ethnic province located in the southwest of China and is an area where the incidence rate for thalassemia is high.
In most clinical laboratories within China, detection of common mutant phenotypes in the population is by first-line techniques after blood phenotypic testing. If the test result is negative, then second-line techniques are used to detect rare or unknown mutations [Citation4, Citation5]. For thalassemia gene detection, the first-line techniques include Gap-Polymerase Chain Reaction (Gap-PCR), real-time PCR, reverse dot blot (RDB) and probe melting curve analysis (PMCA), while second-line techniques include multiplex ligation-dependent probe amplification (MLPA), array-CGH, and Sanger sequencing[Citation4, Citation5]. The main drawbacks of these methods are that they are cumbersome, labor-intensive and have a risk of missed diagnoses, limited resolution, and able to detect a limited range of mutations [Citation6]. In recent years, next-generation sequencing (NGS) technology has also been used for the molecular diagnosis of thalassemia[Citation7, Citation8]. However, disadvantages of the NGS technology include too short read length, introduction of PCR amplification errors, GC bias [Citation9] and high costs[Citation8, Citation10]. In addition, the NGS technology cannot effectively detect repetitive regions and structural variants within the human genome[Citation11]. The third-generation sequencing (TGS) technology developed by Pacific Biosciences (PacBio) effectively solves this problem whereby sequencing accuracy is high, especially HiFi Reads generated by the Circular Consensus Sequencing (CCS) mode with an accuracy of up to 99.999% [Citation11]. This sequencing platform uses a simplified sample preparation procedure and reduces sequencing time and costs [Citation12]. Taken together, TGS provides a solid technology for large-scale clinical applications.
Some previous studies confirmed that the SMRT technology for genetic diagnosis of thalassemia was advantageous in terms of wide detection range, high efficiency and high accuracy [Citation1, Citation13]. However, no related study has been undertaken on using the TGS technology for thalassemia gene detection in the population of Guizhou province. In this study, 176 individuals from Guizhou province with suspected thalassemia were screened using the TGS technology and conventional PCR-RDB simultaneously, while Sanger sequencing was used to evaluate the diagnostic performance of the TGS technology. Subsequently, 53 couples with at least one positive thalassemia phenotype were tested using SMRT to screen for couples with high-risk pregnancies and advised to perform prenatal diagnosis.
Materials and methods
Subjects
The first group (n = 176) of subjects was aged between 3 months to 80 years old and made up of 67 males and 109 females who presented at Guizhou Provincial People's Hospital from January 2020 to December 2021 and assessed to have a positive thalassemia phenotype. The second group of subjects consisted of 53 couples that presented at the same hospital between January 2022 and June 2022 to undergo genetic screening for thalassemia in preparation for pregnancy or during the first trimester, with at least one member of each couple having a positive thalassemia phenotype. The positive hematological phenotype of thalassemia met at least one of the following inclusion criteria: (1) routine hematology examination showed abnormal mean corpuscular volume (MCV) ≤ 82 fL and/or a mean corpuscular hemoglobin (MCH) ≤ 27 pg; (2) hemoglobin electrophoresis showed HbA2 ≥ 3.5%, elevated Hemoglobin F (Hb F) or abnormal hemoglobin. This study was approved by the Ethics Committee of Guizhou Provincial People ‘s Hospital (approval number 2022-05), and all study subjects or their legal guardians signed an informed consent form.
Hematologic screening
All samples were screened for the thalassemia phenotype using routine hematological methods[Citation13]. An automatic blood cell analyzer (Sysmex XN-100-4, Japan) was used to analyze red blood cell parameters and an automatic electrophoresis analytic system (Hydrasys LC; Sebia Electrophoresis, Evry, France) was used for hemoglobin analysis.
Extraction of genomic DNA
The magnetic bead-beating method[Citation1] with the NP968 Nucleic Acid Extraction System (Xi'an Tianlong Science and Technology, Xìan, China) was used to extract genomic DNA from the blood samples. The concentration and purity of extracted DNA were assessed with a NanoDrop spectrophotometer (Thermo Scientific, USA). The ratio of absorbance values at OD260 nm/280 nm for the extracted DNA was between 1.5 and 2.5, and the concentration was 20-40 ng/μl. The extracted DNA was stored at −20°C.
PCR-RDB for detection of α- and β-thalassemia
Five microliters of individual extracted DNA samples were used for PCR amplification. The amplification products were tested using the α/β -thalassemia gene detection kits (Hybriobio Limited, Guangzhou, China) in an automatic nucleic acid molecular hybridizer (HBHM-3000S, Hybriobio Limited, Guangzhou, China). The method was used to detect three common α-thalassemia deletions (–SEA, -α3.7, -α4.2) in the Chinese population, three non-deletional α-thalassemia variants (Hb Constant Spring (CS), Hb Quong Sze (QS), Hb Westmead (WS)), and 19 known β-thalassemia variants at 17 loci which were c.126-129delCTTT, c.130G > T, c.316-197C > T, c.52A > T, c.45_46insG, c.−78A > G, c.−79A > G, c.216_217insA, c.79G > A, c.92 + 1G > A, c.92 + 1G > T, c.84_85insC, c.315 + 5G > C, c.−50A > C, c.−11_−8delAAAC, c.2T > G, c.94delC, c.−80T > C and c.−82C > A. The assays were performed according to the manufacturer’s protocol [Citation14, Citation15].
TGS for detection of α- and β-thalassemia variants
The extracted genomic DNA samples were tested for variants within the α- and β-thalassemia genes using the single-molecule real-time (SMRT) technology. Briefly, the target regions were amplified by long-range multiplex PCR, and then the PCR fragments were end-repaired and ligated to the PacBio barcoded adapters to form individual dumbbell-shaped pre-libraries. Then equal mass of individual pre-libraries were pooled and mixed with the sequencing primer and DNA polymerase to form the PacBio sequencing library. Sequencing was performed using the PacBio Sequel II platform (PacBio, Menlo Park, CA, USA) under the circular consensus-sequencing (CCS) mode with a run time of 30 hours. The average polymerase read length was on average between 70-80 kb. The SMRT Link system provided by PacBio was used to convert raw subreads into high-fidelity CCS reads, which were divided into individual samples based on different barcodes, and then aligned to the genome build hg38. FreeBayes1.3.4 (Biomatters, San Diego, CA) was used to analyze single-nucleotide variations (SNVs) and indels. The SNVs, indels, large deletions and structural variations were annotated according to HbVar database of hemoglobin variants (https://globin.bx.psu.edu/), Ithanet public resource on Hb disorders (https://www.ithanet.eu/) and Leiden Open Variation database (LOVD) (https://www.lovd.nl/).
Validation for discordant thalassemia variants
Discordant variants identified by PCR-RDB and TGS were verified using Sanger sequencing. The primers for amplifying the β-globin gene were: F: 5’AACTCCTAAGCCAGTGCCAGAAGAGC3’ and R: 5’ATGCACTGACCTCCCACATTCCC3’.
Results
Concordance between conventional RDB and TGS for thalassemia genetic testing
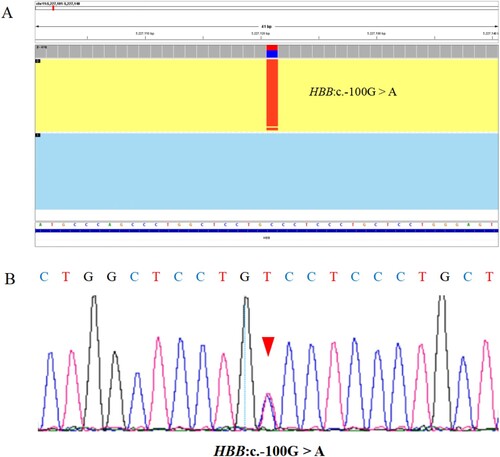
Of the 176 samples, 175 had consistent genotypes between the two assays, comprising of 63 normal individuals and 112 α- and/or β-thalassemia gene carriers, with a concordance rate of 99.43% (175/176) (). A total of 15 types of variants (122 alleles) were detected in 176 samples by TGS, including six types of α-thalassemia variant (58 alleles) and nine types of β-thalassemia variant (64 alleles) (). An additional rare β-thalassemia variant c.−100G > A in one allele was detected by TGS (). The detection rate for thalassemia variants in subjects with positive hematological phenotypes was 64.20% (113/176). The frequencies of the 14 common variants were consistent with those reported in the literature [Citation16, Citation17].
Table 1. Concordance between conventional RDB and TGS for thalassemia genetic testing in 176 samples.
Table 2. Thalassemia variants of 176 samples identified by TGS and conventional PCR-RDB technology.
Clinical phenotype of the patient with a rare β-thalassemia variant
The rare variant c.−100G > A in the HBB gene detected by TGS was validated by Sanger sequencing (). c.−100G > A was identified as a likely rare β+ thalassemia variant [Citation18–20]. The patient with heterozygous c.−100G > A variant was an 18-year-old male with no clinical symptoms, and he also carried a heterozygous –SEA deletion. Red blood cell indices showed that the cells were microcytic but the hemoglobin level in the cells was normal (Hb 136 g/L, MCV 68.8 fl, MCH 21.1 pg, MCHC 307 g/L).
Figure 1. The inconsistent sample as detected by TGS and conventional PCR-RDB technology was verified by Sanger sequencing. (A) The Integrative Genomics Viewer plot of HBB:c.−100G > A variant detected by TGS but not PCR-RDB, and the red boxed area indicates the position of this variant. (B) The Sanger sequencing output profile of the HBB:c.−100G > A variant sequence, and the red arrow indicates the position of this variant.
Genotyping of 53 couples screened for thalassemia
TGS was performed for both couples if one of them had positive hematological parameters. Of the 53 couples screened, TGS detected 13 couples without α- and β-thalassemia gene variants, 13 couples with only one partner carrying α-thalassemia variants, 12 couples with only one partner carrying β-thalassemia variants, 8 couples carrying different types of thalassemia, and 7 couples carrying the same type of thalassemia. Of the 7 couples, five were homotypic α-thalassemia carriers while the other two couples were homotypic β-thalassemia carriers (). While two couples were both α-thalassemia silent carriers and did not have a high pregnancy risk, the other five couples were at risk of having children with moderate or severe forms of thalassemia and required prenatal diagnosis. The five couples accounted for 9.43% (5/53) of couples with at least one partner who was positive for the thalassemia hematological phenotype.
Table 3. Genotyping of 53 couples screened for thalassemia.
Four variants of unknown significance (VUSs) in the HBB gene were detected by TGS in five individuals from the two groups, including two heterozygous carriers of the c.341T > A variant, one heterozygous carrier of the c.316-45G > C variant, one heterozygous carrier of the c.315 + 308delA variant, and one heterozygous carrier of the c.316-3C > T variant. The individual with the c.316-3C > T mutation also carried a c.126-129delCTTT mutation and was a 17-year-old male with no history of blood transfusions. Hematological examination showed mild microcytic hypochromic anemia (Hb 114 g/L, MCV 63.8fl, MCH 18.6 pg, MCHC 290.8 g/L), and Hb electrophoresis showed elevated HbA2 (HbA 92.1%, HbF 1.3%, HbA2 6.6%), consistent with the β-thalassemia minor phenotype.
Discussion
In the past, conventional genetic testing methods for thalassemia were mainly aimed at detecting common variants in specific populations and could not detect rare variants. Several provinces in southern China have launched the ‘zero birth’ program for fetuses with severe thalassemia, which requires clinical laboratories to conduct tests that are able to detect rare thalassemia variants. TGS technology has only just begun to be used clinically, over the last two years, for thalassemia genetic testing. The TGS platform has shown advantages over conventional detection methods and the NGS technology, in terms of detection rates. In this study, TGS technology was used for thalassemia genetic testing and prenatal genetic screening in Guizhou Province, China, to evaluate its potential value for clinical application.
The results of this study have shown that the detection rate for thalassemia gene mutations in individuals with a positive thalassemia hematological phenotype was 64.20% (113/176). In 53 couples with at least one positive hematological phenotype, TGS detected homotypic thalassemia genes in 5 couples accounting for 9.43% (5/53) of the couples tested and these couples required prenatal diagnostic tests. These results propose that Guizhou Province could be an area of high thalassemia incidence with a high positive detection rate for variant thalassemia genes in the population. Nearly one-tenth of the couples with at least one positive hematological phenotype were at risk of giving birth to a child with thalassemia major and therefore, required prenatal diagnosis. Taken together, it is necessary to strengthen thalassemia screening of the reproductive age population in Guizhou province and adopt the thalassemia gene detection methods suitable for the local population to effectively prevent the birth of children with severe thalassemia.
Among 176 individuals with positive hematological phenotypes of thalassemia, the concordance rate between conventional PCR-RDB and TGS for the detection of thalassemia genes was 99.43% (175/176), which was significantly higher than that reported elsewhere. Luo et al[Citation1] reported that the positive detection rate of TGS was 9.91% higher than that of conventional techniques in 434 enrolled cases. Zhuang et al[Citation21] reported a 7.14% incremental yield in rare α-and β-globin gene variants by TGS technology as compared to the conventional detection methods. The difference in concordance rates may be due to our small sample size, but another reason is that we only took definite causative variants into consideration, while other studies included VUSs for statistical analysis. For example, Luo et al[Citation1] included likely benign variants such as c.316-45G > C and c.315 + 308delA in the statistical analysis. As far as pathogenic variants are concerned, the detection rates for both methods are highly concordant. Therefore, conventional PCR-RDB is still an inexpensive and effective genetic test for thalassemia in economically underdeveloped areas.
Hundreds of α- and β-globin gene variants contribute to thalassemia, and these defective types and their combinations are complex. The TGS platform is able to cover the full length of both α- and β-globin genes due to its capacity to produce ultra-long reads[Citation11, Citation22]. Previous studies have shown that the advantage of TGS for thalassemia genetic screening is not only in detecting rare SNVs and Indel-type mutations, but also the ability to detect structural rearrangements of the α-globin gene cluster, including the α-globin gene triplication and HongKong αα (HKαα) allele[Citation23]. In addition, TGS adopts the haplotype analysis mode, which facilitates further understanding of the range of sequence diversity, and TGS can also determine whether the compound heterozygous mutations are in cis or trans configuration[Citation21, Citation24]. Despite our small sample size, TGS detected an additional rare β-thalassemia gene mutation c.−100G > A, indicating that TGS has an advantage in the detection of rare variants. We suggest that TGS can be used as a second-line technique for samples with positive thalassemia hematological phenotypes but negative mutation detection using conventional methods. TGS can also be used directly as a first-line detection procedure for the thalassemia gene in laboratories able to run routine TGS analysis to improve the detection rate of rare mutations.
As the TGS technology is highly sensitive and has a wide detection range, many VUSs will certainly be detected. In our subjects with a small sample size, five individuals were identified as carriers of four types of VUSs. In a number of previous reports on the application of NGS and TGS for thalassemia detection, identification of these VUSs illustrated the high sensitivity of the two detection methods[Citation1, Citation21]. However, when applied in a clinical setting, these VUSs were identified but seemed to be of unknown clinical significance. Hence, detection of these variants in clinical laboratories may lead to misdiagnosis by clinicians. This is particularly critical in prenatal diagnosis where these VUSs should be interpreted with caution. For example, we found that a male subject carried both a rare nonpathogenic variant c.316-3C > T and a common pathogenic variant c.126-129delCTTT within the HBB gene, which easily led clinicians to misdiagnose him as β-thalassemia intermedia, but in fact, he presented with only clinical phenotypes of mild β-thalassemia. Therefore, we recommend that clinical laboratories should not list such VUSs in test reports to avoid confusion and unnecessary stress on the patients. At the same time, laboratory personnel and clinicians should use both genotype and phenotype data to make a comprehensive patient diagnosis and avoid any misdiagnosis.
The TGS-derived raw reads have high error rates resulting from the single-molecule sequencing approach. However, with high-fidelity CCS reads generated from multiple raw reads and a high sequencing depth are expected to reduce the occurrence of random sequencing errors. Previous studies have also demonstrated that with HiFi reads and more than 60× sequencing depth, PacBio sequence reads provide a high degree of accuracy for variant analysis[Citation13, Citation21, Citation24]. Although the total cost of library preparation and sequencing reagents per sample is low, the PacBio instrument is very expensive, which makes it difficult to apply this technology in smaller clinics. Developing a less-expensive benchtop platform could be a solution to increase the clinical application of TGS in areas with a high prevalence of thalassemia.
In conclusion, our study is the first to demonstrate the value of TGS in genetic testing for thalassemia in the Guizhou population. TGS can effectively detect both common and rare thalassemia variants with high accuracy and efficiency, and should be widely used for genetic testing and prenatal thalassemia genetic screening in areas with high incidence of thalassemia.
Ethics statement
Informed consent was obtained according to the Declaration of Helsinki.
Acknowledgements
The authors thank tall individuals for their participation in this study. The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data in this study are shown in the figures and tables.
Additional information
Funding
References
- Luo S, Chen X, Zeng D, et al. The value of single-molecule real-time technology in the diagnosis of rare thalassemia variants and analysis of phenotype-genotype correlation. J Hum Genet. 2022;67:183–195.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391:155–167.
- Wu H, Huang Q, Yu Z, et al. Molecular analysis of alpha- and beta-thalassemia in Meizhou region and comparison of gene mutation spectrum with different regions of southern China. J Clin Lab Anal. 2021;35:e24105.
- Shang X, Zhang X F, Xu X. Clinical practice guidelines for α-thalassemia. Chin J Med Genet. 2020;37:235–236.
- Xuan S, Wu X, Zhang X, et al. Clinical practice guidelines for β-thalassemia. Chin J Med Genet. 2020;37:243–244.
- Liang H, Li L, Yang H, et al. Clinical validation of a single-tube PCR and reverse dot blot assay for detection of common α-thalassaemia and β-thalassaemia in Chinese. J Int Med Res. 2022;50:1410680931.
- Zhao J, Li J, Lai Q, et al. Combined use of gap-PCR and next-generation sequencing improves thalassaemia carrier screening among premarital adults in China. J Clin Pathol. 2020;73:488–492.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. Ebiomedicine. 2017;23:150–159.
- Ardui S, Ameur A, Vermeesch JR, et al. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018;46:2159–2168.
- Munkongdee T, Chen P, Winichagoon P, et al. Update in laboratory diagnosis of thalassemia. Front Mol Biosci. 2020;7:74.
- Wenger AM, Peluso P, Rowell WJ, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37:1155–1162.
- Verma M, Kulshrestha S, Puri A. Methods in molecular biology. Methods Mol Biol. 2017;1525:3–33.
- Liang Q, Gu W, Chen P, et al. A more universal approach to comprehensive analysis of thalassemia alleles (CATSA). J Mol Diagn. 2021;23:1195–1204.
- Lin M, Zhong TY, Chen YG, et al. Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PLoS ONE. 2014;9:e101505.
- Lin X, Cheng B, Cai Y, et al. Establishing and evaluating an auto-verification system of thalassemia gene detection results. Ann Hematol. 2019;98:1835–1844.
- Huang S, Liu X, Li G, et al. Spectrum of β-thalassemia mutations in Guizhou Province, PR China, including first observation of codon 121 (GAA > TAA) in Chinese population. Clin Biochem. 2013;46:1865–1868.
- Huang SW, Xu Y, Liu XM, et al. The prevalence and spectrum of α-thalassemia in Guizhou Province of South China. Hemoglobin. 2015;39:260–263.
- Tan M, Bai Y, Zhang X, et al. Early genetic screening uncovered a high prevalence of thalassemia among 18 309 neonates in Guizhou, China. Clin Genet. 2021;99:704–712.
- Yamsri S, Singha K, Prajantasen T, et al. A large cohort of β+-thalassemia in Thailand: molecular, hematological and diagnostic considerations. Blood Cells Mol Dis. 2015;54:164–169.
- Li D, Liao C, Xie X, et al. A novel mutation of −50 (G→A) in the direct repeat element of the β-globin gene identified in a patient with severe β-thalassemia. Ann Hematol. 2009;88:1149–1150.
- Zhuang J, Chen C, Fu W, et al. Third-generation sequencing as a new comprehensive technology for identifying rare a- and b-globin gene variants in thalassemia alleles in the Chinese population. Arch Pathol Lab Med. 2022;undefined:undefined), doi:10.5858/arpa.2021-0510-OA.
- Rhoads A, Au KF. Pacbio sequencing and its applications. Genomics Proteomics Bioinf. 2015;13:278–289.
- Long J, Sun L, Gong F, et al. Third-generation sequencing: a novel tool detects complex variants in the α-thalassemia gene. Gene. 2022;822:146332.
- Jiang F, Mao A, Liu Y, et al. Detection of rare thalassemia mutations using long-read single-molecule real-time sequencing. Gene. 2022;825:146438.