
1. Introduction
Many drugs are cleared from the body via biotransformation, thereby generating circulating and/or excreted drug metabolites. These metabolites are usually more hydrophilic than the parent drug as a result of oxidative metabolism, reduction, or conjugation with sugars or amino acids. In some cases, hydrolysis or oxidative dealkylation of the drug can result in cleavage into a number of fragments of the native parent drug. In each of these cases, the resultant metabolite may possess a similar or unique safety profile relative to the parent drug.
Because of species-specific expression and homology of many drug-metabolizing enzymes, metabolism profiles can vary between species. Differences in metabolite abundance are often observed between animals and humans. Less commonly, humans may generate a novel metabolite that is not present in any nonclinical test species. A critical component of the drug development process is the nonclinical safety assessment of a new chemical entity in relevant animal species. These nonclinical studies are conducted at doses in excess of the intended therapeutic dose in humans in order to establish safety margins to undesirable effects. Consequently, the safety of such novel metabolite(s) would not be adequately explored in animals by toxicology studies conducted with the parent drug even at much higher doses [Citation1,Citation2].
The Guidance for Industry on Safety Testing of Drug Metabolites published by the US Food and Drug Administration in 2008 (and recently revised in 2016), as well as the ICH M3(R2) guidance, collectively provide recommendations on how and when to characterize the nonclinical safety of drug metabolites of so-called small molecule (i.e. MW <900 g/mol) investigational agents [Citation3–Citation5]. The topic has been reviewed in depth from an industry perspective [Citation6–Citation10]. The currently accepted prevalence threshold for a metabolite of interest is ≥10% of estimated total drug-related exposure at steady state. In addition, while pharmacologic activity of the metabolite is an important factor for safety coverage, pharmacologically inactive metabolites can still be associated with toxicity and must be considered as well in the overall safety evaluation.
Given that 2017 began the tenth year of implementation of the recommendations outlined in the Guidance for Industry on Safety Testing of Drug Metabolites across the pharmaceutical industry, it seems timely to revisit some key concepts of the metabolite safety guidance and share learning from both industry and regulatory perspectives.
2. What is ‘safety coverage’ for a disproportionate human metabolite and are certain metabolites exempt?
A common question arising during drug development is the required ratio between animal exposure of a metabolite at a no-observed-adverse-effect-level and its human exposure at a pharmacological dose. While the FDA guidance implies that similar exposure of the metabolite in human and animals is needed, the ICH M3(R2) Questions and Answers [Citation5] indicate that characterization of metabolite toxicity would be considered adequate when animal exposure is at least 50% of its human exposure. In general, the assessment of safety coverage is made based on total exposure (rather than unbound) in animals versus humans. However, when a metabolite comprises the majority of total circulating drug-related material, animal exposure should equal or exceed the human exposure. Nonetheless, a safety coverage approach will be determined on a case-by-case basis in consultation with FDA.
As stated in guidance documents [Citation3,Citation4], not all disproportionate human metabolites (i.e. present only in humans or present at higher plasma concentrations in humans than in the animals used in nonclinical studies) must be covered in animal studies. While simple O-glucuronides, O-sulfates, and quaternary N+-glucuronides may be considered benign from a human safety perspective, animal coverage of acyl glucuronides could still be warranted because of reactivity concerns. Alternatively, a stable glutathione or N-acetylcysteine conjugate might pose a lesser safety concern than its corresponding short-lived electrophilic metabolite (e.g. epoxide), but this must be considered within the context of all nonclinical safety data. In cases where simple glucuronide and sulfate conjugates are unique to humans, circulate at relatively high levels, and/or possess long half-lives, justification should be provided to explain why such human metabolites may not need further safety evaluation. For example, a long-lived disproportionate carbamoyl N-glucuronide metabolite with indeterminate reactivity was subjected to scrutiny by Center for Drug Evaluation and Research (CDER) and the Sponsor during the drug development process.
While not specifically addressed in the guidance and on a case-by-case basis, some downstream metabolites exceeding the 10% threshold may not require safety assessment when the primary metabolites have been formed at adequate levels in the animal species to cover human exposure, and those metabolites are readily eliminated from the body within a short period of time.
3. Are semi-quantitative or fit-for-purpose methods successful in establishing metabolite safety coverage?
Knowledge of the metabolic profile of the drug in humans and animals can focus efforts and resources in a scientific, staged approach to drug development. Since publication of the metabolite safety testing guidance, an increasing number of sponsor submissions have provided some assessment of drug metabolism in the first-in-human study protocol, consistent with prediction of potential major human metabolites based on in vitro metabolism. Unfortunately, in vitro metabolic profiles from liver microsomes and/or hepatocyte incubations can be poor predictors of in vivo circulating major human metabolites [Citation11,Citation12]. Sometimes the exact structure of seemingly major human metabolite(s) cannot be ascertained or a metabolite standard cannot be easily synthesized, so nontraditional approaches to establish metabolite coverage are needed.
Over the past decade, a number of creative fit-for-purpose methods have been developed to address these gaps. Some of these approaches include mixed matrix LC-MS/MS peak area comparisons across species [Citation13], NMR-based techniques [Citation14,Citation15], and bioanalytical methods with radiocalibrants [Citation16]. LC-MS peak area comparisons between animals and humans are routinely used by some sponsors to demonstrate that metabolite concentrations in animals exceed plasma concentrations in humans. In such cases, the nominal circulating concentration of the metabolite in the animal species is irrelevant as long as the LC-MS peak area clearly exceeds that of human. Alternatively, a modified radiocalibration semi-quantitative method developed by Lilly was used to establish rat and dog safety coverage of a disproportionate human metabolite for which a reference standard could not be synthesized in stable form [Citation17]. Either way, if semi-quantitative methods suggest lack of coverage, it may be prudent to plan for safety assessment of the metabolite after the human disposition study confirms that the metabolite exceeds the 10% prevalence threshold.
4. How common is direct safety testing of a disproportionate human metabolite?
The most common scenario is the presence of the major human metabolite at adequate concentrations in one animal species. Therefore, general toxicity studies with the parent drug, including reproductive development, should demonstrate adequate metabolite coverage. In the event that the selected toxicology species do not provide coverage of a particular metabolite, alternative animal species should be explored. While not frequent, standalone metabolite safety qualification in animals has been required. In a typical year, the Executive Carcinogenicity Assessment Committee of CDER reviews at most one rodent carcinogenicity study involving a major human metabolite. The need for a carcinogenicity study with a disproportionate metabolite would likely be driven by a confirmed genetic toxicity risk, as shown in the following example.
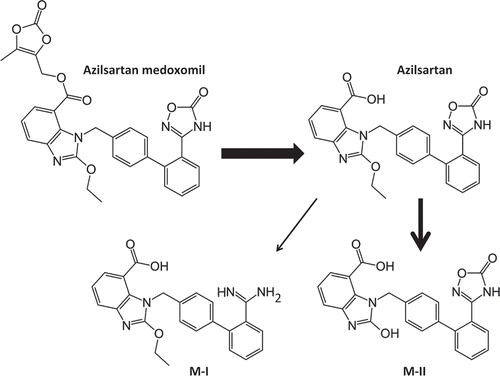
In 2011, the prodrug azilsartan medoxomil (Edarbi®) was approved for treatment of hypertension. Azilsartan is metabolized to two primary metabolites (). The major metabolite in plasma (M-II) is formed by O-dealkylation primarily via CYP2C9 and the minor metabolite (M-I) is formed by decarboxylation primarily via CYP2C8. Systemic exposures to these metabolites in humans were approximately 50% and <1% of azilsartan, respectively. M-I and M-II do not contribute to the pharmacologic activity of the drug. Metabolite M-II was studied in 13-week rat and dog repeat-dose toxicity studies and in reproduction/developmental studies. However, the M-II metabolite was positive for structural aberrations in the Chinese hamster lung cytogenetic assay without metabolic activation. Other genetic toxicity assays for this metabolite were negative. Ultimately, M-II was found to be not carcinogenic when assessed in a 26-week Tg.rasH2 mouse study and in a 2-year rat study. The highest doses tested produced exposures that were, on average, about 30 (mice) and 7 (rats) times the average exposure to M-II in humans at the maximum recommended human dose [Citation18].
Figure 1. Structures of azilsartan medoxomil prodrug, azilsartan, and two primary metabolites (M-I and M-II).
5. When is the appropriate time to establish metabolite safety coverage?
Adopting proactive approaches to the demonstration of safety coverage of human metabolites is the key to enabling a smooth regulatory path to US submission and approval. In some cases, comprehensive in vivo metabolism assessment in animals is determined and submitted as early as Phase 2a. Because some candidate attrition is anticipated in early development, it is not uncommon for sponsors to defer investigation of metabolism until at least Phase 2b. One common mitigation adopted by several companies is the exploratory profiling (or ‘metabolite scouting’) of samples from Phase 1 studies [Citation19]. While resource sparing, the consequences of deferring an exploration of human metabolism until an advanced stage could impede drug development with a potential clinical hold and/or delayed drug approval. In a recent CDER example, a major human circulating metabolite with a very long half-life was identified during later development. This metabolite was also observed in animals; however, due to its poor characterization, comparison of exposure and long-term safety evaluation could not be conducted in animals in a timely manner. Therefore, this led to delay in FDA review and approval because further assessment both in humans and animals was required to address potential safety concerns.
6. Expert opinion
Industry sponsors are encouraged to interact with FDA throughout development, with a special emphasis on data sharing at the End-of-Phase 2 meeting that routinely precedes initiation of large Phase 3 trials. The FDA has provided helpful recommendations on a case-by-case basis, especially when metabolite standards are not available and when metabolite exposure estimates must be limited to fit-for-purpose bioanalytical approaches [Citation20]. Although many sponsors have become familiar with metabolite safety testing guidelines over more than a decade, only proactive and ongoing communication with regulators can prevent delays.
Finally, it must be emphasized that standalone evaluation of disproportionate and unique human metabolites remains an uncommon but important aspect of nonclinical safety assessment. The few examples that have been publicly documented demonstrate that metabolites may possess their own inherent toxicity profile, and this information should be considered by sponsors and regulators to address the safety risks of new investigational drugs.
Declaration of interest
Debra Luffer-Atlas is a full-time employee of Eli Lilly & Co. Aisar Atrakchi is a full-time employee of the U.S. Food and Drug Administration. The views and opinions expressed in this document are solely those of the authors and are not the official policy of the FDA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Davis-Bruno KL, Atrakchi A. A regulatory perspective on issues and approaches in characterizing human metabolites. Chem Res Toxicol. 2006;19:1561–1563.
- Atrakchi A. Interpretation and considerations on the safety evaluation of human drug metabolites. Chem Res Toxicol. 2009;22:1217–1220.
- The US FDA. Guidance for industry: safety testing of drug metabolites. 2016. [Cited 2017 May 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079266.pdf
- ICH M3(R2). Guidance for industry: nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. 2010. [Cited 2017 May 15]. Available from: http://www.fda.gov/downloads/drugs/guidances/ucm073246.pdf
- ICH M3(R2). Questions and answers (R2) on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. 2013. [Cited 2017 May 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292340.pdf2
- Anderson S, Knadler MP, Luffer-Atlas D. Overview of metabolite safety testing from an industry perspective. Bioanalysis. 2010;2:1249–1261.
- Baillie TA. Approaches to the assessment of stable and chemically reactive drug metabolites in early clinical trials. Chem Res Toxicol. 2009;22:263–266.
- Leclercq L, Cuyckens F, Mannens GSJ, et al. Which human metabolites have we MIST? Retrospective analysis, practical aspects, and perspectives for metabolite identification and quantification in pharmaceutical development. Chem Res Toxicol. 2009;22:280–293.
- Smith DA, Obach RS. Metabolites in safety testing (MIST): considerations of mechanisms of toxicity with dose, abundance, and duration of treatment. Chem Res Toxicol. 2009;22:267–279.
- Luffer-Atlas D. The early estimation of circulating drug metabolites in humans. Expert Opin Drug Metab Toxicol. 2012;8:985–997.
- Anderson S, Luffer-Atlas D, Knadler MP. Predicting circulating human metabolites: how good are we? Chem Res Toxicol. 2009;22:243–256.
- Loi C-M, Smith DA, Dalvie D. Which metabolites circulate? Drug Metab Dispos. 2013;41:933–951.
- Gao H, Deng S, Obach RS. A simple liquid chromatography-tandem mass spectrometry method to determine relative plasma exposures of drug metabolites across species for metabolite safety assessments. Drug Metab Dispos. 2010;38:2147–2156.
- Walker GS, Ryder TF, Sharma R, et al. Validation of isolated metabolites from drug metabolism studies as analytical standards by quantitative NMR. Drug Metab Dispos. 2011;39:433–440.
- Vishwanathan K, Babalola K, Wang J, et al. Obtaining exposures of metabolites in preclinical species through plasma pooling and quantitative NMR: addressing metabolites in safety testing (MIST) guidance without using radiolabeled compounds and chemically synthesized metabolite standards. Chem Res Toxicol. 2009;22:311–322.
- Yu CP, Chen CL, Gorycki FL, et al. A rapid method for quantitatively estimating metabolites in human plasma in the absence of synthetic standards using a combination of liquid chromatography/mass spectrometry and radiometric detection. Rapid Commun Mass Spectrom. 2007;21:497–502.
- Yi P, Luffer-Atlas D. A radiocalibration method with pseudo internal standard to estimate circulating metabolite concentrations. Bioanalysis. 2010;2:1195–1210.
- Edarbi (azilsartan kamedoxomil) drug label. [Cited 2017 May 15]. Available from: https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=52b27c75-9f5a-4816-bafd-dace9d7d2063&type=display#section-12
- Sharma R, Litchfield J, Bergman A, et al. Comparison of the circulating metabolite profile of PF-04991532, a hepatoselective glucokinase activator, across preclinical species and humans: potential implications in metabolites in safety testing assessment. Drug Metab Dispos. 2015;43:190–198.
- Gao H, Jacobs A, White RE, et al. Safety assessment of human metabolites: what’s really necessary to ascertain exposure coverage in safety tests? AAPS J. 2013;15:970–973.