

ABSTRACT
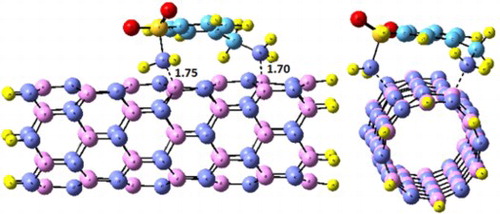
In this study, the adsorption behavior of boron nitride nanotubes (BNNTs) and their Al- and Si-doped BNNTs toward sulfonamide (SA) drugs is explored using density functional theory (DFT) calculations. Total energies, geometry optimizations, frontier molecular orbital (FMO) and density of state (DOS) analyses were obtained using M06-2X level of theory and the 6-311G(d) basis sets. In the most stable conformation, adsorption energies of the sulfonamide molecule over pristine BNNTs, Al-BNNTs, Si-BNNTs are 25.9, 59.7 and 51.7 kcal/mol, respectively. The HOMO–LUMO gap, Eg, which corresponds to sensing ability, is the most for pristine BNNTs toward SA indicating that BNNTs can be a promising candidate for sensing SA. On the other hand, Al-BNNTs and Si-BNNTs cannot used for sensing SA.
GRAPHICAL ABSTRACT
Acknowledgment
The authors thank Prof. Lakshmaiah Sreerama, Department of Chemistry and Earth Sciences, Qatar University, Doha, Qatar for critically editing this manuscript for English language.
Disclosure statement
No potential conflict of interest was reported by the authors.