
ABSTRACT
The MCL1 inhibitors are undergoing clinical testing for multiple leukemia. However, because that MCL1 inhibition has on-target hematopoietic, hepatic and cardiac toxicities, there is substantial interest in finding agents can sensitize leukemia cells to the MCL1 inhibitors. Here we describe that the AKT inhibitors MK-2206 and Gsk690693 sensitize multiple leukemia cells to the MCL1 inhibitor S63845. Further experiments demonstrate that MK-2206 and Gsk690693 sensitize S63845 through the mitochondrial apoptosis pathway. Moreover, MK-2206 downregulates the anti-apoptotic protein BCLXL and induces the BH3-only pro-apoptotic protein BAD dephosphorylation and mitochondrial translocation. Knockdown of BAD significantly inhibits MK-2206-induced sensitization to S63845. Thus, our results suggest that MK-2206 sensitizes multiple leukemia cells to S63845-induced apoptosis, with the mechanisms involving BAD dephosphorylation and BCLXL downregulation.
KEYWORDS:
Introduction
The mitochondrial apoptosis pathway is controlled by BCL2 family proteins, which include: the multidomain anti-apoptotic proteins, such as BCL2, BCLXL and MCL1; the multidomain pro-apoptotic effectors, BAX and BAK; and the BH3-only pro-apoptotic proteins, such as BIM, PUMA and BAD [Citation1–7]. Overexpression of the anti-apoptotic members, including MCL1, not only relates to the development of acute leukemia and lymphoma, but also causes resistance to multiple anti-cancer drugs [Citation8–14]. Thus, several MCL1 inhibitors, including S63845 (analog of S64315), AMG 176, AMG 397, AZD5991, ABBV-467 and PRT1419, have been developed to induce cancer cell apoptosis and to overcome drug resistance [Citation15–23].
These compounds have sub-nanomolar affinities and high selectivity for MCL1 and have been undergoing clinical testing in acute leukemia, lymphoma and multiple myeloma as a single agent or in combinations (clinical trials NCT04702425, NCT03797261, and NCT03465540). Previous studies have indicated on-target side effects of MCL1 inhibition, such as hematopoietic, hepatic and cardiac toxicities [Citation24–28]. These side effects, such as cardiac toxicities, have already been observed during clinical trials of the MCL1 inhibitors and slowed down their clinical usage [Citation29]. Thus, a possible way to solve the paradox is to find drugs that can sensitize cancer cells to the MCL1 inhibitors in order to reduce the drug doses while maintaining the efficacy [Citation30].
The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway plays an important role in regulating cellular protein translation, autophagy, metabolism, and cell survival. Activated by growth factor stimulation, this pathway regulates cellular functions through downstream molecules including mTOR, glycogen synthase kinase 3 beta (GSK3b), forkhead box protein O1 (FOXO1), or nuclear factor kappa beta (NF-kB) [Citation31–33]. Additionally, hyperactivation of the PI3K/AKT pathway has been shown to play important roles in tumorigenesis and drug resistance in both solid tumors and hematological malignancies [Citation32–37]. Quite a few AKT inhibitors have been developed to treat cancer, including Gsk690693 and MK-2206, while MK-2206 is now under clinical testing in several cancers (clinical trials NCT01231919, NCT01253447 and NCT01369849), including relapsed or refractory chronic lymphocytic leukemia (CLL) [Citation31,Citation33–36]. These inhibitors have been shown to induce apoptosis of acute leukemia cells through AKT inhibition and down-regulation of MCL1 [Citation38,Citation39]. In addition to single-agent function, PI3K inhibitor PI-103 or AKT inhibitor MK-2206 sensitizes multiple leukemia cell lines and primary patient samples to the BCL2/BCLXL/BCLw inhibitor ABT-737 or the selective BCL2 inhibitor venetoclax through MCL1 downregulation or BAX activation [Citation40]. In a Phase I/II clinical trial, MK-2206 showed synergistic effects in combination with bendamustine and rituximab in relapsed or refractory CLL patients [Citation41].
In the present study, we report that both AKT inhibitors Gsk690693 and MK-2206 sensitize multiple acute leukemia cell lines to the MCL1 selective inhibitor S63845. Additional observations indicate that the sensitization involves the BAD dephosphorylation and translocation to mitochondria.
Material and methods
Reagents and antibodies
Reagents were obtained from the following suppliers: MK-2206 (#HY-108232), S63845 (#HY-100741), Gsk690693 (#HY-10249) from MedChemExpress; Propidium Iodide (PI) (#P8080) from Solarbio; Annexin V-FITC/PI double stain kit (#556547) from BD Biosciences. Antibodies were purchased from the following suppliers: BAK (#06-536) from Upstate; GAPDH (#A00227) from Boster; PARP (#9542S), BCL2 (#15071S), MCL1 (#94296S), BCLXL (#2764S), BIM (#2933S), BAD (#9292S), p-BAD (Ser136) (#4366S), AKT (#4691S), and p-AKT (Ser473) (#4060S) from Cell Signaling Technology;β-tubulin (#EM0103) from Huabio. The secondary antibodies including horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (#7074S) and HRP-conjugated goat anti-mouse IgG (#7076S) were purchased from Cell Signaling Technology.
Cell lines
All leukemia cell lines, including Jurkat, U937, Molt3, Molt4, THP.1 and HL-60 cells were maintained at densities below 106 cells/mL in RPMI-1640 containing 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin G, 100 μg/mL streptomycin, and 2 mM glutamine. BAK−/−- Jurkat cells were generated as previously described [Citation42]. Cell lines were validated by short tandem repeat profiling and assayed for mycoplasma contamination about every 6 months.
Evaluation of apoptosis
Sub-G1 assay and Annexin V/PI staining were used to evaluate apoptotic cells. To assay for sub-G1 DNA content, cells were collected after treatments and stained with propidium iodide in 0.1% (w/v) sodium citrate containing 0.1% (w/v) Triton X-100. Alternatively, FITC-labeled Annexin V/PI staining was used. After staining, cells were subjected to flow cytometry on a Becton Dickinson FACSCanto II and analyzed using Becton Dickinson CellQuest software [Citation11,Citation43].
Calculation of combination index
The combination index was calculated by CalcuSyn software (Biosoft, Cambridge, UK). The apoptosis data were analyzed by the median effect method under the assumption that effects of the two agents are mutually exclusive, which is equivalent to isobologram analysis [Citation44]. According to this analysis, a combination index (CI) < 1 indicates antagonism.
RNA extraction
After the indicated treatments, the cells were harvested and frozen in liquid nitrogen. Total RNA was isolated using the EasyPure RNA Kit (Transgen Biotech, Beijing, China). The RNA quality and quantity were measured using an Implen NanoPhotometer NP80 (Implen GmbH, Munich, Germany) and gel electrophoresis, respectively.
Transcriptome sequencing and analysis
For transcriptome sequencing, cells were treated as indicated for 4 h. Each treatment was prepared in three independent replicates. The RNA concentration and integrity were further determined by the Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). The RNA-seq libraries 5 were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, MA, USA). The quality of the libraries was analyzed by the Agilent 2100 bioanalyzer and ABI StepOnePlus Real-Time PCR System (Thermo Scientific, Wilmington, USA). The index-coded samples were clustered following the instructions of the cBot Cluster Generation System and then sequenced on the Illumina HiSeq 2500 platform by Novogene Technology (Beijing, China).
DEG and KEGG analysis
The paired-end reads were mapped to the human reference genome. The expression of the genes was analyzed by using the R Subread package (version 2.0.3) [Citation45]. The differentially expressed genes (DEGs) were analyzed using Corset [Citation46]. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed using the R Pathview package [Citation47].
siRNA effects
The siRNA sequence for BAD is 5′-GGTTTAACCGTTGCGTCAC-3′. Log phase Jurkat cells growing in antibiotic-free medium were transiently transfected with the indicated siRNAs using Lipofectamine 2000 (Thermo Fisher). Beginning 24 h after electroporation, cells were treated with the indicated reagents for another 24 h, and measured sub-G1 population to evaluate apoptotic cells.
Immunoblotting
After individual treatments, cells were collected, lysed in sample buffer (62.5 mM Tris-HCl, pH 6.8, 4 M urea, 2% SDS, 1 mM EDTA), and sonicated. Proteins concentrations were then determined by BCA assay. Samples with equal amounts of protein were then separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were then blocked and incubated with primary antibodies at 4°C overnight. The next day, membranes were washed, incubated with HRP-conjugated secondary antibodies and visualized by enhanced chemoluminescence.
Cell fractionation
Jurkat cells that were treated with indicated drugs or DMSO for 24 h were washed, incubated with hypotonic buffer (25 mM HEPES, 5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, pH 7.4) for 20 min on ice, and lysed with 25 strokes in a tight-fitting Dounce homogenizer. NaCl was then added to 150 mM and the nuclear fractions were removed by centrifugation at 2500 rpm for 15 min. After the postnuclear supernatant was sedimented at 8000 rpm for 15 min, the supernatant (crude cytosol fraction) and the pellet (crude mitochondrial fraction) were prepared for immunoblotting.
Statistical analysis
All experiments were independently repeated three times unless otherwise noted. Results analyzed using GraphPad Prism 7 software are shown as mean ± standard deviation (SD). Differences between samples were evaluated by one-way ANOVA with Tukey’s multiple comparison test. P > 0.05 was considered not significant (ns). Significant values are denoted as *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
The AKT inhibitor MK-2206 promotes the MCL-1 inhibitor S63845-induced apoptosis in multiple leukemia cells
Building on a previous observation that the AKT inhibitor MK-2206 sensitizes B-cell precursor acute lymphoblastic leukemia cells to the BCL-2 selective antagonist venetoclax [Citation40], we assessed whether AKT inhibitors also sensitize MCL1 inhibitor-induced cell death in leukemia cell lines. The AKT inhibitor MK-2206 and the MCL1 inhibitor S63845 were first used. Morphologically, MK-2206 itself did not induce Jurkat cell death, however, it increased S63845-induced cell death, as more cells showed fragmentation and apoptotic bodies ((A)), suggesting that apoptosis was induced. To assess the percentage of apoptotic cells induced by the MK-2206/S63845 combinations, we measured the sub-G1 population, which reflects the cells having fragmented DNA. The results showed that MK-2206 significantly enhanced the apoptosis induced by S63845 ((B)). In particular, 128 nM MK-2206 increased 12.5 nM S63845-induced Jurkat apoptosis from ∼12% to 42% ((B)). Synergistic effects were observed with the two agents (right panel, (B)). We then used Annexin V and PI double staining assay to assess the apoptotic cells and found that MK-2206 also increased the percentage of Annexin V + and/or PI + cells induced by S63845, similar to DNA fragmentation assay ((C)). To further confirm that apoptosis was induced, we evaluated PARP cleavage, a marker often used for caspase activation, and found that MK-2206 significantly increased the PARP cleavage induced by S63845 in Jurkat cells (left panel, (D)). Taken together, these data suggested that MK-2206 sensitized S63845-induced apoptosis in Jurkat cells.
Figure 1. MK-2206 promotes S63845-induced apoptosis in multiple leukemia cell lines. (A) After Jurkat cells were treated with DMSO (Con), S63845 (12.5 nM), MK-2206 (128 nM), or S63845 (12.5 nM) + MK-2206 (128 nM), the images of the cells were taken under microscopy. (B, E–H) After Jurkat cells (B), Molt4 (E), U937 (F), THP.1 (G) or HL-60 cells (H) were treated with indicated concentrations of S63845 in combination with MK-2206 for 24 h, the cells were subjected to flow cytometry analysis for sub-G1 cells. Summary data from three independent experiments are shown. Right panels, combination indexes were calculated. (C, I-J) After Jurkat cells (C), U937 (I) or THP.1 cells (J) were treated with indicated concentrations of S63845 in combination with MK-2206 for 24 h, the cells were subjected to Annexin V/PI double staining and assessed by flow cytometry. Summary data from three independent experiments (left panels) and a representative experiment (right panels) are shown. (D) After Jurkat (left) or U937 (right) cells were incubated with control, S63845 (12.5 nM) alone, MK-2206 (128 nM) alone, or their combination for 24 h, whole cell lysates were prepared and subjected to immunoblotting with antibodies against PARP and GAPDH. Data in B, C and E–J represent mean ± SD from three independent experiments. Differences between groups in this figure, the following figures and supplementary figures were determined by ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ns, not significant.
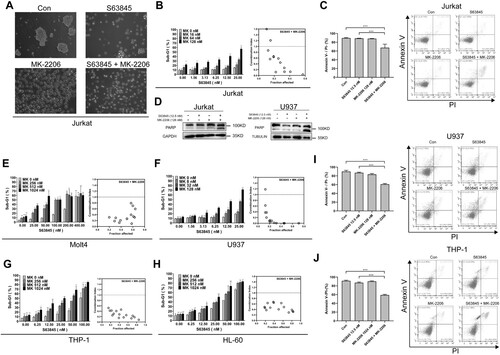
To test whether MK-2206 synergize with S63845 in other leukemia cell lines, we treated several other leukemia cell lines with S63845/MK-2206 combinations. These experiments suggested that MK-2206 and S63845 have synergistic effects in the induction of apoptosis (evaluated by Sub-G1) of multiple leukemia cell lines, including another ALL cell line Molt4 ((E)), and three AML cell lines U937 ((F)), THP.1 ((G)) and HL-60 ((H)). Moreover, the combination of MK-2206 with S63845 also induced more Annexin V + and/or PI + cells than S63845 monotherapy in U937 and THP.1 ((I,J)). To further test whether apoptosis was induced, we also evaluated PARP cleavage induced by S63845 and MK-2206 combination in U937 cells, and again a significant PARP cleavage was induced after S63845 combined with MK-2206 (right panel, (D)). Taken together, these data suggested that MK-2206 sensitized S63845-induced apoptosis in multiple leukemia cell lines.
The AKT inhibitor Gsk690693 promotes the S63845-induced apoptosis in multiple leukemia cells
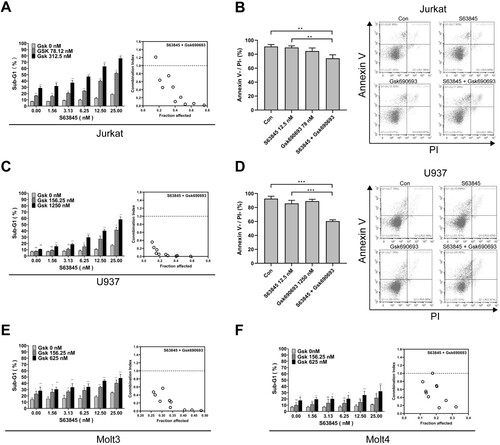
To test whether other AKT inhibitors also synergize S63845 to induce leukemia cell apoptosis, we used another AKT inhibitor Gsk690693 to combine with S63845. The results indicated that Gsk690693 synergized with S63845 to induce apoptosis in Jurkat cells, as assessed by the percentage of sub-G1 cells ((A)). Gsk690693 also promoted S63845 to induce Annexin V + and/or PI + cells in Jurkat ((B)). Moreover, Gsk690693 showed synergy with S63845 to induce the AML cell line U937 apoptosis ((C,D)). In addition, Gsk690693 synergized with S63845 to induce apoptosis in the ALL cell lines Molt3 and Molt4 ((E,F)).
Figure 2. Gsk690693 promotes S63845-induced apoptosis in multiple leukemia cell lines. (A, C, E–F) After Jurkat cells (A), U937 (C), Molt3 (E) or Molt4 cells (H) were treated with indicated concentrations of S63845 in combination with Gsk690693 (Gsk) for 24 h, the cells were subjected to flow cytometry analysis for sub-G1 cells. Summary data from three independent experiments are shown. Right panels, combination indexes were calculated. (B, D) After Jurkat cells (B), or U937 (D) were treated with indicated concentrations of S63845 in combination with Gsk690693 for 24 h, the cells were subjected to Annexin V/PI double staining and assessed by flow cytometry. Summary data from three independent experiments (left panels) and a representative experiment (right panels) are shown.
Transcriptome sequencing analysis suggests that S63845/MK-2206 combination regulates mitochondrial pathways
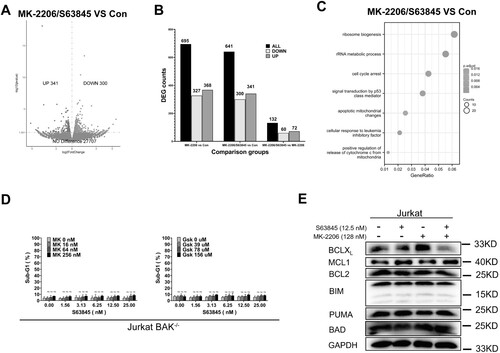
To find out the mechanism by which MK-2206 sensitizes S63845-induced apoptosis, comparative transcriptome sequencing of Jurkat cells after DMSO, MK-2206 or S63845/MK-2206 combination treatments was carried out. Compared to control (DMSO), 368 genes were upregulated and 327 genes were downregulated after MK-2206 only treatment, whereas 341 genes were upregulated and 300 genes were downregulated upon S63845/MK-2206 combination treatment ((A,B), and Supplemental Figure 1A). Compared to MK-2206 treatment, S63845/MK-2206 combination only upregulated 72 genes and downregulated 60 genes ((B)), suggesting that MK-2206 induced much more transcriptional changes than S63845. Further analysis by KEGG of these differential expression genes (DEGs) indicated that several signaling pathways were enriched upon S63845/MK-2206 combination treatment, including apoptotic mitochondrial changes, positive regulation of apoptotic signaling pathway, cell cycle arrest signaling pathway and some other signaling pathways (Supplemental Figure 1B). While most of these pathways were essential in the regulation of mitochondrial apoptotic pathway, our transcriptome sequencing results suggested that MK-2206 probably sensitizes Jurkat cells to S63845-induced cell death through the change of mitochondrial apoptosis pathway.
Figure 3. MK-2206 sensitizes to S63845-induced apoptosis through the mitochondrial apoptotic pathway. (A) After Jurkat cells were treated with solvent (Con), MK-2206 (128 nM), or S63845 (12.5 nM)/MK-2206 (128 nM) combination, cells were collected and subjected to transcriptome analysis. Volcano plot of upregulated and downregulated differentially expressed genes (DEGs) of MK-2206/S63845 vs Con were shown. (p value < 0.05 was used to select the DEGs.) (B) DEGs between MK-2206 vs. Con group, MK-2206/S63845 combination vs. Con group, and MK-2206/S63845 combination vs. MK-2206 group were shown. (C) KEGG enrichment analysis using the changed DEGs from S63845/MK-2206 combination vs Con. (D) After BAK−/− Jurkat cells were treated with of indicated concentration of S63845 in combination with MK-2206 (left panel) or Gsk690693 (right panel) for 24 h, the cells were subjected to flow cytometry analysis for sub-G1. (E) After Jurkat cells were incubated with control, S63845 (12.5 nM) alone, MK-2206 (128 nM) alone, or their combination for 24 h, whole cell lysates were subjected to immunoblotting with the indicated antibodies. A representative experiment from three independent experiments is shown. Data in D are presented as mean ± SD from three independent experiments.
MK-2206 sensitizes S63845 through the mitochondrial apoptotic pathway and partially through BCLXL downregulation
In further experiments, we set up experiments to validate whether the mitochondrial apoptotic pathway was essential in MK-2206-induced sensitization to S63845. In Jurkat cells, which have about 8 times more BAK than BAX [Citation48], BAK gene knockout not only abolished the apoptosis induced in Jurkat cells by treatment with S63845/MK-2206 ((D)), but also abolished the apoptosis induced by S63845/Gsk690693 combination, suggesting that cell death induced by the combinations is through the mitochondrial apoptotic pathway.
To identify the BCL2 family protein(s) responsible for the synergistic effects of the combination, we used western blotting to examine BCL2 family protein expressions. In Jurkat cells, we observed BCLXL downregulation after treatment with MK-2206 alone or in combination with S63845, but not other BCL2 family proteins, such as the anti-apoptotic proteins MCL1 and BCL2, and the pro-apoptotic proteins BIM, PUMA and BAD ((E)). Given the fact that S63845 inhibits MCL1 and downregulation of BCLXL might sensitize S63845-induced apoptosis, these data suggested that BCLXL downregulation might at least partially account for MK-2206-induced sensitization of S63845 in Jurkat cells.
MK-2206 induces BAD dephosphorylation and translocation to mitochondria
Because a previous study suggested that MK-2206 induces the BH3-only protein BAD dephosphorylation [Citation49] and BAD dephosphorylation have been suggested to play an important role in the induction of apoptosis [Citation50–56], we performed immunoblotting analysis of AKT phosphorylation and phosphorylation of its downstream substrate BAD. This analysis indicated that MK-2206 treatment decreased phosphorylation of AKT at Ser473 and BAD phosphorylation at Ser99 ((A)). The AKT dephosphorylation induced by MK-2206 is consistent with previous studies [Citation57]. Dose-dependent dephosphorylation of AKT was observed after MK-2206 treatment but not S63845 treatment ((B)). Further experiments indicated that MK-2206 treatment but not S63845 treatment significantly increased BAD translocation to mitochondria in Jurkat cells ((C)). MK-2206 but not S63845 also increased BAD translocation to mitochondria in U937 cells ((D)), suggesting similar sensitization mechanisms by MK-2206 in different leukemia cell lines.
Figure 4. BAD dephosphorylation and translocation induced by MK-2206 is essential for MK-2206-induced sensitization to S63845 in Jurkat cells. (A) After Jurkat cells were incubated with control, S63845 (12.5 nM) alone, MK-2206 (128 nM) alone, or their combination for 24 h, whole cell lysates were prepared and subjected to immunoblotting with antibodies against AKT, p-AKT (Ser473), p-BAD (Ser99) and GAPDH. (B) After Jurkat cells were incubated with increased concentrations of S63845 or MK-2206, whole cell lysates were prepared and subjected to immunoblotting with antibodies against p-AKT (Ser473) and GAPDH.(C-D) 24 h after Jurkat cells (C) or U937 cells (D) were treated with control, S63845 (12.5 nM) alone, MK-2206 (128 nM) alone, or their combination, the indicated cellular fractions were isolated and subjected to immunoblotting for BAD, TOM20 or GAPDH. (E) 24 h after Jurkat cells were transiently transfected with Control or BAD siRNAs, whole cell lysates were subjected to immunoblotting. (F) 24 h after Jurkat cells were transfected with Control siRNA or BAD siRNA (E), cells were treated with 128 nM MK-2206 with or without S63845 (0–25 nM) for another 24 h, then cells were subjected to flow cytometry analysis for sub-G1. Data in F is presented as mean ± SD from three independent experiments.
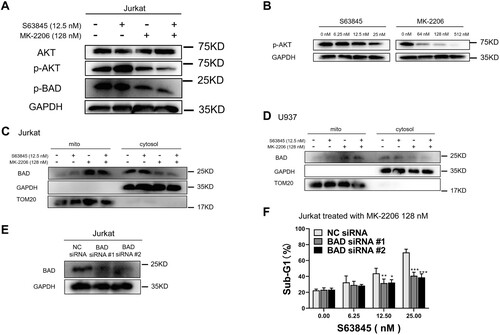
To investigate whether BAD plays a role in MK-2206-induced sensitization, we knocked down BAD using siRNA ((E)). This analysis indicated that BAD knockdown significantly reduced apoptosis induced by S63845/MK-2206 combination ((F)), suggesting that BAD dephosphorylation and translocation to mitochondria play an important role of MK-2206 sensitization to S63845.
Discussion
Here we report that AKT inhibitors MK-2206 and Gsk690693 sensitize acute leukemia cell lines to the MCL1 inhibitor S63845. Further experiments indicate that MK-2206 induces downregulation of BCLXL, which accounts partially for its sensitization to S63845. Moreover, MK-2206 induces BAD dephosphorylation and translocation to mitochondria, and knockdown of BAD inhibits MK-2206 sensitization to S63845, suggesting that MK-2206-induced BAD dephosphorylation enhances killing by S63845.
Hematological malignancies are derived from cells of the hematologic system. Overexpression of the anti-apoptotic BCL2 family proteins including MCL1 is related to tumorigenesis of several hematological malignancies, such as AML and lymphoma. Moreover, MCL1 are often upregulated after relapse and contributes to drug resistance [Citation2,Citation5,Citation7–11]. Although MCL1 has been targeted for quite a long time, none of the MCL1 inhibitors has been approved yet to treat cancer either as single agent or in combinations. One of the reasons is the on-target side effects of the MCL1 inhibition, including hematopoietic, hepatic and cardiac toxicities [Citation24–28]. Recent clinical studies have also indicated cardiac toxicities of MCL1 inhibitors in patients. Thus, combination to reduce the usage of MCL1 inhibitors is of potential interest in the treatment of cancer, especially leukemia.
PI3K/AKT signaling plays important role in regulating cellular functions. Moreover, activation of PI3K/AKT/mTOR signaling contributes to the pathogenesis of many cancer types. For example, PI3Ks have been reported to be involved in cell growth, proliferation, differentiation and intracellular trafficking, which in turn contribute to cancer development [Citation58]. AKT is functionally activated or deactivated through phosphorylation or dephosphorylation, resulting in different consequences in controlling cell metabolism, growth, proliferation, and survival [Citation59]. The activated AKTs affect cellular proliferation or survival through multiple downstream signaling pathways, such as activating the pathway for the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), or suppression of the p53 pathway [Citation60]. Gsk690690 and MK-2206 are AKT inhibitors that effectively inhibit AKT1, AKT2, and AKT3. In particular, MK-2206 is an allosteric inhibitor of AKT with activity against all three AKT isoforms but has more inhibition against AKT1 and AKT2 [Citation61]. MK-2206 has been under clinical trials for endometrial cancer [Citation62], uterine serous carcinoma [Citation63], and breast cancer [Citation64].
Gsk690690 or MK-2206 induces or sensitizes cell death through the growth inhibition pathways and cell cycle arrest Pathways. Because of the possible sensitization effects of the AKT inhibitors, and also because a previous study showed that MK-2206 sensitizes the B-cell leukemia cells to venetoclax [Citation40], we started our study to evaluate the AKT inhibitors to sensitize MCL1 inhibitors in leukemia cell lines. Our study found AKT inhibitors MK-2206 and Gsk690693 sensitize multiple leukemia cells to the MCL1 inhibitor S63845.
To find out the potential mechanism, we used transcriptome sequencing. After analyzed the transcriptome sequencing data with KEGG program, we identified that the mitochondrial apoptotic pathway is the potential pathway that is involved in the MK-2206 sensitization. We further confirmed the importance of mitochondrial apoptosis pathway by using BAK knockout cells and found that BAK knockout totally abolished both S63845/MK-2206 combination-induced and S63845/Gsk690693 combination-induced apoptosis. Because the mitochondrial apoptotic pathway is controlled by BCL2 family proteins, we therefore analyzed BCL2 family protein expressions upon S63845, MK-2206 monotherapies or their combinations. While a previous study has indicated that MK-2206 downregulates MCL1 [Citation39], however, in our study, we did not find MK-2206 downregulates MCL1. Instead, we found BCLXL downregulation after MK-2206 treatment, which at least partially contributes to its sensitization of the MCL1 inhibitor S63845. In addition, previous studies have also suggested MK-2206 induces BAD dephosphorylation [Citation49], which plays an important role in regulating apoptosis [Citation50–56], we therefore investigated BAD phosphorylation. Our experiments not only found MK-2206 induces BAD dephosphorylation but also indicated that BAD translocate to mitochondria upon MK-2206 treatment. Previous studies have found after BAD translocation to mitochondria, BAD can bind to and inhibit the anti-apoptotic protein BCLXL [Citation49,Citation65], and the inhibition of BCLXL can sensitize MCL1 inhibitor [Citation66]. Our experiments found BAD knockdown inhibited MK-2206 sensitization to S63845, in agreement with the possibility of BCLXL inhibition by BAD and underscoring the essential role of BAD in MK-2206 sensitization to the MCL1 inhibitor.
In summary, the present study shows that the AKT inhibitor MK-2206 sensitizes multiple acute leukemia cells to the MCL1 inhibitor S63845. Further experiments suggest that MK-2206 downregulates BCLXL and dephosphorylates BAD, both of which play important roles in MK-2206 sensitization.
Supplemental Material
Download PDF (185.9 KB)Acknowledgements
Conceptualization, H.D.; Investigation, Y.L., L.D., K.Y., X.S., L.H., S.G.; Writing, H.D. and Y.L. The manuscript has been reviewed by all the authors.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
This study includes no data deposited in external repositories. All the raw data reported in this paper will be shared by the lead contact upon request.
Additional information
Funding
References
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi:10.1038/nrc883.
- Chipuk JE, Moldoveanu T, Llambi F, et al. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi:10.1016/j.molcel.2010.01.025.
- Czabotar PE, Lessene G, Strasser A, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi:10.1038/nrm3722.
- Moldoveanu T, Follis AV, Kriwacki RW, et al. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014;39:101–111. doi:10.1016/j.tibs.2013.12.006.
- Correia C, Lee S-H, Meng XW, et al. Emerging understanding of Bcl-2 biology: implications for neoplastic progression and treatment. Biochim Biophys Acta. 2015;1853:1658–1671. doi:10.1016/j.bbamcr.2015.03.012.
- Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. doi:10.1038/cdd.2017.186.
- Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193. doi:10.1038/s41580-018-0089-8.
- Zhou P, Levy NB, Xie H, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood. 2001;97:3902–3909. doi:10.1182/blood.v97.12.3902.
- Xiang Z, Luo H, Payton JE, et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest. 2010;120:2109–2118. doi:10.1172/JCI39964.
- Kaufmann SH, Karp JE, Svingen PA, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91:991–1000. doi:10.1182/blood.V91.3.991.
- Meng XW, Lee S-H, Dai H, et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem. 2007;282:29831–29846. doi:10.1074/jbc.M706110200.
- Satta T, Grant S. Enhancing venetoclax activity in hematological malignancies. Exp Opin Invest Drugs. 2020;29:697–708. doi:10.1080/13543784.2020.1789588.
- Reed JC. Bcl-2–family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi:10.1182/blood-2007-09-078162.
- Sarosiek KA, Chonghaile TN, Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol. 2013;23:612–619. doi:10.1016/j.tcb.2013.08.003.
- Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi:10.1038/nature19830.
- Szlavik Z, Csekei M, Paczal A, et al. Discovery of S64315, a potent and selective Mcl-1 inhibitor. J Med Chem. 2020;63:13762–13795. doi:10.1021/acs.jmedchem.0c01234.
- Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Dis. 2018;8:1582–1597. doi:10.1158/2159-8290.CD-18-0387.
- Caenepeel S, Karen R, Belmontes B, et al. Discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res. 2020;80:6218–6218. doi:10.1158/1538-7445.AM2020-6218.
- Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9:5341. doi:10.1038/s41467-018-07551-w.
- Cory S, Roberts AW, Colman PM, et al. Targeting BCL-2-like proteins to kill cancer cells. Trends Cancer. 2016;2:443–460. doi:10.1016/j.trecan.2016.07.001.
- Roberts AW, Wei AH, Huang DC. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood. 2021;138:1120–1136. doi:10.1182/blood.2020006785.
- Montero J, Haq R. Adapted to survive: targeting cancer cells with BH3 mimetics adaptation to BH3 mimetics. Cancer Dis. 2022: OF1–OF16. doi:10.1158/2159-8290.CD-21-1334.
- Diepstraten ST, Anderson MA, Czabotar PE, et al. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22:45–64. doi:10.1038/s41568-021-00407-4.
- Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi:10.1126/science.1106114.
- Wang X, Bathina M, Lynch J, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi:10.1101/gad.215855.113.
- Thomas RL, Roberts DJ, Kubli DA, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365–1377. doi:10.1101/gad.215871.113.
- Hikita H, Takehara T, Shimizu S, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50:1217–1226. doi:10.1002/hep.23126.
- Brennan MS, Chang C, Tai L, et al. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood. 2018;132( ):1573–1583. doi:10.1182/blood-2018-06-859405.
- A study of venetoclax and AMG 176 in patients with relapsed/refractory hematologic malignancies, https://clinicaltrials.gov/ct2/show/results/NCT03797261.
- Wei AH, Roberts AW, Spencer A, et al. Targeting MCL-1 in hematologic malignancies: rationale and progress. Blood Rev. 2020;44:100672. doi:10.1016/j.blre.2020.100672.
- Song M, Bode AM, Dong Z, et al. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–1031. doi:10.1158/0008-5472.CAN-18-2738.
- Liu R, Chen Y, Liu G, et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11:797. doi:10.1038/s41419-020-02998-6.
- Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk. Lymph. 2018;59:2524–2534. doi:10.1080/10428194.2017.1421760.
- Bertacchini J, Heidari N, Mediani L, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72:2337–2347. doi:10.1007/s00018-015-1867-5.
- Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Ann Rev Med. 2016;67:11–28. doi:10.1146/annurev-med-062913-051343.
- Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Dis. 2009;8:627–644. doi:10.1038/nrd2926.
- Brazzatti J, Klingler-Hoffmann M, Haylock-Jacobs S, et al. Differential roles for the p101 and p84 regulatory subunits of PI3Kγ in tumor growth and metastasis. Oncogene. 2012;31:2350–2361. doi:10.1038/onc.2011.414.
- Levy DS, Kahana JA, Kumar R, et al. GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood. 2009;113:1723–1729. doi:10.1182/blood-2008-02-137737.
- Lu J-W, Lin Y-M, Lai Y-L, et al. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med Oncol. 2015;32:1–9. doi:10.1007/s12032-015-0650-7.
- Richter A, Fischer E, Holz C, et al. Combined application of Pan-AKT inhibitor MK-2206 and BCL-2 antagonist venetoclax in B-cell precursor acute lymphoblastic leukemia. Int J Mol Sci. 2021;22:2771. doi:10.3390/ijms22052771.
- Larsen JT, Shanafelt TD, Leis JF, et al. Akt inhibitor MK-2206 in combination with bendamustine and rituximab in relapsed or refractory chronic lymphocytic leukemia: results from the N1087 alliance study. Am J Hematol. 2017;92:759–763. doi:10.1002/ajh.24762.
- Dai H, Ding H, Peterson KL, et al. Measurement of BH3-only protein tolerance. Cell Death Differ. 2018;25:282–293. doi:10.1038/cdd.2017.156.
- Dai H, Smith A, Meng XW, et al. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011;194:39–48. doi:10.1083/jcb.201102027.
- Berenbaum MC. What is synergy? Pharmacol Rev. 1989;41:93–141.
- Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi:10.1093/nar/gkz114.
- Davidson NM, Oshlack A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15:410. doi:10.1186/s13059-014-0410-6.
- Luo W, Pant G, Bhavnasi YK, et al. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017;45:W501–W508. doi:10.1093/nar/gkx372.
- Dai H, Meng XW, Lee SH, et al. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem. 2009;284:18311–18322. doi:10.1074/jbc.M109.004770.
- Mai Z, Sun H, Yang F, et al. Bad is essential for Bcl-xL-enhanced Bax shuttling between mitochondria and cytosol. Int J Biochem Cell Biol. 2023;155:106359. doi:10.1016/j.biocel.2022.106359.
- Pandey V, Zhu T, Ma L, et al. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018;415:177–186. doi:10.1016/j.canlet.2017.11.017.
- Pandey V, Wang B, Mohan CD, et al. Discovery of a small-molecule inhibitor of specific serine residue BAD phosphorylation. Proc Natl Acad Sci U S A. 2018;115:E10505–E10514. doi:10.1073/pnas.1804897115.
- Bui NL, Pandey V, Zhu T, et al. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018;415:177–186. doi:10.1016/j.canlet.2017.11.017.
- Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27(Suppl 1):S53–S70. doi:10.1038/onc.2009.44.
- Datta SR, Dudek H, Tao X, et al. Greenberg, Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi:10.1016/s0092-8674(00)80405-5.
- Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi:10.1016/s0092-8674(00)81383-5.
- Zha J, Harada H, Yang E, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628. doi:10.1016/s0092-8674(00)81382-3.
- Konopleva MY, Walter RB, Faderl SH, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myeloid leukemia. Clin Cancer Res. 2014;20:2226–2235. doi:10.1158/1078-0432.CCR-13-1978.
- Yuan T, Cantley L. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi:10.1038/onc.2008.245.
- Qi CL, Huang ML, Zou Y, et al. The IRF2/CENP-N/AKT signaling axis promotes proliferation, cell cycling and apoptosis resistance in nasopharyngeal carcinoma cells by increasing aerobic glycolysis. J Exp Clin Cancer Res. 2021;40:390. doi:10.1186/s13046-021-02191-3.
- Ma Y, Sender S, Sekora A, et al. The inhibitory response to PI3K/AKT pathway inhibitors MK-2206 and buparlisib is related to genetic differences in pancreatic ductal adenocarcinoma cell lines. Int J Mol Sci. 2022;23:4295. doi:10.3390/ijms23084295.
- Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs In vitro and In vivoMK-2206 sensitizes tumors to chemotherapy. Mol Cancer Ther. 2010;9:1956–1967. doi:10.1158/1535-7163.MCT-09-1012.
- Myers AP, Konstantinopoulos PA, Barry WT, et al. Phase II, 2-stage, 2-arm, PIK3CA mutation stratified trial of MK-2206 in recurrent endometrial cancer. Int J Cancer. 2020;147:413–422. doi:10.1002/ijc.32783.
- Stover EH, Xiong N, Myers AP, et al. A phase II study of MK-2206, an AKT inhibitor, in uterine serous carcinoma. Gynecol Oncol Rep. 2022;40:100974. doi:10.1016/j.gore.2022.100974.
- Xing Y, Lin NU, Maurer MA, et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019;21:1–12. doi:10.1186/s13058-019-1154-8.
- Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi:10.1016/j.molcel.2004.12.030.
- Lee EF, Harris TJ, Tran S, et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis. 2019;10:342. doi:10.1038/s41419-019-1568-3.