
ABSTRACT
Introduction
The increasing incidence of mental illnesses and neurodegenerative diseases results in a high demand for drugs targeting the central nervous system (CNS). These drugs easily reach the CNS, have a high affinity for CNS targets, and are prone to cause seizures as an adverse drug reaction. Current seizure liability assessment heavily depends on in vivo or ex vivo animal models and is therefore ethically debated, labor intensive, expensive, and not always predictive for human risk.
Areas covered
The demand for CNS drugs urges the development of alternative safety assessment strategies. Yet, the complexity of the CNS hampers reliable detection of compound-induced seizures. This review provides an overview of the requirements of in vitro seizure liability assays and highlights recent advances, including micro-electrode array (MEA) recordings using rodent and human cell models.
Expert opinion
Successful and cost-effective replacement of in vivo and ex vivo models for seizure liability screening can reduce animal use for drug development, while increasing the predictive value of the assays, particularly if human cell models are used. However, these novel test strategies require further validation and standardization as well as additional refinements to better mimic the human in vivo situation and increase their predictive value.
1. Introduction
Mental illnesses such as depression, anxiety, bipolar disorders, or schizophrenia strongly affect the quality of life. Estimates from the World Health Organization indicate that mental disorders affect one in four people, placing these disorders amongst the leading causes of ill-health worldwide [Citation1]. Other disorders that affect the brain include neurodegenerative diseases like Parkinson, Huntington’s or Alzheimer’s diseases. The prevalence of these diseases is increasing, partially due to the extension of human lifespan. Currently, Alzheimer’s disease is the most prevalent neurodegenerative disease, with an estimated 47 million patients worldwide [Citation2], but the overall number of patients suffering from mental illnesses and neurodegenerative disorders is much larger. These patients often require medication to keep their condition stable and to reduce the burden of disease. Hence, the increasing number of patients suffering from such diseases, combined with the high burden of disease, results in an increasing demand for drugs that target the central nervous system (CNS).
However, drug research and development is a very costly process that takes around 15 years [Citation3]. Out of over 1000 compounds, 250 make it to the preclinical studies of which five enter clinical trials, potentially resulting in only one approved drug. Before drugs can enter the market, they need to meet safety requirements set by regulatory authorities, such as the European Medicine Agency (EMA) for Europe and the US Food and Drug Administration (FDA) for the United States of America. These regulatory authorities rely heavily on animal studies in their requirements. One of the legal requirements for all CNS drugs is that they must pass neurotoxicity studies [Citation4], in which the potential of the drug to harm the nervous system or interfere with its function is investigated. Potential functional impairments such as learning and memory deficiency and behavioral changes are assessed using in vivo experiments with associated neuropathological endpoints. In addition to these neurotoxicity studies, potential new drugs must also undergo seizure liability assessment.
Drug-induced seizures and convulsions are the most frequently encountered CNS-related adverse drug reaction during pre-clinical development, followed by gait abnormalities, tremors emesis, and sedation [Citation5]. Seizures are life-threatening events in which there is a temporary dysfunction of the brain characterized by periods of excessive synchronous neuronal discharge [Citation6]. During a seizure, neurons fire abnormally and in a hyper-synchronized manner, exhibiting uncontrolled hyper-excitability [Citation7,Citation8]. Especially CNS drugs are likely to cause seizures as these drugs easily reach the brain and have a high affinity for nervous system targets [Citation9].
Despite the frequent occurrence and severity of drug-induced seizures, there are no official test guidelines. Current seizure liability assessment largely relies on low-throughput in vivo experiments [Citation7] or ex vivo hippocampal brain slice recordings [Citation5]. These assessments are often carried out late in the pre-clinical drug discovery process, making failure a very costly setback. Hence, there is a clear need for cost-effective alternative strategies that have increased throughput, are less dependent on animal experimentation and have sufficient predictive value.
This review will therefore provide a background on neuronal communication, seizures, and the neurotransmitter systems involved to evaluate the requirements of seizure liability assays. Additionally, this review highlights the major achievements obtained with current state of the art cellular imaging and electrophysiological techniques using rodent primary cortical cultures, their major caveats, and recommendations for future in vitro seizure liability assessment, including the use of human-induced pluripotent stem cells (hiPSCs).
2. Neuronal function and seizures
2.1. Healthy neuronal function
The CNS consists of millions of neurons and supporting cells that send, receive, and integrate signals from all over the body. While intercellular signaling largely relies on neurotransmitters, intracellular signaling strongly depends on action potentials. The generation and propagation of action potentials relies on depolarization and repolarization by voltage-dependent sodium channels (NaVs) and voltage-dependent potassium channels (KVs), respectively. At the presynaptic terminal, the action potential triggers the opening of voltage-gated calcium channels (VGCCs) to trigger the fusion of neurotransmitter-filled vesicles with the presynaptic membrane, thereby initiating chemical neurotransmission [Citation10,Citation11] (). Upon release and diffusion through the synaptic cleft, activation of postsynaptic neurotransmitter receptors can result in neuronal excitation or inhibition. The most abundant inhibitory neurotransmitter in the mammalian brain are γ-aminobutyric acid (GABA) [Citation12,Citation13] and glycine [Citation14], whereas glutamate is the main excitatory neurotransmitter in the CNS [Citation12]. Acetylcholine (ACh) is the main excitatory neurotransmitter in the peripheral nervous system, but like many other neurotransmitters it has a modulatory role in the CNS, resulting either in excitation or inhibition depending on the receptor (sub)types involved [Citation15].
Figure 1. Schematic representation of neuronal signaling. Integration of dendritic input results in generation of an action potential that travels via the axon to the presynaptic terminal, where the neuron makes contact with the postsynaptic neuron (left side of picture). Arrows depict travel direction of action potential. When an action potential reaches the presynaptic terminal, Ca2+ influx via voltage-gated calcium channels (VGCC) triggers the fusion of vesicles loaded with neurotransmitter with the cell membrane, thereby releasing neurotransmitter in the synaptic cleft (right side of picture, top part). Neurotransmitters can bind to ionotropic receptors that undergo a confirmation change upon binding allowing for the passage of ions through the channel (right bottom half, left receptor). Neurotransmitters can also bind to metabotropic receptors (right receptor). Upon binding, this receptor activates a G-protein complex that then activates an enzyme. This enzyme either activates a second messenger system that triggers cellular responses or opens an ion channel. Most of these processes can be subject to modulation by drugs, potentially resulting in seizure induction
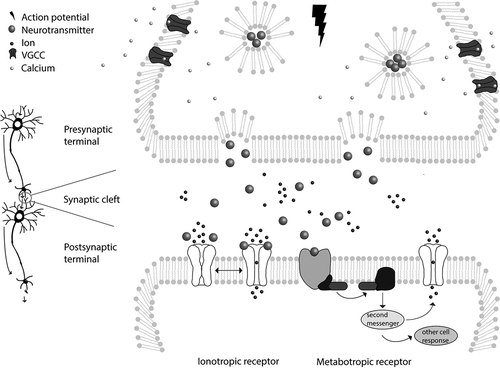
To maintain normal inter- and intracellular signaling, proper function of these ion channels and neurotransmitter receptors is vital. The balance between the diverse excitatory and inhibitory signals must be precisely regulated and failure to maintain this strict balance can result in severe neurological symptoms, including life-threatening seizures.
2.2. Physiology of seizures
At the most elementary basis, seizures can be seen as a result of a disruption in the delicate balance between neuronal excitation and inhibition [Citation16,Citation17]. Particularly the GABA and glutamate neurotransmitter systems play a major role in the development of seizures, although perturbations in the intracellular ion balance via other neurotransmitter receptors, ion channels, ion pumps and ion or neurotransmitter transporters can also increase neuronal excitability and subsequently increase seizure risk.
Changes in extracellular ion concentration can also affect excitability and seizure risk. As reviewed by Antonio et al. (2016) [Citation18], an increase in extracellular [K+] or decreases in extracellular [Ca2+], [Mg2+] or [Cl−] are all able to induce seizure-like events. A slight decrease in extracellular [Ca2+] can already affect the balance between the coupling of the excitatory and inhibitory systems. Also, a decrease in extracellular [Ca2+] affects the threshold for voltage-gated ion channels and facilitates the activation of the NMDA-R. Additionally, activation of this receptor is facilitated by a decrease in extracellular [Mg2+]. Lower extracellular [Mg2+] increases the amount of neurotransmitter that is released. A reduction in extracellular [Cl−] affects the transmembrane Cl− gradient and a strong reduction can ultimately induce seizurogenic activity. Increases in extracellular [K+] lead to neuronal depolarization and thus increased neurotransmitter release as well as prolongation of action potential duration and increased bursting behavior of neurons [Citation18].
Increased seizure risk can further result from astrocyte dysfunction. Astrocytes play an important role in homeostasis of extracellular ion concentrations [Citation19] and are involved in, for example, potassium clearance [Citation20]. Such alterations in extracellular ion levels can enhance neurotransmitter release from nearby neurons. Moreover, astrocytes are important glutamate scavengers. Activation of astrocytes by seizurogenic compounds can interfere with extracellular glutamate homeostasis and may even result in additional glutamate release from astrocytes [Citation19], resulting in (further) activation of the excitatory system. Because astrocytes contain glutamine synthetase, they play an important role in the GABA – glutamate – glutamine cycle (for review see [Citation21–23]). Low activity of astrocytic glutamine synthetase results in downregulation of this cycle and an astrocytic metabolic shutdown. This in turn results in GABA depletion due to reduced GABA-recycling. It also creates low levels of tissue glutamine [Citation24]. A deficiency in glutamine synthetase is thus an important risk factor for the development of seizures [Citation25].
The increased excitability and the excessive discharge of (excitatory) neurotransmitters in itself is not sufficient to induce seizures. As a seizure is characterized by hyper-synchronized firing of the neurons involved, neuronal network synchronization is an essential step in the development of seizures [Citation7,Citation8]. Seizurogenic compounds typically create a paroxysmal depolarizing shift: a network-driven burst that enhances synchronization [Citation26,Citation27]. Another way in which networks can be synchronized is via gap-junctions. They create a current flow from one cell to another [Citation17]. Via this gap-junction current, neurons can synchronize rapidly, thereby facilitating the onset of seizures.
Summarizing, there are numerous factors and targets that affect seizure risk, including changes in extracellular ion concentrations and modulation of neurotransmitter and ion channel function (see also ). Major risk factors include increased excitation and reduced inhibition. Increased excitation is caused mainly by agonists for the NMDA-R, AMPA-R, kainate-R and nACh-R. A decrease in inhibition of the neuronal network is primarily caused by antagonists of GABA and/or glycine receptors. Compounds that can either activate NaVs or inhibit KVs can also affect seizure risk by increasing neuronal firing. A selection of frequently used known seizurogenic compounds with their main mode of action is listed in .
Table 1. Overview of a selection of known seizurogenic compounds with their targets and potential to cause seizures in rodents and/or humans. Adapted from [Citation28]
3. Traditional assays for seizure liability assessment
Besides in vivo experiments, the ex vivo rat hippocampal brain slice assay is the most frequently used technique for seizure liability testing [Citation5]. This assay accurately mimics the in vivo organization of the brain with active and intact networks and different cell types [Citation52]. However, the life span of ex vivo brain slices is relatively short [Citation53]. Importantly, the brain slices are still of animal origin and recordings require specific expertise and equipment, and are labor intensive, thereby limiting high-throughput screening [Citation52].
Some of the concerns associated with the ex vivo rat hippocampal brain slice assay can be overcome by
using organotypic slice assays. These slices are usually derived from neonatal rodents and can be cultured in vitro for weeks [Citation54]. Although there are concerns that this model may not resemble the adult brain, the organotypic slices resemble the in vivo structure containing most neuronal subtypes present in the brain and thereby retaining intrinsic properties of brain tissue. Due to the long culture duration, recovery from preparation insults is possible and necrotic cells and debris disappear over time [Citation54].
However, both in vivo experiments and ex vivo and organotypic hippocampal brain slice assays are costly, time-consuming, ethically debated and relying on specialized expertise. Alternative assays are therefore urgently needed. Since seizures disrupt nervous system physiology, often in the absence of morphological changes, seizure liability tests should have a temporal resolution that is sufficient to detect action potentials. Consequently, seizure liability tests should be designed to assess changes in ion channel or neurotransmitter receptor function, intracellular calcium changes or network responses with (sub)millisecond resolution.
Patch-clamp techniques are accurate and efficient for measuring ionic currents across the membrane [Citation55] and can thus provide in-depth information regarding ion channel and neurotransmitter receptor function. However, these techniques are technically demanding and the procedure perturbs cell physiology. Moreover, patch-clamp techniques are poorly amenable for high-throughput screens, despite the development of automated patch-clamp recordings [Citation56,Citation57], as the numerous ion channels and receptors potentially involved in seizure liability would all have to be investigated separately.
Preferably, assays to assess seizure liability are minimally invasive and based on an integrated readout that reflects the nett effect of the numerous targets on cellular or network excitability, for a review on different assays see [Citation52]. Fluorescent imaging techniques that monitor changes in intracellular calcium levels or membrane potential with high temporal resolution are less invasive than patch-clamp techniques. These techniques enable visualization of free intracellular Ca2+ and membrane potential, respectively. As the intracellular Ca2+ concentration and the membrane potential reflect the nett effect of changes in underlying effects on ion channels and neurotransmitter receptors, these techniques can be used to detect electrical and oscillatory activity as well as network activity and synchronicity [Citation58,Citation59]. Changes in these parameters following exposure to test compounds can be used to assess seizure liability. However, the throughput of such imaging techniques is hampered by the need for a high temporal resolution, largely excluding plate-reader-based imaging approaches [Citation60,Citation61].
4. Micro-electrode array (MEA) recordings to assess seizurogenicity in vitro
Micro-electrode array (MEA) measurements are noninvasive, and changes in activity following exposure can be measured in real-time. MEAs allow for simultaneous extracellular recordings of local field potentials from multiple locations within an in vitro neuronal network [Citation62]. MEAs consist of a surface area with an integrated array of micro-electrodes on top of which neuronal cells can be cultured (, left). MEA measurements are noninvasive and provide a broad range of data on parameters that describe the state of the neuronal network. Changes in these parameters following drug exposure can be measured in real-time with millisecond temporal resolution. MEA recordings thereby reflect the physiologically relevant effects on the full network. For example, a drug-induced increase in the number of action potentials, thus an increase in activity, can be seen as an increased spike frequency. Increased activity can also be reflected in increased bursting, whereas network synchronicity is reflected in an increased network burst activity (, right). An overview of different metric parameters that can be derived from MEA measurements and that are important for in vitro seizure liability assessment can be found in .
Figure 2. MEA plates have an electrode grid on the bottom on top of which (neuronal) cells can be cultured (left) for noninvasive recordings of electrical activity. Recorded activity can be depicted in a raster plot (right) that illustrates the major MEA metric parameters. The example raster plot depicts the activity of a human iPSC-derived neuronal co-culture at 16 electrodes (horizontal lines) in a single well, where each tick mark (red circle) depicts one spike in a ~ 100 s recording window. An example of a burst is encircled in green and network burst in orange. Burst duration and network burst duration are depicted with a green and orange arrow, respectively, whereas an inter-burst-interval (IBI) is marked with a purple arrow. The cumulative trace above the raster plots indicates the synchronized activity between the different electrodes. The blue circle thus represents the level of synchronicity
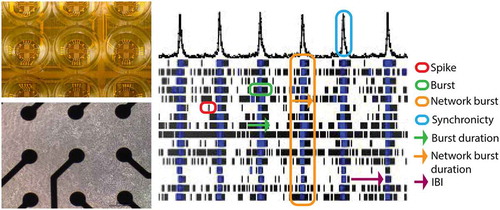
Table 2. Different metric parameters obtained from MEA measurements. Adapted from [Citation77]
While capturing and defining an in vitro seizure is complex due to the number of cellular mechanisms that can be involved, certain changes in in vitro neuronal activity can be indicative of in vivo seizures. In particular changes in parameters related to activity, organization of spikes in (network) bursts and synchronicity can be used to detect a hyperactive and/or hyper-synchronized state of the network, which is an indication for seizure-like events in the in vivo situation [Citation40,Citation51]. Examples of key parameters for the detection of seizurogenicity are spike rate (network) burst rate, percentage of spikes incorporated in (network) bursts, burst duration and synchronicity parameters; see also .
4.1. MEA recordings using rat primary cortical cultures
Ideally, an in vitro model captures the in vivo situation as closely as possible with sufficient complexity necessary to answer the research question, without overcomplicating the model [Citation63]. For seizure liability assessment, this translates to a model that forms functional networks composed of various cell types representative for the brain region(s) involved in seizures and with a physiology that mimics the in vivo brain. In such a model, known seizurogenic compounds such as picrotoxin (PTX), 4-aminopyridine (4-AP) and strychnine should induce seizure-like, synchronized activity, as hyper-synchronous bursts in the in vitro network are presumed to be the main trigger for seizures [Citation51].
Rodent primary cortical cultures contain most of the components of the in vivo cortex, including GABA-ergic and glutamatergic neurotransmitter systems as well as supportive astrocytes. Moreover, cortical neurons are involved in seizures [Citation64] and thus play a crucial role in in vitro seizure liability assessment. While many different cell types from different animals can be grown on MEAs, rat primary cortical cultures have become the current gold standard [Citation5]. For preparation of these cultures (see [Citation65]), cortices of, e.g., embryonic stage day 16–18 or postnatal day 0–1 rat pups are dissected and brought into single-cell suspension. Cells are then plated on top of an MEA electrode grid coated with, for example, PEI. Although the structure of the brain is lost during preparation of the culture, primary rat cortical cultures grown on MEAs possess many characteristics of in vivo neuronal networks, including development of spontaneous network activity, bursting, and network bursting [Citation66]. Spontaneous neuronal network activity develops within a week after plating and increases over time [Citation66–68]. Over time, primary rodent cortical neuronal networks become more organized, which is reflected in, for example, an increase in the percentage of spikes in a (network) burst. After 1–2 weeks in culture, networks start to exhibit (network) bursts [Citation66,Citation68]. Importantly, cortical cultures are responsive to various neurotransmitters [Citation69,Citation70], indicating the presence of a wide range of common neurotransmitter receptors. Furthermore, activity in these cultures can be modulated by diverse pharmacological and toxicological agents, including domoic acid, tetrodotoxin (TTX), methylmercury (MeHg), diazepam, and amphetamine [Citation69–73]. Notably, rat cortical cultures grown on MEAs show reproducibility across different laboratories [Citation74,Citation75] and provide high sensitivity and specificity in screening approaches [Citation70,Citation73].
Rat cortical cultures grown on MEAs have been successfully used for seizure liability assessment. Several studies have shown that these cultures can efficiently be used to detect seizures in vitro as increased spiking (network) bursting activity and/or synchronicity [Citation28, Citation37, Citation76, Citation77]. The GABA antagonists pentylenetetrazol (PTZ), PTX, bicuculline, gabazine, endosulfan and amoxapine all have been shown to increase neuronal network activity as reflected in increased spike, burst, and network burst rates as well as enhanced synchronicity [Citation28, Citation40, Citation67, Citation70, Citation71, Citation77, Citation78]. Just like PTX, pilocarpine is used to model epilepsy in vivo. Pilocarpine increases activity in rat primary cortical cultures grown on MEAs [Citation28,Citation77]. Rat cortical cultures grown on MEAs exposed to linopirdine also exhibit seizurogenic activity [Citation28,Citation40]. Additionally, seizure-like activity is observed following exposure of rat cortical cultures to strychnine [Citation79], although decreased activity has been reported as well [Citation40,Citation77], suggesting that the exposure concentration and duration are critical. This is also observed using glutamate as an endogenous excitatory stimulus. Low concentrations evoke a short-lived hyper-excitation, whereas high concentrations can completely inhibit neuronal activity [Citation69].
4.2. MEA recordings using human-induced pluripotent stem cell-derived neuronal cultures
Despite the major achievements obtained with assessing seizure liability using primary rat cortical cultures, interspecies differences may hamper translation to the human situation. Humans and rodents have very different brain structures [Citation80], with, for example, far less developed prefrontal and temporal cortices in rodents compared to human [Citation81]. Moreover, neurons from different species, even closely related ones, exhibit different electrophysiological behavior [Citation82]. The forthcoming problem of interspecies translation is reflected in the fact that despite all drug development guidelines and recommendations, potential drug candidates frequently fail due to CNS-related adverse drug reactions that are not detected in preclinical studies [Citation83]. On top of that, 10% of the drug attrition rate is due to CNS-related adverse drug reactions and safety concerns [Citation84]. Such concerns regarding the predictive value and utility of in vivo and in vitro animal models as well as associated animal welfare concerns have triggered the development of more predictive alternative test strategies based on human cells.
The discovery that pluripotent stem cells could be generated from adult fibroblasts through the addition of reprogramming factors [Citation85] changed the stem cell field. It led to the introduction of different cell types derived from human-induced pluripotent stem cells (hiPSCs) that are free from the legal and ethical issues surrounding the use of human embryonic stem cells (hESCs). Human iPSCs exhibit the same characteristics as hESCs, including self-renewal, a stable genetic background and the potential to differentiate into any other somatic cell type [Citation86]. Through a combination of growth factors and cell culture conditions, hiPSCs can be differentiated into neuroprogenitor cells (NPCs) [Citation81] that can give rise to dopaminergic [Citation87], glutamatergic [Citation88], cholinergic [Citation89], serotonergic [Citation90] and GABA-ergic [Citation91] neurons as well as supportive cells such as astrocytes [Citation92,Citation93] and microglia [Citation94].
Culturing hiPSC-derived neurons and astrocytes on MEAs, and knowing in advance when they exhibit mature activity is less straightforward than for rodent primary cortical cultures. There are some pitfalls for high-throughput neurotoxicity screening, including batch to batch variation [Citation95,Citation96] and reproducibility, as small changes in growth conditions can result in unwanted differentiation. Moreover, differentiation protocols to derive neurons and astrocytes from iPSCs can take up to months [Citation95,Citation97] making it a labor intensive and expensive process for researchers. Although efforts are made to reduce culture duration [Citation98], the recent introduction of commercially available hiPSC-derived neurons that are produced in large purity and quality-controlled batches may make these cells more attractive for efficient screening. Additionally, using commercially available hiPSC-derived neurons shortens the culture duration significantly. On top of that, researchers do not necessarily have to check for the ratio of neurons to astrocytes as is essential when cells are grown and differentiated from scratch by the researcher. In the case of commercially available cells, the supplier provides a certificate of analysis that can be used for determining how to mix the different cell types in the desired ratio. Some suppliers even deliver their cells pre-mixed. Exactly when the cultures start to exhibit spontaneous network activity on the MEA depends on the protocol used to derive the cells, or in the case of commercially available cells, on the stage of maturation at time of freezing the cells. Commercially available cells generally reach sufficient maturity for toxicity testing within 2–4 weeks of culture [Citation79].
Cultures from hiPSC-derived neurons have been shown to form complex neuronal networks with excitatory and inhibitory neurons. These cultures contain different types of neurons, such as GABAergic and glutamatergic neurons, but no supporting cells unless explicitly added. When grown on MEAs, they develop spontaneous neuronal activity following days or weeks in culture. While the onset and level of activity depend on the type of iPSC-derived neurons used [Citation79,Citation99–101], the level of activity is low compared to rat primary cortical cultures and (network) bursting behavior is limited [Citation63,Citation102,Citation103]. Despite the lower level of activity [Citation104], human cultures behave comparable to rat primary cortical cultures following pharmacological and physiological modulation, indicating their potential for neurotoxicity testing [Citation63].
Importantly, addition of astrocytes to neuronal cultures enhances synaptic maturation and transmission [Citation105] and increases firing frequency and bursting behavior [Citation59,Citation102,Citation106,Citation107]. In contrast to pure neuronal cultures, the co-cultures containing astrocytes also develop pronounced network bursting behavior. These co-cultures with astrocytes exhibit a pattern of development that is comparable to rat primary cortical cultures with increased spiking and (network) bursting activity over time and a higher number of spikes occurring in (network) bursts. Notably, it appears that the co-culture model with the highest percentage of astrocytes is also the model with highest level of synchronization [Citation79]. Astrocytes may also influence the sensitivity of cultures toward toxicological insults [Citation108,Citation109]. This is especially important when it comes to excitotoxicity, an event that is associated with the development of seizures.
Co-culture models described in literature differ in seeding density and ratio of neurons to astrocytes. It appears that models with higher seeding densities exhibit higher network activity [Citation79]. Also, the ratio of excitatory glutamatergic to inhibitory GABAergic neurons can greatly influence behavior of neuronal cultures. Networks with a higher ratio of glutamatergic to GABAergic neurons display higher network activity [Citation110]. The fraction of inhibitory neurons in the network and/or the degree of excitability present in the network may also be critical for the chemosensitivity as it appears that a co-culture with high neuronal activity is more sensitive toward neurotoxicants that decrease neuronal activity [Citation109]. Additional factors that may influence chemical sensitivity of neuronal networks include the degree of neuronal differentiation and network maturation as well as network complexity (e.g., the number of contacts that the different cell types establish). Considering that different hiPSC-derived neuronal models can also differ in receptor expression, it becomes clear that spontaneous activity and drug responses of different neuronal co-culture models are dictated by a complex interplay of a large number of model-specific properties. Thorough knowledge of the composition and characteristics of the model system is therefore essential to understand potential differences in chemical sensitivity and to interpret the data with respect to human seizure risk.
Several studies indicate that hiPSC-derived models have potential for in vitro seizure liability assessment because exposure to seizurogenic compounds, such as PTX, 4-AP and strychnine, affects activity and synchronicity [Citation79,Citation101,Citation111]. Importantly, hiPSC-derived neuronal co-cultures detect seizures at the same or even a lower level than rat primary cortical cultures [Citation77]. In a comparable study, an hiPSC-derived neuronal co-culture model was exposed to nine additional known seizurogenic (PTZ, amoxapine, enoxacin, amoxicillin, pilocarpine, linopirdine, and CPZ) and non-seizurogenic compounds (phenytoin and acetaminophen) and the effects on neuronal activity were compared to rat primary cortical cultures. Overall, both models react upon exposure to seizurogenic compounds, although with different profiles. While the hiPSC-derived neuronal co-culture responds to all tested compounds, the rat primary cortical culture is not affected by amoxicillin. Sensitivities are comparable for amoxapine, linopirdine, and pilocarpine. However, another study comparing hiPSC-derived neuronal co-cultures and rodent primary cortical cultures reported excitability at lower concentrations on the human model following exposure to linopirdine and pilocarpine as compared to the rodent model [Citation111]. The hiPSC-derived neuronal co-culture however is more sensitive for exposure to PTZ, PTX, strychnine, enoxacin, 4-AP and CPZ. Remarkably, in hiPSC-derived neuronal co-cultures (network) burst parameters are more sensitive for excitatory changes, whereas spike parameters are more affected in rodent cultures [Citation77]. The importance of the addition of astrocytes to the hiPSC-derived neuronal culture became evident by the finding that epileptiform activity was detected following exposure to the convulsant drugs gabazine and kaliotoxin in co-cultures but not in mono-cultures [Citation59]. However, the presence of astrocytes may not always be required to detect seizurogenic activity as exposure of mono- and co-cultures to PTZ and 4-AP led to an increase in activity in both models, although the effect was stronger in co-cultures [Citation101]. Exposure to the negative control, acetaminophen, did not affect network behavior [Citation77]. Exposure to the anti-epileptic compound phenytoin increased spiking and bursting activity at low concentrations, but at higher test concentrations (>10 µM) activity was not affected [Citation77]. Phenytoin thus exhibits a bi-phasic effect. However, these experiments were performed in a healthy neuronal network without seizure activity. To mimic the in vivo situation more closely, to reveal the anti-epileptic properties of phenytoin and to enhance translatability, co-exposures should be performed in which seizures are evoked with, for example, the seizurogenic agent PTZ or other conditions that increase excitability (see section 2.2) followed by an exposure to the anti-epileptic agent such as phenytoin. It was previously shown that seizures induced in in vitro hiPSC-derived neuronal networks by PTZ could be suppressed by phenytoin. Remarkably, seizures induced by 4-AP could not be stopped by this agent [Citation101]. The effectivity of anti-epileptic drugs may thus depend on the type of seizure. On top of that, the biphasic effect of phenytoin indicates the importance of in vivo drug blood and serum concentrations for in vitro evaluation of the (anti-)seizurogenic properties of test compounds.
These combined studies indicate that hiPSC-derived neuronal co-cultures can already be used as a first screen in in vitro seizure liability assessment, although some differences exist between the human and rodent neuronal models as well as between the different human models.
5. Future directions and challenges in in vitro seizure liability assessment
5.1. Challenges in hiPSC-derived neuronal culturing
Generation and differentiation of hiPSCs is a relatively new technique [Citation85]. Many researchers and companies started to produce hiPSC-derived neurons, under slightly different conditions and with slightly different protocols. Those on first sight small differences, can have a substantial impact on the resulting batch of hiPSC-derived neurons, thereby potentially hampering reproducibility. Maintenance of hiPSCs requires technical skills since the undifferentiated state is instable and cells are sensitive for mechanical stress [Citation112]. Also, slight changes in culture and growth conditions can result in unwanted differentiation [Citation95], thereby creating batch to batch variation. A study in which the same iPSC line was tested across five different laboratories detected poor cross-site reproducibility and identified the laboratory as the largest source of variation [Citation113]. However, with advances in technology such as automated robots for culturing [Citation112] and culture media with growth factors that can maintain growth for multiple days reducing the number of handling moments may (partly) eliminate this concern. Besides enhancing reproducibility, automation has as additional benefit that it strongly reduces labor costs and intensity [Citation114].
In the case of commercially available hiPSC-derived neurons, researchers do not differentiate the cells themselves, saving valuable time. Also, these cells come in large, quality-controlled batches [Citation95], ensuring that researchers can perform a complete study on the same cells.
Commercial suppliers often provide cells in full packages with supplements and sometimes even complete culture medium. Though this strongly enhances the ease of use of commercially available hiPSC-derived neuronal cells, it hampers comparison between models. Comparison is further hampered by differences in manufacturer guidelines, including seeding density, culture duration and composition of the medium. For example, it has been shown that concentrations of ions and serum (or serum-replacement factors) in media can directly affect neuronal network activity [Citation115] and the effects of toxic insults on hiPSC-derived neurons [Citation116]. It is therefore difficult to elucidate the origin of potential different outcomes between (commercial) models.
5.2. Opportunities and improvements for hiPSC-derived neuronal models
Several studies compared hiPSC-derived neuronal data with rodent primary cortical data (e.g. [Citation75,Citation77]. However, human and rodent brain structures are very different [Citation80] and it is thus likely that the in vitro behavior of neuronal networks also differs between species. Thus, instead of optimizing the hiPSC-derived neuronal co-cultures in a way that they resemble rat primary cortical cultures, other points of optimization might be of more importance, such as comparison of human in vitro data with human in vivo data derived from, for example, pharmacovigilance reports.
Following differentiation, hiPSC-derived neurons can be cryopreserved and stored without losing functionality [Citation117,Citation118] or affecting the neuronal differentiation program [Citation119]. However, there is likely an optimal maturation stage at which neurons can be frozen for optimal recovery, as is the case for hiPSC-derived cardiomyocytes [Citation120]. Characteristics of thawed hiPSC-derived neurons are thus likely affected by the maturation stage at which they were frozen. While laborious, this also implies it is essential to thoroughly characterize the culture model prior to its implementation in neurotoxicity testing.
A cell model should mimic the in vivo situation as closely as possible, whilst keeping the model as simple and reproducible as possible [Citation61]. However, a frequently raised critique concerns the 2D environment of current models. Due to the absence of organized cell layers, 2D models lack the complexity and hierarchical structure, connectivity, and matrix environment found in 3D and the in vivo brain. Organoids and spheroids from hiPSC-derived neurons have been shown to capture early development and complexity of the human brain [Citation121–123]. However, it is still challenging to couple these cultures to MEAs, since electrodes are located at the bottom of the plate and do not penetrate 3D cultures. Moreover, every step taken to add complexity may increase the variability and cause problems with reproducibility, particularly with self-organizing organoids. Nevertheless, successful efforts have been made to model focal seizures in vitro in a microfluidic device integrated with MEA [Citation124]. In this device, three separate networks with synchronized bursting can be grown that make network-to-network contact via axonal connections through microtunnels. When one network is exposed to the convulsant kainate, the resulting seizure is local and does not spread to the other networks, thus allowing modeling of focal seizures [Citation124].
Human iPSCs can be differentiated in dopaminergic [Citation87], glutamatergic [Citation88], cholinergic [Citation125], serotonergic [Citation90] and GABAergic neurons [Citation91] as well as several types of supportive cells. By mixing these different types of neurons and supportive cells in the right ratios, specific brain regions can be modeled thereby potentially increasing the predictive value. In regards of seizure liability assessment, the neocortex, hippocampus, and amygdala would be of major interest [Citation126].
In the in vivo situation, drugs have to cross the blood-brain-barrier (BBB). Thus, to better assess the seizurogenic potential of compounds, addition of a BBB to the in vitro model can increase the predictive value. BBB models can be cultured with hiPSC-derived neurons and astrocytes [Citation127] and can even be incorporated on microfluidic chips [Citation128] allowing for direct assessment of the effect of an administered dose on the network. Similarly, coupling a BBB model to the MEA test system may be of great benefit for in vitro neurotoxicity assessment and safety pharmacology.
5.3. Challenges for risk and safety assessment using alternative strategies
Human iPSC-derived neuronal co-cultures can thus be used as a first screen in in vitro seizure liability assessment. Yet, in order to move toward animal-free neurotoxicity testing, the predictive value of results obtained with hiPSC-derived neuronal co-cultures in comparison to in vivo outcomes must be elucidated. However, in vitro exposures often do not match one to one with in vivo exposures. This is because in the in vitro situation, exposure is directly to the neuronal networks, in contrast to the (human) in vivo situation, where processes of absorption, distribution, metabolism, and excretion (ADME) are present and can alter the actual target concentration. Thus, an in vitro hit or threshold concentration is not necessarily a concentration that causes adverse drug reactions or health concerns in real life. To translate the laboratory (in vitro) setting to the real in vivo world, an in vitro to in vivo extrapolation (IVIVE) must be performed. Extrapolation of in vitro to in vivo results can be based on physiologically based pharmacokinetic (PBPK) models [Citation129,Citation130]. For such models, values for parameters such as body weight, brain volume, brain to blood partition coefficient and liver to blood partition coefficient must be used as input [Citation131]. These values can be derived from either intoxication or pharmacovigilance case reports or, if no human data is available, from animal experiments.
Marketed drugs, such as pilocarpine and amoxapine, have gone through thorough testing during the drug development pipeline before they were allowed to enter the market. Therefore, a broad range of data must be available from pharmaceutical companies that could be used as input for PBPK models. The outcome of such PBPK models can provide information about the predictive value of hiPSC-derived models for seizure liability and (general) neurotoxicity. Unfortunately, until today these data are not easily accessible for researchers, needlessly hampering validation of hiPSC-models. This also hampers replacement of in vivo studies with in vitro alternatives. Future efforts should therefore focus on making these data from approved drugs publicly (or at least for research purposes) available as both academia and industry would benefit from such a cooperative environment.
The challenge on the in vitro side for PBPK modeling and risk assessment in general is to decide which parameter of the MEA-analysis to include. MEA-recordings yield a wealth of data on many metric parameters, although there is still no consensus on which MEA parameter is most predictive. Moreover, modeling approaches frequently rely on in vitro EC50 values [Citation131,Citation132]. However, with seizurogenicity data often an increase in activity is followed by a decrease, probably as a result of over-excitation of the network. The shape of the resulting curve hampers proper EC50 calculations and use in PBPK models. Instead, a threshold approach such as LOEC or NOEC could be used. A challenging factor here is that NOEC, LOEC and EC50 values depend on the MEA parameter chosen. One approach could be to use the most sensitive parameter and to decide this on a compound to compound basis.
Human iPSC-derived neurons are often clonally derived [Citation133,Citation134]. Cells from one batch come from one donor and thus display low genetic variation and will likely respond in a similar way to chemical exposures. Whereas iPSC-derived neurons obtained from one donor provide useful information, the genotype of the donor is of paramount importance for interpretation of the results. It is unlikely that the genotype of this donor is representative for the general population, thus raising questions with regards to risk assessment. Consequently, it could be argued that for proper assessment of seizure risk, a pooled sample of hiPSC-derived neurons from multiple donors must be used. A downside of pooling could be that specific effects are leveled out as a part of the sample population could be extremely sensitive and another part could be relatively insensitive.
Recent insights on sex-specific differences in drug efficacy and adverse drug reactions [Citation135], indicating that women suffer more adverse drug reactions than men [Citation136], argue in favor of a test strategy with male and female hiPSC-derived neurons tested separately.
Human iPSC-derived neurons from different disease backgrounds can also help provide clues about differences in vulnerability toward certain drugs. Patient-derived hiPSC-derived neurons have already been successfully cultured from patients suffering from neurodegenerative diseases, such as Huntington’s [Citation137], Alzheimer’s [Citation138] or Parkinson’s disease [Citation139]. Pharmaceutical companies could integrate these hiPSC-derived disease models in their test battery to identify promising novel treatments that alleviate or reverse disease symptoms [Citation95,Citation140].
Additionally, the use of patient-derived hiPSC-derived neurons paves the road toward personalized medicine and safety testing. Implementation of these neurons in healthcare could significantly reduce adverse drug reactions because susceptibility and vulnerability of the patient may be established on forehand. Tests can be performed to deduct optimal dosing and to test for cross-reactions. Although experiments with such patient-derived hiPSC-derived neurons will be very labor intensive and time-consuming with the currently available technology, it holds great promise for the future and paves the way to disease-specific neurotoxicity and safety testing.
6. Conclusion
Recent efforts have demonstrated that hiPSC-derived neuronal co-cultures are a promising alternative for rodent primary cortical cultures. However, before these models can be incorporated in seizure liability risk assessment strategies, a thorough characterization of the (new) culture model must take place. When performing in vitro seizure liability assessment assays, for example, using MEA, it must be kept in mind that astrocytes greatly influence the level and pattern of neuronal activity and sensitivity toward a toxicant. This sensitivity also depends on the ratio of GABAergic to glutamatergic neurons present in the model. Therefore, the characterization starts with immunocytochemistry to confirm the presence of different types of neurons and supportive cells. Preferably, the ratio between neurons and astrocytes is established as well as the ratio of the different neurons (i.e. inhibitory to excitatory). Immunocytochemistry also allows for visualization of the development of the in vitro network structure. After establishing that networks are grown and mimicking in vitro structures, MEA recordings can confirm development of spontaneous network activity and these recordings can then be used to determine the optimal window for drug exposure. Presence of relevant neurotransmitter receptor pathways can be confirmed with MEA recordings, whereas presence of specific functional neurotransmitter pathways can be assessed separately using targeted approaches such as single-cell calcium imaging. Following this minimal required level of characterization, hiPSC-derived neuronal co-cultures can be used as an initial screen for in vitro seizure liability assessment. Several studies have shown that hiPSC-derived neuronal models predict seizure liability just as well or even better than primary rodent cortical cultures. Therefore, these models may contribute to the reduction of the number of test animals needed and may in the future be able to help replacing animal models, while moving toward personalized safety assessment.
7. Expert opinion
Human iPSC-derived neuronal co-cultures grown on MEAs hold great potential for in vitro seizure liability assessment. These co-cultures show increased activity following exposure to known seizurogenic compounds, whereas no seizurogenic activity is detected following exposure to non-seizurogenic compounds. Because these human-based models perform just as well or even better than the currently used rat primary cortical culture, hiPSC-derived neuronal co-cultures can be incorporated in the drug developmental pipeline for neurotoxicity screening. Incorporation of these models has as benefit that it reduces the amount of test animals needed in several ways. First, by eliminating the need for use of rodent primary cortical cultures as an in vitro test strategy. Further reduction of test animals is ensured by the fact that only compounds that do not induce seizures in human models are selected for further study. As became clear that some compounds do not exhibit seizurogenic activity in rodent models, but do so in human models, the number of potential drug candidates for subsequent testing is further reduced. Another benefit of this strategy is that pharmaceutical companies will save money on drug development in the long run, since adverse drug reactions that are normally only detected during clinical trials in humans, when a lot of money has been invested, might now be filtered out in the in vitro test.
However, before these models can be incorporated in the drug development pipeline, the test set of chemicals must be expanded for model validation. So far, only a limited set of compounds, mainly GABAA receptor antagonists, has been used. In order to understand the predictive value of hiPSC-derived neuronal co-cultures, many known seizurogenic and non- or anti-seizurogenic compounds with different modes of actions must be tested. This will give a good overview of the rate of true positive and negatives as well as the false positives and negatives. As is the case with any model, including animal models, hiPSC-derived neuronal co-cultures will give false positive and false negative outcomes. Consequently, some promising drug candidates will not go to the next stage in drug development due to a false-positive seizure-risk outcome of the iPSC model, resulting in a missed opportunity. On the other hand, compounds falsely detected as negative will move to the next round of testing where they at a later stage might show seizure liability. At this stage, hiPSC-derived neuronal models may not yet be able to fully replace animal models for seizure liability testing. However, they can already be used as a first screening tool. In order to move forward, the false positive and false negative rates of rodent in vivo models and hiPSC-derived neuronal co-cultures should be compared. An additional challenge is that false positive and false negatives are now often based on comparison with earlier in vivo animal experiments. However, these in vivo results itself might be false positive or negative. Although hampered by the scarce availability of human data, it would be best to compare results obtained with hiPSC-derived neuronal models to in vivo findings in humans, for example, derived from clinical observations and pharmacovigilance reports.
An extensive test library also allows for a thorough comparison with data obtained from rat primary cortical cultures to determine the differences between these two types of models. This extensive set of compounds should be tested on the same hiPSC-derived model across different laboratories to ensure reproducibility of test results. Batch-to-batch variation is of concern; test should therefore preferably be performed on the same cell batch and differences between batches should be monitored.
Another concern results from the increasing amount of hiPSC-derived neurons that are (commercially) available. It is of uttermost importance that a thorough characterization and standardization takes place. Besides the fact that this gives a clear idea of targets present in the model, it will abet regulatory authorities to include these models in their regulatory framework.
Although the great variety of available cells allows for mimicking different brain regions, it also hampers reproducibility of results. Minor changes in culture conditions, media composition, or cell ratios greatly influence experimental outcomes. Therefore, we would argue that a centralized database detailing the culture protocol, cell types and ratios used in which experiments would be greatly beneficial for the progress of the field. This will also expedite acceptance of these models in risk assessment strategies as authorities can specify which model types are required for which types of study.
Research in the field of seizure liability using hiPSC-derived neuronal co-cultures, and hiPSC-derived co-cultures in general holds great potential for the future. Co-cultures can be further optimized to better mimic different brain regions in order to model specific diseases.
Acceptance of the model(s) in the regulatory framework is a main goal. However, upon regulatory acceptance the model(s) can still develop further to enhance the predictive value and specificity. Also, the model(s) could be coupled to an in vitro blood-brain-barrier to help determine whether a compound could reach the brain. If this coupling works, a further reduction of compounds that need to be tested in vivo can be realized since compounds that are unable to cross the BBB are unlikely to pose a seizure risk.
Although full acceptance and incorporation of hiPSC-derived neuronal co-cultures will require time, it is a realistic expectation. In the near future, models mimicking specific brain regions can be developed and added to a database, allowing for enhanced reproducibility. Recommendations for models connected to different adverse outcomes may further enable incorporation of hiPSC-derived neurons in drug developmental pipelines.
Article highlights
Current seizure liability assessment still heavily depends on in vivo or ex vivo animal models, although there is an urgent need for novel in vitro test strategies that are cost-effective and predictive for the human situation.
Characteristics of the in vitro cell model should closely resemble in vivo brain conditions, including the ratio of glutamatergic to GABAergic neurons as well as the presence of astrocytes, which play a critical role in (network) bursting behavior and synchronicity.
Human iPSC-derived neuronal co-cultures grown on micro-electrode arrays (MEAs) can be used as a first screening tool for in vitro neurotoxicity testing and seizure liability assessment.
Thorough characterization of new hiPSC-derived neuronal models is critical before incorporating these models in in vitro seizure liability assessment strategies.
Human iPSC-derived neuronal co-cultures grown on MEAs perform comparable or, in some cases, better than rodent primary cortical cultures in in vitro seizure liability assessment and offer great opportunities for improvement of drug safety and risk assessment.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- World Health Organization. WHO | mental disorders affect one in four people. WHO; 2013. Available at: https://www.who.int/news/item/28-09-2001-the-world-health-report-2001-mental-disorders-affect-one-in-four-people [Accessed October 22, 2019].
- Prince M, Wimo A, Guerchet M, et al. World alzheimer report 2015: the global impact of dementia - an analysis of prevalence, incidence, cost and trends. Alzheimer’s Dis Int. 2015;84. DOI:10.1111/j.0963-7214.2004.00293.x..
- Roses AD. Pharmacogenetics in drug discovery and development: a translational perspective. Nat Rev Drug Discov. 2008;7(10):807–817.
- ICH. ICH Topic S 7 A safety pharmacology studies for human pharmaceuticals step 5 note for guidance on safety pharmacology studies for human pharmaceuticals date for coming into operation. 2001. Available at http://www.emea.eu.int [Accessed October 23, 2019.
- Authier S, Arezzo J, Delatte MS, et al. Safety pharmacology investigations on the nervous system: an industry survey. J Pharmacol Toxicol Methods. 2016;81:37–46.
- Gotman J. A few thoughts on “what is a seizure?”. Epilepsy Behav. 2011;22(SUPPL. 1):S2.
- Easter A, Bell ME, Damewood JR, et al. Approaches to seizure risk assessment in preclinical drug discovery. Drug Discov Today. 2009;14(17–18):876–884.
- Jiruska P, de Curtis M, Jefferys JGR, et al. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. 2013;591(4):787–797.
- Gao Z, Chen Y, Cai X, et al. Predict drug permeability to blood–brain-barrier from clinical phenotypes: drug side effects and drug indications. Bioinformatics. 2016;33(6):713.
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59(6):861–872.
- Pang ZP, Südhof TC. Cell biology of ca2+-triggered exocytosis. Curr Opinion Cell Biol. 2010 August;22(4):496–505. . Curr Opin Cell Biol.
- Petroff OAC. GABA and glutamate in the human brain. Neuroscientist. 2002;8(6):562–573.
- D’Hulst C, Atack JR, Kooy RF. The complexity of the gabaa receptor shapes unique pharmacological profiles. Drug Discov Today. 2009;14(17–18):866–875. .
- Cleland JC, Griggs RC. Channelopathies of the nervous system. Neurobiol Disease. 2007:pp 319–332. Elsevier. DOI:10.1016/B978-012088592-3/50033-5..
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76(1):116–129.
- Lerche H, Jurkat-Rott K, Lehmann-Horn F. Ion channels and epilepsy. Am J Med Genet - Semin Med Genet. 2001;106(2):146–159.
- Scharfman HE. The neurobiology of epilepsy. Curr Neurol Neurosci Rep. 2007 July;7(4):pp 348–354. .
- Antonio LL, Anderson ML, Angamo EA, et al. In vitro seizure like events and changes in ionic concentration. J Neurosci Methods. 2016;260:33–44.
- Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures? Elsevier Ltd Trend Molecul Med. 2005;1112:pp 530–533. .
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36(4–5):291–300.
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. GLIA. 2012 August;60(8):1215–1226. . Glia.
- Schousboe A. Metabolic signaling in the brain and the role of astrocytes in control of glutamate and GABA neurotransmission. Neuroscience letters. Elsevier Ireland Ltd:2019 Jan 10 11–13. DOI:10.1016/j.neulet.2018.01.038..
- Walls AB, Waagepetersen HS, Bak LK, et al. The glutamine–glutamate/ GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism In: Springer New York LLC: Neurochemical research; 2014. p. 402–409. DOI:10.1007/s11064-014-1473-1..
- Eid T, Behar K, Dhaher R, et al. Roles of glutamine synthetase inhibition in epilepsy. Neurochem Res. 2012 November;37(11):2339–2350. . Neurochem Res.
- Chan F, Lax NZ, Voss CM, et al. The role of astrocytes in seizure generation: insights from a novel in vitro seizure model based on mitochondrial dysfunction. Brain. 2019;142(2):391–411.
- Johnston D, Brown TH. The synaptic nature of the paroxysmal depolarizing shift in hippocampal neurons. Ann Neurol. 1984;16(1 S):S65–S71.
- Kubista H, Boehm S, Hotka M. The paroxysmal depolarization shift: reconsidering its role in epilepsy, epileptogenesis and beyond. Int J Mol Sci. 2019 Feb 1;20(3):577. . MDPI AG.
- Tukker AM, Wijnolts FMJ, de Groot A, et al. Vitro techniques for assessing neurotoxicity using human IPSC-derived neuronal models. New York, NY: Humana, 2019:17–35. DOI:10.1007/978-1-4939-9228-7_2..
- Mackenzie L, Medvedev A, Hiscock JJ, et al. Picrotoxin-induced generalised convulsive seizure in rat: changes in regional distribution and frequency of the power of electroencephalogram rhythms. Clin Neurophysiol. 2002;113(4):586–596.
- Dhir A. Pentylenetetrazol (PTZ) kindling model of epilepsy. In: Current protocols in neuroscience. Vol. Chapter 9. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2012. p. Unit9.37. .
- Singh T, Yadav S. Role of micrornas in neurodegeneration induced by environmental neurotoxicants and aging. Ageing Res Rev. 2020;60:p 101068. Elsevier Ireland Ltd July 1. .
- Salazar P, Tapia R, Rogawski MA. Effects of neurosteroids on epileptiform activity induced by picrotoxin and 4-aminopyridine in the rat hippocampal slice. Epilepsy Res. 2003;55(1–2):71–82.
- Kumlien E, Lundberg PO. Seizure risk associated with neuroactive drugs: data from the WHO adverse drug reactions database. Seizure. 2010;19(2):69–73.
- De Sarro A, Zappalá M, Chimirri A, et al. Quinolones potentiate cefazolin-induced seizures in DBA/2 mice. Antimicrob Agents Chemother. 1993;37(7):1497–1503.
- Kawakami J, Yamamoto K, Asanuma A, et al. Inhibitory effect of new quinolones on GABAA receptor-mediated response and its potentiation with felbinac inxenopusoocytes injected with mouse-brain mrna: correlation with convulsive potencyin vivo. Toxicol Appl Pharmacol. 1997;145(2):246–254.
- Raposo J, Teotónio R, Bento C, et al. Amoxicillin, a potential epileptogenic drug. Epileptic Disord. 2016;18(4):454–457.
- Fan J, Thalody G, Kwagh J, et al. Assessing seizure liability using multi-electrode arrays (MEA). Toxicol Vitro. 2019;55:93–100.
- Alcoreza O, Tewari BP, Bouslog A, et al. Sulfasalazine decreases mouse cortical hyperexcitability. Epilepsia. 2019;60(7):1365–1377.
- Müller S, Guli X, Hey J, et al. Acute epileptiform activity induced by gabazine involves proteasomal rather than lysosomal degradation of KCa2.2 channels. Neurobiol Dis. 2018;112:79–84.
- Bradley JA, Strock CJ. Screening for neurotoxicity with microelectrode array. Curr Protoc Toxicol. 2019;79(1):e67.
- Park HR, Song P, Lee JJ, et al. Endosulfan-induced prolonged super-refractory status epilepticus. J Epilepsy Res. 2018;8(2):93–96.
- Alachkar A, Łażewska D, Latacz G, et al. Studies on anticonvulsant effects of novel Histamine H3R antagonists in electrically and chemically induced seizures in rats. Int J Mol Sci. 2018;19(11):3386.
- Boyd-Kimball D, Gonczy K, Lewis B, et al. Classics in chemical neuroscience: chlorpromazine. acschemneuro ACS Chem Neurosci. 2018;10(1):8b00258. .
- Marchi N, Oby E, Batra A, et al. In Vivo and In Vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48(10):1934–1946.
- Zimmerman TJ. 4. Pilocarpine. Ophthalmology. 1981;88(1):85–88.
- Li F, Liu L. Comparison of kainate-induced seizures, cognitive impairment and hippocampal damage in male and female mice. Life Sci. 2019. DOI:10.1016/j.lfs.2019.116621.
- Peña F, Tapia R. Seizures and neurodegeneration induced by 4-aminopyridine in rat hippocampus in Vivo: role of glutamate- and GABA-mediated neurotransmission and of ion channels. Neuroscience. 2000;101(3):547–561.
- Codadu NK, Graham RT, Burman RJ, et al. Divergent paths to seizure-like events. Physiol Rep. 2019;7(19). DOI:10.14814/phy2.14226.
- Maslarova A, Salar S, Lapilover E, et al. Increased susceptibility to acetylcholine in the entorhinal cortex of pilocarpine-treated rats involves alterations in KCNQ channels. Neurobiol Dis. 2013;56:14–24.
- Qiu C, Johnson BN, Tallent MK. K + M-Current regulates the transition to seizures in immature and adult hippocampus. Epilepsia. 2007;48(11):2047–2058.
- Ishii MN, Yamamoto K, Shoji M, et al. Human induced pluripotent stem cell (hipsc)-derived neurons respond to convulsant drugs when co-cultured with hipsc-derived astrocytes. Toxicology. 2017;389:130–138. .
- Grainger AI, King MC, Nagel DA, et al. In vitro models for seizure-liability testing using induced pluripotent stem cells. Front Neurosci. 2018;12:590.
- Buskila Y, Breen PP, Tapson J, et al. Extending the viability of acute brain slices. Sci Rep. 2015;4(1):5309.
- Lein PJ, Barnhart CD, Pessah IN. Acute hippocampal slice preparation and hippocampal slice cultures. Methods Mol Biol. 2011;758:115–134.
- Sakmann, B.; Neher, E. Patch Clamp Techniques for Studying Ionic Channels in Excitable Membranes. Annu. Rev. Physiol., 1984, 46 (1), 455–472. https://doi.org/10.1146/annurev.physiol.46.1.455.
- Annecchino LA, Schultz SR. Progress in automating patch clamp cellular physiology. Brain Neurosci Adv. 2018;2:239821281877656.
- Suk HJ, Boyden ES, van Welie I. Advances in the automation of whole-cell patch clamp technology. J Neurosci Methods. 2019 Oct 1;326:108357. Elsevier B.V .
- Smetters D, Majewska A, Yuste R. Detecting action potentials in neuronal populations with calcium imaging. Method A Compan Method Enzymol. 1999;18(2):215–221.
- Tukker AM, De Groot MWGDM, Wijnolts FMJ, et al. Is the time right for in vitro neurotoxicity testing using human IPSC-derived neurons? ALTEX. 2016;33(3):261–271.
- Heusinkveld HJ, Westerink RHS. Caveats and limitations of plate reader-based high-throughput kinetic measurements of intracellular calcium levels. Toxicol Appl Pharmacol. 2011;255(1):1–8.
- Meijer M, Hendriks HS, Heusinkveld HJ, et al. Comparison of plate reader-based methods with fluorescence microscopy for measurements of intracellular calcium levels for the assessment of in vitro neurotoxicity. Neurotoxicology. 2014;45:31–37.
- Johnstone AFM, Gross GW, Weiss DG, et al. Microelectrode arrays: a physiologically based neurotoxicity testing platform for the 21st century. NeuroToxicology. 2010;31(4):331–350. .
- Westerink RHS. Do we really want to REACH out to in vitro? Neurotoxicology. 2013;39:169–172.
- Martín-López D, Jiménez-Jiménez D, Cabañés-Martínez L, et al. The role of thalamus versus cortex in epilepsy: evidence from human ictal centromedian recordings in patients assessed for deep brain stimulation. Int J Neural Syst. 2017;27(7):1750010.
- Tukker AM, Wijnolts FMJ, de Groot A, et al. Applicability of hipsc-derived neuronal co-cultures and rodent primary cortical cultures for in vitro seizure liability assessment. Toxicol Sci. 2020;178(1):71–87. .
- Cotterill E, Hall D, Wallace K, et al. Characterization of early cortical neural network development in multiwell microelectrode array plates. J Biomol Screen. 2016;21(5):510–519.
- Dingemans MML, Schütte MG, Wiersma DMM, et al. Chronic 14-day exposure to insecticides or methylmercury modulates neuronal activity in primary rat cortical cultures. Neurotoxicology. 2016;57:194–202.
- Hyvärinen T, Hyysalo A, Kapucu FE, et al. Functional characterization of human pluripotent stem cell-derived cortical networks differentiated on Laminin-521 substrate: comparison to rat cortical cultures. Sci Rep. 2019;9(1). DOI:10.1038/s41598-019-53647-8.
- Hondebrink L, Verboven AHA, Drega WS, et al. Neurotoxicity screening of (Illicit) drugs using novel methods for analysis of microelectrode array (MEA) recordings. Neurotoxicology. 2016;55:1–9.
- McConnell ER, McClain MA, Ross J, et al. Evaluation of multi-well microelectrode arrays for neurotoxicity screening using a chemical training set. Neurotoxicology. 2012;33(5):1048–1057.
- Hogberg HT, Sobanski T, Novellino A, et al. Application of micro-electrode arrays (MEAs) as an emerging technology for developmental neurotoxicity: evaluation of domoic acid-induced effects in primary cultures of rat cortical neurons. Neurotoxicology. 2011;32(1):158–168.
- Nicolas J, Hendriksen PJM, van Kleef RGDM, et al. Detection of marine neurotoxins in food safety testing using a multielectrode array. Mol Nutr Food Res. 2014;58(12):2369–2378.
- Valdivia P, Martin M, LeFew WR, et al. Multi-well microelectrode array recordings detect neuroactivity of toxcast compounds. Neurotoxicology. 2014;44:204–217.
- Novellino A, Scelfo B, Palosaari T, et al. Development of micro-electrode array based tests for neurotoxicity: assessment of interlaboratory reproducibility with neuroactive chemicals. Front Neuroeng. 2011;4:4..
- Vassallo A, Chiappalone M, De Camargos Lopes R, et al. A multi-laboratory evaluation of microelectrode array-based measurements of neural network activity for acute neurotoxicity testing. Neurotoxicology. 2017;60:280–292.
- Bradley JA, Luithardt HH, Metea MR, et al. In vitro screening for seizure liability using microelectrode array technology. Toxicol Sci. 2018;163(1):240–253. .
- Kreir M, Van Deuren B, Versweyveld S, et al. Do in vitro assays in rat primary neurons predict drug-induced seizure liability in humans? Toxicol Appl Pharmacol. 2018;346:45–57. .
- Mack CM, Lin BJ, Turner JD, et al. Burst and principal components analyses of mea data for 16 chemicals describe at least three effects classes. Neurotoxicology. 2014;40:75–85.
- Tukker AM, Van Kleef RGDM, Wijnolts FMJ, et al. Towards animal-free neurotoxicity screening: applicability of hipsc-derived neuronal models for in vitro seizure liability assessment. ALTEX. 2020;37(1):121–135.
- Clowry G, Molnár Z, Rakic P. Renewed focus on the developing human neocortex. J Anat. 2010;217(4):276–288.
- Dolmetsch R, Geschwind DH. The human brain in a dish: the promise of IPSC-derived neurons. Cell. Cell Press: Jun 10 2011 831–834. DOI:10.1016/j.cell.2011.05.034..
- Steffenhagen C, Kraus S, Dechant F-X, et al. Identity, fate and potential of cells grown as neurospheres: species matters. Stem Cell Rev Reports. 2011;7(4):815–835.
- Arrowsmith J, Miller P. Phase II and phase iii attrition rates 2011–2012. Nat Rev Drug Discov. 2013;12(8):569.
- Fung M, Thornton A, Mybeck K, et al. Evaluation of the characteristics of safety withdrawal of prescription drugs from worldwide pharmaceutical markets-1960 to 1999. Drug Inf J. 2001;35(1):293–317.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676.
- Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295–305.
- Swistowski A, Peng J, Liu Q, et al. Efficient generation of functional dopaminergic neurons from human induced pluripotent stem cells under defined conditions. Stem Cells. 2010;28(10):1893–1904.
- Zeng H, Guo M, Martins-Taylor K, et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One. 2010;5(7):7.
- Hu Y, Qu Z-Y, Cao S-Y, et al. Directed differentiation of basal forebrain cholinergic neurons from human pluripotent stem cells. J Neurosci Methods. 2016;266:42–49.
- Vadodaria KC, Stern S, Marchetto MC, et al. Serotonin in psychiatry: in vitro disease modeling using patient-derived neurons. Cell Tissue Res. 2018;371(1):161–170.
- Liu H, Zhang SC. Specification of neuronal and glial subtypes from human pluripotent stem cells. Cell Mol Life Sci. 2011 December;68(24):pp 3995–4008. .
- Emdad L, D’Souza SL, Kothari HP, et al. Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. 2012;21(3):404–410.
- Juopperi TA, Kim WR, Chiang C-H, et al. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of huntington’s disease patient cells. Mol Brain. 2012;5(1):17.
- Abud EM, Ramirez RN, Martinez ES, et al. IPSC-derived human microglia-like cells to study neurological diseases. Neuron. 2017;94(2):278–293.e9.
- Little D, Ketteler R, Gissen P, et al. Using stem cell–derived neurons in drug screening for neurological diseases. Neurobiol Aging. 2019;78:130–141. .
- Schwartzentruber J, Foskolou S, Kilpinen H, et al. Molecular and functional variation in ipsc-derived sensory neurons. Nat Genet. 2017;50(1):54–61.
- Odawara A, Katoh H, Matsuda N, et al. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Sci Rep. 2016;6(1). DOI:10.1038/srep26181.
- Qi Y, Zhang XJ, Renier N, et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat Biotechnol. 2017;35(2):154–163.
- Hyysalo A, Ristola M, Mäkinen MEL, et al. Laminin Α5 substrates promote survival, network formation and functional development of human pluripotent stem cell-derived neurons in vitro. Stem Cell Res. 2017;24:118–127.
- Kuijlaars J, Oyelami T, Diels A, et al. Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci Rep. 2016;6(1). DOI:10.1038/srep36529.
- Odawara A, Matsuda N, Ishibashi Y, et al. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Sci Rep. 2018;8(1):10416.
- Odawara A, Saitoh Y, Alhebshi AH, et al. Long-term electrophysiological activity and pharmacological response of a human induced pluripotent stem cell-derived neuron and astrocyte co-culture. Biochem Biophys Res Commun. 2014;443(4):1176–1181. .
- Taga A, Dastgheyb R, Habela C, et al. Role of human-induced pluripotent stem cell-derived spinal cord astrocytes in the functional maturation of motor neurons in a multielectrode array system. Stem Cells Transl Med. 2019;8(12):1272–1285.
- Brown JP, Hall D, Frank CL, et al. Evaluation of a microelectrode array-based assay for neural network ontogeny using training set chemicals. Toxicol Sci. 2016;154(1):126–139.
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14(5):pp 311–321.
- Tang X, Zhou L, Wagner AM, et al. Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res. 2013;11(2):743–757.
- Kayama T, Suzuki I, Odawara A, et al. Temporally coordinated spiking activity of human induced pluripotent stem cell-derived neurons co-cultured with astrocytes. Biochem Biophys Res Commun. 2018;495(1):1028–1033.
- Takemoto T, Ishihara Y, Ishida A, et al. Neuroprotection elicited by nerve growth factor and brain-derived neurotrophic factor released from astrocytes in response to methylmercury. Environ Toxicol Pharmacol. 2015;40(1):199–205.
- Tukker AM, Wijnolts FMJ, de Groot A, et al. Human IPSC-derived neuronal models for in vitro neurotoxicity assessment. Neurotoxicology. 2018;67:215–225.
- Iida S, Shimba K, Sakai K, et al. Synchronous firing patterns of induced pluripotent stem cell-derived cortical neurons depend on the network structure consisting of excitatory and inhibitory neurons. Biochem Biophys Res Commun. 2018;501(1):152–157.
- Kreir M, De Bondt A, Van den Wyngaert I, et al. Role of Kv7.2/Kv7.3 and M1 muscarinic receptors in the regulation of neuronal excitability in HiPSC-derived neurons. Eur J Pharmacol. 2019;858:172474.
- Konagaya S, Ando T, Yamauchi T, et al. Long-term maintenance of human induced pluripotent stem cells by automated cell culture system. Sci Rep. 2015;5(1). DOI:10.1038/srep16647.
- Volpato V, Smith J, Sandor C, et al. Reproducibility of molecular phenotypes after long-term differentiation to human ipsc-derived neurons: a multi-site omics study. Stem Cell Reports. 2018;11(4):897–911.
- Jenkins M, Bilsland J, Allsopp TE, et al. Patient-specific HiPSC bioprocessing for drug screening: bioprocess economics and optimisation. Biochem Eng J. 2016;108:84–97.
- Bardy C, Van Den Hurk M, Eames T, et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci U S A. 2015;112(20):E2725–E2734.
- Joshi P, Bodnya C, Ilieva I, et al. Huntington’s disease associated resistance to Mn neurotoxicity is neurodevelopmental stage and neuronal lineage dependent. Neurotoxicology. 2019;75:148–157.
- D’Aiuto L, Zhi Y, Kumar Das D, et al. Large-scale generation of human IPSC-derived neural stem cells/early neural progenitor cells and their neuronal differentiation. Organogenesis. 2014;10(4):365–377.
- Wakeman DR, Hiller BM, Marmion DJ, et al. Cryopreservation maintains functionality of human ipsc dopamine neurons and rescues parkinsonian phenotypes in vivo. Stem Cell Reports. 2017;9(1):149–161.
- Nishiyama Y, Iwanami A, Kohyama J, et al. Safe and efficient method for cryopreservation of human induced pluripotent stem cell-derived neural stem and progenitor cells by a programmed freezer with a magnetic field. Neurosci Res. 2016;107:20–29.
- Preininger MK, Singh M, Xu C. Cryopreservation of human pluripotent stem cell-derived cardiomyocytes: strategies, challenges, and future directions. Adv Exp Med Biol. 2016;Vol. 951:pp 123–135. Springer New York LLC. .
- Gabriel E, Gopalakrishnan J. Generation of Ipsc-derived human brain organoids to model early neurodevelopmental disorders. J Vis Exp. 2017;2017(127):122.
- Leite PEC, Pereira MR, Harris G, et al. Suitability of 3D human brain spheroid models to distinguish toxic effects of gold and poly-lactic acid nanoparticles to assess biocompatibility for brain drug delivery. Part Fibre Toxicol. 2019;16(1). DOI:10.1186/s12989-019-0307-3.
- Pamies D, Block K, Lau P, et al. Rotenone exerts developmental neurotoxicity in a human brain spheroid model. Toxicol Appl Pharmacol. 2018;354:101–114.
- Pelkonen A, Mzezewa R, Sukki L, et al. A modular brain-on-a-chip for modelling epileptic seizures with functionally connected human neuronal networks. Biosens Bioelectron. 2020;168:112553..
- Muñoz SS, Engel M, Balez R, et al. A simple differentiation protocol for generation of induced pluripotent stem cell-derived basal forebrain-like cholinergic neurons for alzheimer’s disease and frontotemporal dementia disease modeling. Cells. 2020;9(9):9.
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5(7):553–564.
- Linville RM, DeStefano JG, Sklar MB, et al. Human IPSC-derived blood-brain barrier microvessels: validation of barrier function and endothelial cell behavior. Biomaterials. 2019;190–191:24–37.
- Vatine GD, Barrile R, Workman MJ, et al. Human IPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell. 2019;24(6):995–1005.e6.
- Bal-Price AK, Suñol C, Weiss DG, et al. Application of in vitro neurotoxicity testing for regulatory purposes: symposium iii summary and research needs. Neurotoxicology. 2008;29(3):520–531.
- Krewski D, Acosta D, Andersen M, et al. Toxicity testing in the 21st century: a vision and a strategy. J Toxicol Environ Heal Part B. 2010;13(2–4):51–138.
- Croom EL, Shafer TJ, Evans MV, et al. Improving in vitro to in vivo extrapolation by incorporating toxicokinetic measurements: a case study of lindane-induced neurotoxicity. Toxicol Appl Pharmacol. 2015;283(1):9–19.
- Sharma RP, Schuhmacher M, Kumar V. Developing integrated pbpk/pd coupled mechanistic pathway model (mirna-bdnf): an approach towards system toxicology. Toxicol Lett. 2017;280:79–91.
- Hong SG, Dunbar CE, Winkler T. Assessing the risks of genotoxicity in the therapeutic development of induced pluripotent stem cells. Nature Publishing Group Mol Ther. 2013;212:pp 272–281. .
- Willmann CA, Hemeda H, Pieper LA, et al. To clone or not to clone? induced pluripotent stem cells can be generated in bulk culture. PLoS One. 2013;8(5):5.
- Nicolson TJ, Mellor HR, Roberts RRA. Gender differences in drug toxicity. Trends Pharmacol Sci. 2010;31(3):108–114.
- Gochfeld M. Sex Differences in Human and Animal Toxicology: toxicokinetics. Toxicol Pathol. 2017;45(1):172–189.
- Zhang N, An MC, Montoro D, et al. Characterization of human huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2010 OCT;1–11. DOI:10.1371/currents.RRN1193..
- Ochalek A, Mihalik B, Avci HX, et al. Neurons derived from sporadic alzheimer’s disease ipscs reveal elevated tau hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimer’s Res Ther. 2017;9(1). DOI:10.1186/s13195-017-0317-z.
- Schöndorf DC, Aureli M, McAllister FE, et al. IPSC-derived neurons from GBA1-associated parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014;5(1). DOI:10.1038/ncomms5028.
- Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nature Publishing Group March 1 Nat Rev Mol Cell Biol. 2016;173:pp 170–182. .