
ABSTRACT
Objectives
Clinical manifestations of patients with Hemoglobin E/beta-thalassemia vary from mild to severe phenotypes despite exhibiting the same genotype. Studies have partially identified genetic modifiers. We aimed to study the association between rare variants in protein-coding regions and clinical severity in Thai patients.
Methods
From April to November 2018, a case–control study was conducted based on clinical information and DNA samples collected from Thai patients with hemoglobin E/beta-thalassemia over the age of four years. Cases were patients with severe symptoms, while patients with mild symptoms acted as controls. Whole exome sequencing and rare variant association study were used to analyze the data.
Results
All 338 unrelated patients were classified into 165 severe and 173 mild cases. Genotypes comprised 81.4% of hemoglobin E/beta-thalassemia, 2.7% of homozygous or compound heterozygous beta-thalassemia, and 0.3% of (δβ)0 thalassemia Hb E while 15.7% of samples were not classified as beta-thalassemia. A novel cis heterozygotes of IVS I-7 (A > T) and codon 26 (G > A) was identified. Six genes (COL4A3, DLK1, FAM186A, PZP, THPO, and TRIM51) showed the strongest associations with severity (observed p-values of <0.05; significance lost after correction for multiplicity). Among known modifiers, KLF1 variants were found in four mild patients and one severe patient.
Conclusion
No rare variants were identified as contributors to the clinical heterogeneity of hemoglobin E/beta-thalassemia. KLF1 mutations are potential genetic modifiers. Studies to identify genetic factors are still important and helpful for predicting severity and developing targeted therapy.
Introduction
Hemoglobin (Hb) E/beta-thalassemia is caused by compound heterozygous mutations of the beta-globin gene. It has a higher prevalence than other beta-thalassemia diseases in Asia. The total number of patients in Thailand is estimated to be around 100,000 cases [Citation1]. Patients with Hb E/beta-thalassemia have a wide spectrum of clinical severity from thalassemia intermedia (never or occasionally requiring blood transfusions) to thalassemia major (requiring regular blood transfusions). From previous studies, some factors can ameliorate the clinical severity by decreasing excessive alpha-globin including types of beta mutations (β+ or β++), coinheritance with alpha-thalassemia, and increasing gamma-globin synthesis [Citation2]. Increased fetal hemoglobin (Hb F) production by single nucleotide polymorphisms (SNPs) at the HBS1L-MYB intergenic region or MYB (on chromosome 6q23), SNPs at BCL11A (on chromosome 2p16), SNPs at position 158 of the HBG2 promoter, or KLF1 mutations have been reported [Citation3–6]. However, Nuinoon et al. found that types of beta mutation or a coinheritance with alpha mutations were associated with milder severity in only 12% of patients with Hb E/beta-thalassemia [Citation7]. Moreover, common SNPs identified by genome-wide association study (GWAS) in three different loci including the beta-globin cluster, the HBS1L-MYB intergenic region, and the BCL11A were significantly associated with disease severity and Hb F levels in 27% of Thai patients [Citation7].
Due to the large proportion of unexplained variance in disease severity in Hb E/beta-thalassemia [Citation7–9], there is an unmet need to identify genetic modulating factors that are useful in predicting the clinical severity during prenatal diagnosis or since early childhood and in developing novel treatments at targeted mutations. Therefore, we studied the rare single nucleotide or rare small structural variants as the genetic factors that may be present within protein-coding regions, possibly affecting protein sequences and function by whole exome sequencing (WES) and rare variant association study (RVAS). Such rare genetic factors may not have been identified by previous GWAS.
Materials and methods
Study design
The study protocols were approved by the Siriraj Hospital Institutional Review Board (protocol no. Si 219/2018). The study was conducted according to the Good Clinical Practice and the Declaration of Helsinki. A case–control study was performed with a case-to-control ratio of 1:1. Cases were Hb E/beta-thalassemia patients with severe phenotype whereas controls were patients with mild phenotype. Adult patients gave written informed consent and patients aged less than 18 years provided assent if they were capable of understanding the explanation and providing assent to participate in the study.
Patients
Clinical information, complete blood count, Hb typing and/or beta-thalassemia mutation testing (if available), and DNA samples of Thai patients from 15 hospitals in the central, northern, and northeastern regions of Thailand were collected between April and November 2018. The scoring system was used to classify patients as mild, moderate, or severe phenotypes (Table S1) [Citation10]. Extreme-Phenotype Sampling was used to increase the chance to identify causal variants in patients at the extreme ends of the distribution [Citation11,Citation12]. Therefore, only patients with mild (scores 0–3.5) and severe phenotype (scores 7.5–10) were enrolled. Inclusion criteria were unrelated individuals aged four years or older with Hb E/beta-thalassemia. The diagnosis was confirmed with medical record and/or Hb typing and/or polymerase chain reaction (PCR) for beta-thalassemia mutations. PCR-based techniques including reverse dot blot (RDB), amplification refractory mutation system (ARMS)-PCR, and Gap-PCR were used to detect known beta mutations in Thailand. Exclusion criteria were patients with moderate phenotype, patients who were treated with drugs that affect the percentage of Hb F (e.g. hydroxyurea), patients with other co-morbidities associated with anemia (e.g. autoimmune hemolytic anemia, chronic kidney disease), patients who received their last red blood cell transfusion less than two weeks before invitation to participate in the study.
Genomic analysis and exome sequencing methods
The DNA was extracted from peripheral blood by a Gentra Puregene Blood Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Each DNA sample was enriched for target regions with IDT xGen Exome Panel before performing WES using an Illumina NovaSeq6000 system (see Supplementary method S1 Text for more details). Sanger sequencing was used to confirm the cis heterozygotes of beta-globin gene (Applied Biosystems SeqStudio Genetic Analyzer, ThermoFisher Scientific, Waltham, MA) and 4 KLF1 variants (Applied Biosystems 3500 Series Genetic Analyzer, ThermoFisher Scientific, Waltham, MA) detected by WES. Six samples coinherited with KLF1 mutations were additionally tested for seven deletional alpha mutations by multiplex PCR [Citation13].
Whole exome sequencing data processing and filtering of candidate variants
Following the best practices Genome Analysis Toolkit (GATK) version 4 workflow for germline variant discovery, the duplicated reads in the raw mapped read BAM files were marked prior to creating the analysis-ready BAM file, which was then used in the subsequent variant calling step as recommended. The WES data were aligned to the human reference genome (GRCh38) using Burrows–Wheeler Aligner-Maximal Exact Match (BWA-mem). Subsequently, single nucleotide variants (SNVs) and small indels were called using the best practices GATK version 4 workflow for germline SNVs and indels. Additional variant quality control was performed using the GATK’s variant quality score log-odds (VQSLOD). The Excess-het filter was used to identify variants which were not in Hardy–Weinberg equilibrium.
The allele frequency (AF) of each variant was calculated by combining the information from Thai patients in this study with the East Asian and South Asian populations from gnomAD using both WES and whole genome sequencing data. The calculated AF was representative of those in general Asian population available in publicly available datasets. The variants with AF of ≤0.005, >0.005 to ≤0.01, >0.01 to ≤0.05, and >0.05 to ≤0.1 were included in the subsequent analyses.
Variants effects were annotated by the variant effect predictor (VEP) [Citation14], meta-logistic regression (meta-LR) [Citation15], sorting intolerant from tolerant (SIFT) [Citation16], and polymorphism phenotyping (PolyPhen) servers [Citation17]. VEP was used to classify the variants as having high, moderate, low, or modifier impact to protein structures and/or function, as well as their consequences (non-sense, frameshift, missense, etc.). Furthermore, meta-LR was used to predict moderate-impact variants as deleterious, tolerated, or unknown variants to improve the ability to enrich for variants with deleterious effects on protein function. Then variants having high impact, moderate impact, and moderate impact with deleterious effects were selected. SIFT and PolyPhen were used to predict variants in genes related to red-blood-cell (RBC) disorders. Sequence variants were interpreted and classified based on the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG) guidelines [Citation18] and were confirmed with the ClinVar database [Citation19].
Rare variant association study
The Optimal Sequence Kernel Association Test was chosen because it has power to analyze variants with different proportions of causal variants and different directions of association [Citation20]. A quantitative outcome (scores 0–3.5 vs 7.5–10) was used instead of qualitative outcome to increase the power to detect the association between predictor and outcome variables. The patients’ phenotypes were non-normally distributed. Subsequently, the Continuous Extreme Phenotype Optimal Sequence Kernel Association Test (CEP-SKAT-O) was used to analyze the association between variants and severity score [Citation11]. Covariates were age, sex, types of beta-thalassemia mutation, coinheritance with non-deletional alpha mutations (Hb Constant spring and Hb Pakse), types of hospitals in which patients were treated, relatedness of individuals detected by principal component analysis 1 and 2. After obtaining all candidate variants, 24 association studies were performed (). Bonferroni-corrected p-values (i.e. observed p-value multiplied by number of comparisons) for familywise error were calculated. A p-value <0.05 was considered statistically significant.
Figure 1. Diagram of the procedure for the 24 rare variant association studies (RVAS’s) between candidate variants and clinical severity in patients with Hb E/beta-thalassemia. Variants with passing the Variant Quality Recalibration Score (Pass) and/or Excess het filter (Excess het), different allele frequencies, and different predictions [high impact, moderate impact to protein by variant effect predictor (VEP), and moderate impact with deleterious effect by meta-logistic regression (meta-LR)] were grouped for analysis.
![Figure 1. Diagram of the procedure for the 24 rare variant association studies (RVAS’s) between candidate variants and clinical severity in patients with Hb E/beta-thalassemia. Variants with passing the Variant Quality Recalibration Score (Pass) and/or Excess het filter (Excess het), different allele frequencies, and different predictions [high impact, moderate impact to protein by variant effect predictor (VEP), and moderate impact with deleterious effect by meta-logistic regression (meta-LR)] were grouped for analysis.](/cms/asset/e2b651cd-9261-4b62-ac96-88c9fb1990e8/yhem_a_2187155_f0001_oc.jpg)
Statistical analysis
For comparison of baseline characteristics of the mild and severe groups, continuous variables were analyzed by the Mann–Whitney U test while proportions were analyzed by the Chi-square or the Fisher’s exact test as appropriate. All statistical analyses were performed using SPSS Statistics for Windows, version 18 (SPSS Inc., Chicago, IL, USA). A two-sided p-value of <0.05 was considered statistically significant.
Results
Clinical and laboratory phenotype of the patients
Clinical information and DNA samples were collected from 338 unrelated patients. One hundred and seventy-three cases had mild phenotype, and were the control group. The remaining 165 cases had severe phenotype, and were the case group. Number of cases to controls from central, northern, and northeastern regions of Thailand was 86–113, 39–20, and 40–40, respectively. Their baseline characteristics are presented in . All parameters were significantly different between two phenotypes except Hb F percentage.
Table 1. Clinical and laboratory information of mild and severe patients were compared according to the scoring system.
Genotype and type of beta-globin mutations in Thailand
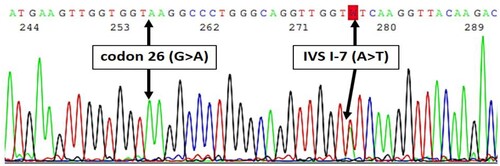
WES detected genotypes of beta-thalassemia in 84.3% (285/338) of patients. Among these 285 patients, 81.4% were Hb E/beta-thalassemia (β0/E 77.5%, β+/E 3.6%, and β+E/E 0.3%) whereas Hb E/(δβ)0 thalassemia and homozygote or compound heterozygote of beta-thalassemia were found in 0.3% and 2.7%, respectively. Comparison of genotypes of beta-thalassemia between mild and severe phenotypes is shown in Table S2. Patients who carried β0-thalassemia mutations were found in 83.6% of severe cases and in 76.9% of mild cases. Codon 41/42 (β0) and codon 17 (β0) were the two most common mutations in severe (56.5% and 26.8%, respectively) and mild patients (56.9% and 27.8%, respectively). A novel cis heterozygotes of IVS I-7 (A > T) (β+) and codon 26 (G > A) (βE) coinherited with in trans codon 26 (G > A) (βE) was identified in 1 patient (Table S3). A 31-year-old woman has been diagnosed with Hb E/beta-thalassemia at the age of 25. She has rarely received blood transfusion and no splenomegaly. Her laboratory revealed Hb 9.1 g/dL and mean corpuscular volume 65.7 fL. Hb typing by high-performance liquid chromatography (HPLC) showed Hb E 66.6% and Hb F 39%. Such mutations were confirmed by Sanger sequencing ().
Figure 2. Sanger sequencing revealed in cis interaction of IVS I-7 (A > T) (β+) and codon 26 (G > A) (βE) coinherited with in trans codon 26 (G > A) (βE).
Thirteen percent (44/338) of the patients had the results of their beta-thalassemia genotyping tested by PCR before their blood samples were analyzed by WES. PCR-based techniques were used to determine known beta mutations including point mutations located outside exons and large deletional mutations while WES cannot accurately identify these types of variants [Citation21]. Therefore, among 15.7% (53/338) of patients without genotypes of beta-thalassemia by WES, 9 patients (9/53) had confirmed Hb E/beta-thalassemia mutations by PCR (). Moreover, 27 patients (27/53) had Hb typing compatible with Hb E/beta-thalassemia. While WES showed heterozygous Hb E in eight patients, PCR revealed compound heterozygote of IVS-II-654 (C > T) (β+) and Hb E in five patients and compound heterozygote of nt-28 (β++) and Hb E in three patients (). IVS-II-654 (C > T) and nt-28 (A > G) were located in second intron and promoter, respectively. Consequently, the read depth in these regions was too low to accurately call the mutations. One patient had a compound heterozygote of 3.48 kb deletion and Hb E by PCR while WES showed homozygous Hb E. Because the large structural variant was deleted from one chromosome, all Hb E variants were then called from the remaining one (). According to the limitations of WES, the remaining samples possibly had non-deletional beta mutations located in introns, promoter, untranslated regions, or large deletional beta mutations.
Figure 3. Diagram showing 53 out of 338 patients with phenotype of thalassemia but no thalassemia genotypes analyzed by whole exome sequencing (WES).
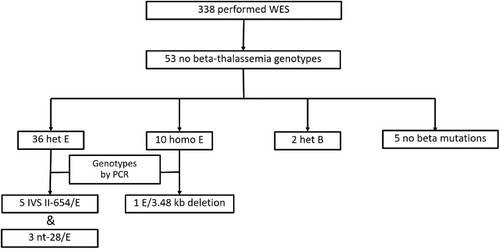
Figure 4. Whole exome sequencing showing the number of reads covered on IVS-II-654 and nt-28 mutations. The number of reads was too low. Therefore, the mutations could not be confidently called by whole exome sequencing. (A) The IVS-II-654 mutation was located in the second intron. (B) The nt-28 mutation was located in the promoter. Both mutations were confirmed by PCR.
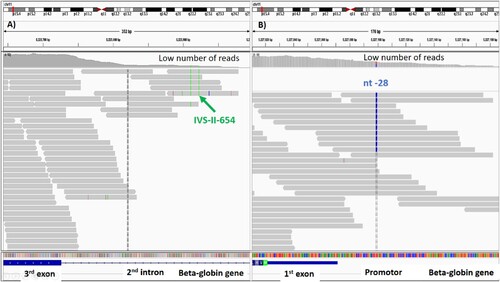
Figure 5. The sample was called as homozygous Hb E by whole exome sequencing, but it was compound heterozygous mutation of 3.48 kb deletion and codon 26 (G > A) (βE) by PCR. Codon 26 (G > A) (βE) from the remaining chromosome was called from all reads.

Results of rare variant association studies
For all the rare variant association tests, we compared rare variants within each gene carried by patients with severe symptoms (who were the cases) to those carried by patients with mild symptoms (who were the controls) to identify the potential genetic modifiers of severity. After analysis, we could not identify significant association between genes and thalassemia severity with the AF threshold of rare variants varying between 0.005 and 0.1 forming 24 sets of RVAS’s (Table S4) (results of each set of 24 RVAS’s are shown in S1 File). Among the genes with nominal significant associations (p < 0.05), none had known biological functions that could explain the severity of beta-thalassemia (Figure S1). The six genes consisting of Collagen type IV alpha 3 chain (COL4A3), Delta-like non-canonical Notch ligand 1 (DLK1), Family with sequence similarity 186 member A (FAM186A), PZP alpha-2-macroglobulin like (PZP), Thrombopoietin (THPO), and Tripartite motif-containing 51 (TRIM51) showed the strongest associations with severity (observed p-values of <0.05). However, the results were not significant after the Bonferroni correction (). COL4A3 encodes protein subunit for type IV collagen. It is linked to Alport syndrome. DLK1 encodes protein involved in the differentiation adipocytes. It is associated with child and adolescent obesity. FAM186A and TRIM51 are mostly expressed in testis. The protein encoded by PZP is elevated in the sera of patients with presymptomatic Alzheimer’s disease. THPO encodes protein involved in megakaryocyte maturation and thrombopoiesis. Its mutation leads to thrombocythemia.
Table 2. Results of 24 rare variant association studies between variants within the top six genes and clinical severity of patients with beta-thalassemia.
Therefore, we focused our attention on the genes involved in RBC disorders, and they also did not show any significant association (Table S5). The RBC-related variants and their predictions are displayed in S2 and S3 Files. The known genes previously reported as genetic modifiers by increasing Hb F production (KLF1, BCL11A, and MYB) are shown in .
Table 3. Number of cases of mild and severe phenotypes with variants in known modified genes of beta-thalassemia.
Five patients with mild symptoms and one patient with severe symptoms had variants in KLF1 gene with observed p-values ranging from 0.17 to 0.47 and adjusted p-values of 1.0. One variant in BCL11A gene was not a modifier because it was found in both mild and severe patients with an observed p-values of 0.14 and an adjusted p-value 1.0. No variant in MYB gene was identified. Then we further examined six patients with KLF1 variants. Five patients had β0/βE genotype while the remaining had Hb E heterozygote. No deletional and non-deletional alpha mutations were detected in all of them. Clinical and molecular information of the patients are shown in and . Four KLF1 mutations were confirmed by Sanger sequencing (Figures S2–S7).
Table 4. Clinical and laboratory information of the six patients with heterozygous KLF1 variants.
Table 5. Predicted consequences and allele frequencies of the four KLF1 variants.
Discussion
The aim of this study was to identify genetic modifiers of beta-thalassemia severity. WES was chosen to investigate rare SNVs or small insertions/deletions in the entire coding sequence that have the possibility to be modifying factors. However, WES is not an appropriate technique to diagnose beta-thalassemia because some beta mutations are SNVs located outside exons or large structural variants [Citation22,Citation23]. Moreover, it is not cost-effective to sequence the whole exome to identify mutations in beta genes only [Citation21]. Therefore, diagnostic yield of beta-thalassemia by WES in this study was 84.3%.
The association between phenotype and genetic factors
Most types of beta-thalassemia are inherited by autosomal recessive pattern including homozygote or compound heterozygote. However, patients who carried the same beta genotype do not express the same phenotype. There are other genetic modifiers affected clinical severity [Citation2]. In this study, number of patients who carried β0/βE or β0/β0 was comparable in both the mild and severe groups (76.9% vs 83.6%). Previous studies using the same scoring system to classify phenotypes of Thai patients with Hb E/beta-thalassemia similarly reported a higher proportion of β0 mutations in both the mild and severe groups (76.1% vs 84.5% by Sripichai et al. [Citation10] and 91.9% vs 100% by Nuinoon et al. [Citation7]). Both studies tested alpha mutations by PCR. They found that around one-quarter of the mild patients carried alpha-thalassemia heterozygote that can alleviate beta-thalassemia severity. Our study used WES, so deletional alpha mutations, which are more prevalent than point mutations, could not be revealed. However, due to quite similar patients’ samples, if the minority of our mild patients carried alpha-thalassemia heterozygote and/or β+ mutations, we still cannot explain the observed majority with mild phenotype.
Musallam et al. reported that patients with β0/β0 had similar morbidity and mortality rate to those of patients with β0/β+ or β+/β+ [Citation24]. Higher Hb F levels can ameliorate clinical severity [Citation2] but this study and Nuinoon et al. [Citation7] showed that patients in both mild and severe groups had no significant difference of Hb F levels.
For modifiers determined by GWAS, SNPs in beta-globin gene cluster, HBS1L-MYB intergenic region, and BCL11A contributed to disease severity in only 27% of patients with Hb E/beta-thalassemia [Citation7]. Hariharan et al. analyzed the effect of several modifiers (beta and alpha mutations, variants in gamma-globin promoters, SNPs in BCL11A, HBS1L-MYB intergenic region, and KLF1) in patients with homozygous beta-thalassemia. They found that patients who carried more ameliorating alleles tend to delay median age of presentation but no statistical significance [Citation25].
It still has the knowledge gap of disease modifiers. We then firstly performed WES and RVAS’s for detecting rare genetic modifiers on exome in patients with beta-thalassemia. Somewhat surprisingly, we did not find significant associations. The reasons might be explained by first, the effect size of rare causal variants is too small to be detected by the analyzed method. Second, genetic association studies usually need huge sample size, our sample size may have been quite small to achieve significant power to detect rare variants associated with disease severity, although we used the extreme-phenotype sampling approach. Third, the scoring system might not perfectly classify patients with mild from severe phenotype. The scores might be affected by patients’ recall bias (i.e. age at presentation or age at receiving first transfusion) or by different transfusion protocols (transfusion frequency) in each participating hospital. Moreover, some patients with severe symptoms at the beginning who received a hypertransfusion regimen were classified as moderate phenotype and excluded from the analysis. Finally, we could not test other factors that could be the severity modifiers of clinical variation, such as epigenetics, interactions among genes, or gene-environment interactions.
Types of beta-globin mutations in Thailand
In Thailand, Codon 41/42(-TTCT) (β0) was the most common mutation found throughout the country (32–53.8%) [Citation22]. Codon 17 (A > T) (β0) mutation was the second most common mutation in the central, northern, and northeastern regions (10.3%–34.4%) [Citation22,26,Citation27] with the exception of IVS I-5 (G > C) (β+) being the second most common mutation in southern region (22.3%) [Citation28]. In the present study in which more than half the patients were from the central region, codon 41/42 (-TTCT) and Codon 17 (A > T) were the first and second most common mutations identified. Mutations of nt-28 (A > G) (β++) in the promoter and IVS-II-654 (C > T) (β+) in second intron were reported previously as the common mutations in Thailand (1.4%–22.5% and 0.4%–11.2%, respectively) [Citation22]. However, they could not be called by WES in the present study due to their locations far from exons. Using PCR-based technique with allele-specific primers is more appropriate to detect prevalent mutations in our country.
Mutations causing beta-thalassemia trait or disease are usually a single mutation in each beta-globin gene. Currently, more than 200 alleles have been reported worldwide. Whereas, less than 40 of them are double mutations occurring in the same beta gene [Citation29]. For in cis interaction with Hb E, only three mutations have been reported worldwide, i.e. Hb Corbeil [codon 104 (G > C) with Hb E] [Citation30], Hb T-Cambodia [codon 121 (G > C) with Hb E] [Citation31], and Hb E-Udon Thani [IVS I-7 (A > G) with Hb E] [Citation32]. Our patient had cis heterozygotes of IVS I-7 (A > T) and Hb E coinherited with in trans Hb E. Her Hb typing was not different from those of patients with Hb E/beta-thalassemia having single mutation in each beta gene. Therefore, the double mutation can be only detected by DNA analysis.
Genetic modifiers by KLF1 variants
KLF1 is an erythroid-specific-transcription factor that regulates many RBC genes. It activates genes encoding globins, heme synthesis enzymes, structural membrane, etc. [Citation33]. KLF1 also has a role in Hb switching by directly activating beta-globin and indirectly suppression of gamma-globin by activation of BCL11A [Citation34,Citation35]. Therefore, loss-of-function heterozygous and compound heterozygous mutations of KLF1 lead to the high production of Hb F [Citation6,Citation36,Citation37], which possibly ameliorates the severity of beta-thalassemia.
In the present study, four KLF1 mutations were identified in five patients with the β0/βE and αα/αα genotypes. Three variants were identified in patients with mild symptoms. First, a frameshift variant of c.519_525dupCGGCGCC disrupts protein before DNA binding domains [Citation38]. Second, a missense variant of c.1057G > A was located in third exon involving the zinc finger domain [Citation39]. It possibly reduces protein function. A third missense variant of c.629C > G was classified as a variant with no functional consequences [Citation33]. Therefore, the patient’s symptoms may have been alleviated by other modifiers that were not tested yet. A severe case had a missense variant of c.809C > G located in second exon involving the transactivation domain [Citation40]. The inability to ameliorate thalassemia severity of some KLF1 mutations may be explained by their wide range of regulatory functions influencing other essential erythroid genes and coordinated transcription factors [Citation34].
Most studies reported that patients who carried β0/β0 with KLF1 heterozygote had milder clinical severity than those without such mutation [Citation6,Citation41–44]. However, one study identified KLF1 mutations more frequently in patients with severe symptoms [Citation45]. The reason might be that these variants were located in non-coding sequences or classified as a neutral polymorphism [Citation33]. Therefore, they did not affect protein function. To confirm the effect of these variants to protein, further experiments are needed to clarify their functions. We described and summarized KLF1 mutations in patients with beta-thalassemia in Table S6.
Conclusion
Rare variants could not be determined to explain the heterogeneity of beta-thalassemia symptoms. KLF1 mutations are possibly the potential factor to ameliorate thalassemia severity. Further studies are needed to focus on KLF1 mutations and their effect on protein function, epigenetics, interactions among genes, or gene-environment interactions.
Supplemental Material
Download Zip (7.4 MB)Acknowledgements
The authors gratefully acknowledge Associate Professor Vijay G. Sankaran (Division of Hematology/Oncology, Boston Children’s Hospital, Harvard Medical School) for advising us to use WES and RVAS to identify genetic modifiers in patients with Hb E/beta-thalassemia and requesting a grant to perform WES, Jeffrey M. Verboon (computer analyst at Sankaran Lab, Boston Children’s Hospital) for advising and helping us analyze and interpret the genetic studies, Assistant Professor Sanan Visuthisakchai (Head of Division of Hematology), Associate Professor Theera Ruchutrakool (Subhead), and staff (Division of Hematology of the Faculty of Medicine Siriraj Hospital, Mahidol University) for all their support, Professor Suthat Fucharoen (senior consultant at the Thalassemia Research Center, Institute of Molecular Biosciences, Mahidol University) for advising us about how to apply the scoring system, Claudia Fiorini (PhD Staff scientist at Sankaran Lab, Boston Children’s Hospital) for preparing DNA samples for WES, Chompunut Kanjanakorn (scientist at Siriraj Thalassemia Center, Siriraj Hospital) for sequencing the beta-globin gene, Professor Vip Viprakasit (Department of Pediatrics and the Thalassemia Center, Faculty of Medicine Siriraj Hospital, Mahidol University) for advising us about KLF1 mutations identified in Thai patients, Ms. Suchada Riolueang (Chief Technical Officer at the ATGenes company) for sequencing the KLF1 gene, and Professor Peter Hokland (Department of Hematology, Aarhus University, Denmark) for critical review of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Flint J, Harding RM, Boyce AJ, et al. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998;11(1):1–51.
- Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2018;32(2):177–191.
- Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197–1199.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842.
- Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A. 2007;104(27):11346–11351.
- Liu D, Zhang X, Yu L, et al. KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of β-thalassemia. Blood. 2014;124(5):803–811.
- Nuinoon M, Makarasara W, Mushiroda T, et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum Genet. 2010;127(3):303–314.
- Winichagoon P, Fucharoen S, Chen P, et al. Genetic factors affecting clinical severity in beta-thalassemia syndromes. J Pediatr Hematol Oncol. 2000;22(6):573–580.
- Nuntakarn L, Fucharoen S, Fucharoen G, et al. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42(1):32–35.
- Sripichai O, Makarasara W, Munkongdee T, et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83(6):482–484.
- Barnett IJ, Lee S, Lin X. Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet Epidemiol. 2013;37(2):142–151.
- Peloso GM, Rader DJ, Gabriel S, et al. Phenotypic extremes in rare variant study designs. Eur J Hum Genet. 2016;24(6):924–930.
- Tan AS, Quah TC, Low PS, et al. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98(1):250–251.
- McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122.
- Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24(8):2125–2137.
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814.
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.
- Landrum MJ, Lee JM, Benson M, et al. Clinvar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868.
- Lee S, Abecasis GR, Boehnke M, et al. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95(1):5–23.
- Brancaleoni V, Di Pierro E, Motta I, et al. Laboratory diagnosis of thalassemia. Int J Lab Hematol. 2016;38(Suppl 1):32–40.
- Mankhemthong K, Phusua A, Suanta S, et al. Molecular characteristics of thalassemia and hemoglobin variants in prenatal diagnosis program in northern Thailand. Int J Hematol. 2019;110(4):474–481.
- Panichchob P, Iamdeelert P, Wongsariya P, et al. Molecular spectrum of β-thalassemia mutations in central to eastern Thailand. Hemoglobin. 2021;45(2):97–102.
- Musallam KM, Vitrano A, Meloni A, et al. Primary HBB gene mutation severity and long-term outcomes in a global cohort of β-thalassaemia. Br J Haematol. 2022;196(2):414–423.
- Hariharan P, Gorivale M, Sawant P, et al. Significance of genetic modifiers of hemoglobinopathies leading towards precision medicine. Sci Rep. 2021;11(1):20906.
- Boonyawat B, Monsereenusorn C, Traivaree C. Molecular analysis of beta-globin gene mutations among Thai beta-thalassemia children: results from a single center study. Appl Clin Genet. 2014;7:253–258.
- Traivaree C, Monsereenusorn C, Rujkijyanont P, et al. Genotype-phenotype correlation among beta-thalassemia and beta-thalassemia/HbE disease in Thai children: predictable clinical spectrum using genotypic analysis. J Blood Med. 2018;9:35–41.
- Laosombat V, Fucharoen SP, Panich V, et al. Molecular basis of beta thalassemia in the south of Thailand. Am J Hematol. 1992;41(3):194–198.
- Kountouris P, Lederer CW, Fanis P, et al. Ithagenes: an interactive database for haemoglobin variations and epidemiology. PLoS One. 2014;9(7):e103020.
- Wajcman H, Kister J, Promé D, et al. Interaction of 2 amino acid substitutions within the same beta chain of human hemoglobin: the examples of Hb Corbeil and Hb Villeparisis. C R Acad Sci III. 1995;318(7):785–794.
- Hutt PJ, Fairbanks VF, Thibodeau SN, et al. Hb T-Cambodia, a beta chain variant with the mutations of Hb E and Hb D-Punjab, confirmed by DNA analysis. Hemoglobin. 1997;21(3):205–218.
- Singha K, Fucharoen G, Fucharoen S. Molecular characterisation of haemoglobin E-Udon Thani (HBB:c.[79G > A;92 + 7A > G]): a novel form of Hb E-β-thalassaemia syndrome. J Clin Pathol. 2019;72(4):322–324.
- Perkins A, Xu X, Higgs DR, et al. Krüppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood. 2016;127(15):1856–1862.
- Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42(9):801–805.
- Zhou D, Liu K, Sun CW, et al. KLF1 regulates BCL11A expression and gamma-to beta-globin gene switching. Nat Genet. 2010;42(9):742–744.
- Satta S, Perseu L, Moi P, et al. Compound heterozygosity for KLF1 mutations associated with remarkable increase of fetal hemoglobin and red cell protoporphyrin. Haematologica. 2011;96(5):767–770.
- Huang J, Zhang X, Liu D, et al. Compound heterozygosity for KLF1 mutations is associated with microcytic hypochromic anemia and increased fetal hemoglobin. Eur J Hum Genet. 2015;23(10):1341–1348.
- rs483352838 RefSNP Report – dbSNP – NCBI. Available from: https://www.ncbi.nlm.nih.gov/snp/rs483352838
- rs1328031641 RefSNP Report – dbSNP – NCBI. Available from: https://www.ncbi.nlm.nih.gov/snp/rs1328031641
- rs558942739 RefSNP Report – dbSNP – NCBI. Available from: https://www.ncbi.nlm.nih.gov/snp/rs558942739
- Phanrahan P, Yamsri S, Teawtrakul N, et al. Molecular analysis of non-transfusion dependent thalassemia associated with hemoglobin E-β-thalassemia disease without α-thalassemia. Mediterr J Hematol Infect Dis. 2019;11(1):e2019038.
- Xie XM, Liu YN, Li J, et al. A Krüppel-like factor 1 gene mutation ameliorates the severity of β-thalassemia: a case report. Hemoglobin. 2019;43(2):137–139.
- Hariharan P, Gorivale M, Colah R, et al. Does the novel KLF1 gene mutation lead to a delay in fetal hemoglobin switch? Ann Hum Genet. 2017;81(3):125–128.
- Fanis P, Kousiappa I, Phylactides M, et al. A novel mutation in the erythroid transcription factor KLF1 is likely responsible for ameliorating β-thalassemia major. Hum Mutat. 2019;40(10):1768–1780.
- Khamphikham P, Sripichai O, Munkongdee T, et al. Genetic variation of Krüppel-like factor 1 (KLF1) and fetal hemoglobin (HbF) levels in β(0)-thalassemia/HbE disease. Int J Hematol. 2018;107(3):297–310.