
ABSTRACT
Introduction
Direct oral anticoagulants (DOACs) have overtaken vitamin K antagonists to become the most widely used method of anticoagulation for most indications. Their stable and predictable pharmacokinetics combined with relatively simple dosing, and the absence of routine monitoring has made them an attractive proposition for healthcare providers. Despite the benefits of DOACs as a class, important differences exist between individual DOAC drugs in respect of their pharmacokinetic and pharmacodynamic profiles with implications for dosing and reversal in cases of major bleeding.
Areas Covered
This review summarizes the state of knowledge relating to the pharmacokinetics of dabigatran (factor IIa/thrombin inhibitor) and apixaban, edoxaban and rivaroxaban (factor Xa) inhibitors. We focus on pharmacokinetic differences between the drugs which may have clinically significant implications.
Expert Opinion
Patient-centered care necessitates a careful consideration of the pharmacokinetic and pharmacodynamic differences between DOACs, and how these relate to individual patient circumstances. Prescribers should be aware of the potential for pharmacokinetic drug interactions with DOACs which may influence prescribing decisions in patients with multiple comorbidities. In order to give an appropriate dose of DOAC drugs, accurate estimation of renal function using the Cockcroft-Gault formula using actual body weight is necessary. An increasing body of evidence supports the use of DOACs in patients who are obese, and this is becoming more routine in clinical practice.
1. Introduction
In many countries, the direct oral anticoagulants (DOACs) have overtaken vitamin K antagonists to become the most widely used method of oral anticoagulation for most indications. Their stable and predictable pharmacokinetics combined with relatively simple dosing, and the absence of routine monitoring has made them an attractive proposition for healthcare providers. Despite the benefits of DOACs as a class, important differences exist between individual DOAC drugs in respect of their pharmacokinetic and pharmacodynamic profiles. These differences, coupled with the expanding role of DOACs in the clinical setting, as well as nuances in their dosing, mean that clinicians can be faced with increasingly complex decisions when selecting the most appropriate agent for a given patient [Citation1].
Since their inception, DOACs have been subject to extensive investigation with guidelines surrounding their use changing as new evidence has emerged [Citation1]. It is therefore important for those individuals involved in the prescribing and monitoring of DOACs to be familiar with the pharmacokinetic and pharmacodynamic properties of these agents and this article provides a detailed review of the topic.
1.1. The pharmacodynamic effects of DOACs
DOACs exert their pharmacodynamic effect by interfering with the normal function of clotting factors. In the clotting cascade, the two main targets for DOACs are factor IIa (thrombin) and factor Xa. Apixaban, edoxaban, and rivaroxaban all target factor Xa, whereas dabigatran is a direct thrombin inhibitor (). Betrixaban is also a factor Xa inhibitor but has since been withdrawn by its manufacturer, so is not discussed extensively.
Table 1. Pharmacodynamic properties of DOACs.
Apixaban is a direct and reversible inhibitor of both free and clot bound factor Xa [Citation2,Citation3]. Whilst it has no direct effects on platelet aggregation, it inhibits thrombin-induced platelet activation [Citation2,Citation3] and consequently it is known to prolong prothrombin time, international normalized ratio and activated partial thromboplastin time [Citation3,Citation4]. The impact of apixaban on thrombin generation lasts over 12 hours following oral administration [Citation3]. Anti-factor Xa activity demonstrates a close linear relationship with plasma apixaban concentrations across a wide range of doses [Citation3]. Other markers of hemostasis, such as clotting time, demonstrate a high degree of variability and do not accurately reflect apixaban activity [Citation3].
Edoxaban is a selective and competitive inhibitor of factor Xa. It inhibits thrombin formation and platelet aggregation and causes an increase in prothrombin time, international normalized ratio and activated partial thromboplastin time, inhibiting thrombin generation for up to 24 hours [Citation5]. Plasma edoxaban levels and anti-factor Xa activity demonstrate a linear relationship [Citation6].
Rivaroxaban is a direct, specific, and competitive inhibitor of factor Xa, inhibiting thrombin formation, and prolonging both prothrombin and activated partial thromboplastin time [Citation7,Citation8]. Rivaroxaban induced inhibition of thrombin generation persists beyond 24 hours permitting once daily dosing [Citation8] and prolongation of prothrombin time exhibits a linear correlation with rivaroxaban plasma concentration [Citation7].
Unlike the other DOACs, dabigatran is not a factor Xa inhibitor, and is instead a direct competitive inhibitor of free and fibrin-bound thrombin [Citation9,Citation10]. The effect of dabigatran on thrombin is rapid and reversible and prevents the conversion of fibrinogen into fibrin, preventing the development of a thrombus [Citation9,Citation10]. Dabigatran is known to prolong thrombin time, ecarin clotting time and activated partial thromboplastin time [Citation9,Citation10], with peak prolongation of coagulation corresponding with the maximum plasma concentration (Cmax) and dropping by approximately 50% 12 hours after administration [Citation11].
The dosing regimens associated with the different DOACs have been the subject of much discussion. From a patient-perspective once daily dosing (edoxaban and rivaroxaban) may better promote compliance than twice-daily dosing (apixaban and dabigatran). However it has been suggested that rivaroxaban’s once daily dosing can lead to higher concentration peaks (and therefore greater risk of bleeding). Despite this, a recent meta-analysis, including 12 studies (24 dose comparisons) across a range of indications concluded that the risk of thrombosis and bleeding were similar across once and twice-daily regimens [Citation12].
The routine monitoring of DOACs is generally not considered necessary although it may be of some benefit in certain situations and patient populations such as major bleeding, declining renal function, pre-procedure and [Citation13] treatment failure (breakthrough stroke) [Citation14] which may result from a number of factors, including pharmacogenetic inter-individual differences in drug metabolism [Citation15]. Whilst therapeutic ranges have not been clearly established, correlations between plasma trough levels and clinical effects (bleeding or thromboembolism) have been demonstrated with dabigatran and edoxaban (RE-LY and ENGAGE trials) and the European Heart Rhythm Association Guidelines state that plasma monitoring may be useful in emergency situations or special cases [Citation16]. Point of care tests have recently been studied, which sensitively identified effective thrombin and factor Xa inhibition by DOACs in a real-world cohort of patients presenting at an emergency department [Citation17,Citation18]. Where indicated, apixaban, edoxaban, and rivaroxaban activity can be accurately predicted with anti-factor Xa levels. Heparin anti-Xa levels can be used to identify the presence of a DOAC, and agent-specific Xa-levels can be used in quantification [Citation3,Citation13,Citation19,Citation20]. However, recent studies in elderly patients with atrial fibrillation (AF), multimorbidity, and polypharmacy receiving apixaban/rivaroxaban found great inter-individual variability in plasma concentrations and thrombin generation (TG) [Citation21]. Dabigatran activity can be monitored by assaying thrombin time, dilute thrombin time, ecarin clotting time and activated partial thromboplastin time, although the latter is less sensitive and not appropriate for precise quantification [Citation10].
The International Council for Standardization in Haematology Recommendations for Laboratory Measurement of Direct Oral Anticoagulants documents outline the expected plasma concentrations of drugs associated with specific DOAC doses, and provided important insights into the reliability of assays [Citation22].
In the event of overdose, the administration of activated charcoal has been shown to reduce exposure to, and facilitate the elimination of apixaban, edoxaban, rivaroxaban, and dabigatran although this approach is based on standard medical treatment and not necessarily endorsed by the manufacturers [Citation10,Citation23–29]. In the event of life-threatening or uncontrolled bleeding, andexanet (a recombinant form of human factor Xa) is available as an antidote to apixaban and rivaroxaban [Citation30], and idarucizumab (a monoclonal antibody with an affinity for dabigatran approximately 300 times more potent than that of dabigatran for thrombin) is available as an antidote for dabigatran [Citation31]. Although the use of andexanet for the reversal of edoxaban is not currently licensed in the UK, Europe or America [Citation30,Citation32,Citation33], it has been shown to significantly decrease anti-factor Xa activity and achieve hemostasis in most patients with acute bleeding secondary to edoxaban, and is the subject of an ongoing clinical trial evaluating this further [Citation34,Citation35]. In many parts of the world the availability of andexanet may be limited by cost, and prothrombin complex concentrate is used off-label for DOAC reversal [Citation36]. In the context of reversal agents, it is pertinent to note that they can be employed in patients with hyperacute ischemic stroke who are on anticoagulation, who may otherwise qualify for (and be in the window for) intravenous thrombolysis [Citation37].
1.2. The pharmacokinetic profiles of DOACs
In contrast to the older oral anticoagulants such as warfarin and acencoumarol, DOACs benefit from a stable pharmacokinetic profile which enables standardized dosing without the need for routine monitoring. Because of this, coupled with the evidence of efficacy, DOACs have become the treatment of choice in the management of both venous thromboembolism (VTE) and atrial fibrillation (AF) across Europe and the United States. There are, however, a number of important and clinically relevant differences in the pharmacokinetic profiles of each drug which are discussed below ().
Figure 1. Pharmacokinetic considerations in DOAC prescribing (Figure created with biorender.com).
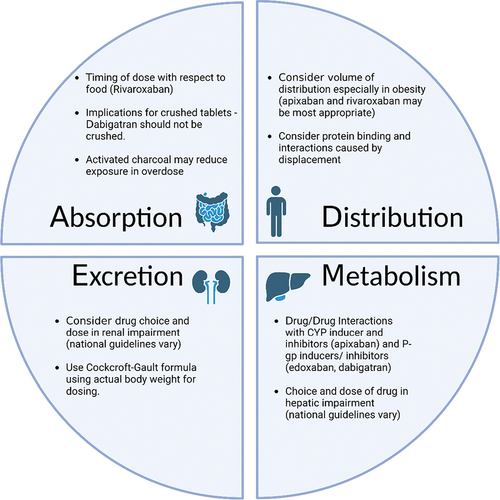
1.3. Characteristics of the available DOACs
1.3.1. Apixaban
Apixaban is primarily absorbed in the upper small intestine with absorption progressively decreasing as it moves through the gastrointestinal tract [Citation38]. Following oral administration, the absolute oral bioavailability of apixaban is approximately 50% [Citation39]. The remainder of the dose is lost through incomplete absorption and hepatic first pass metabolism [Citation3]. Studies on the effect of fasting and fed states on the absorption of apixaban have shown some variation but conclude that food does not impact on bioavailability in any clinically significant way [Citation3,Citation40,Citation41]. Thus, apixaban can be taken with or without food [Citation42–44]. Following oral administration of apixaban, Cmax is achieved after 2.5 to 4 hours [Citation41,Citation45].
Crushing and dissolution of apixaban tablets for administration to patients with dysphasia or via nasogastric tubes appears to have no clinically significant impact on bioavailability [Citation46] and is licensed in the UK and Europe when water, glucose 5%, apple juice or apple puree is used a solvent and the solution administered orally [Citation42]. If administering via a nasogastric tube, the manufacturer recommends 60 mL water or glucose 5% as a solvent [Citation42].
The volume of distribution of apixaban is approximately 21 L, suggesting that it is primarily distributed in the extracellular fluid [Citation42]. Given the limited intracellular distribution of apixaban, extremes of body would be expected to have only limited impact on the pharmacokinetic profile of the drug [Citation39], and this has been confirmed in pharmacokinetic studies [Citation47]. In a study examining the impact of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety, and tolerability of apixaban, there were modest changes in apixaban exposure as a result of both low and high body weight, but this did not justify dose adjustment unless there were other factors that might simultaneously increase exposure [Citation47]. These findings reflect the manufacturer’s recommendations not to reduce the dose based on body weight alone, but in the presence of other factors such as impaired renal function and advancing age [Citation42]. Recent clinical evidence supports the safe use of apixaban in patients who are morbidly obese with a body mass index (BMI) >40 Kg/m2 in the management of both AF and VTE [Citation48–52], and the manufacturers make no recommendation to avoid or alter the dose based on high body weight alone [Citation42]. A recent review of available pharmacokinetic, interventional, and observational data has concluded that obesity does not substantially impact on the efficacy or safety of apixaban in the management of AF or VTE [Citation53].
The plasma protein binding of apixaban is approximately 87% [Citation42,Citation54] suggesting a limited potential for interactions resulting from displacement from plasma proteins [Citation54].
Studies examining the impact of age on the pharmacokinetics of apixaban have found that although Cmax was comparable between younger and older participants, exposure was modestly raised in elderly subjects although this may have simply been the consequence of declining renal function [Citation55]. Population pharmacokinetic analysis suggests that age alone appears to only have a small impact on apixaban exposure [Citation56]. Similarly, although Cmax and exposure have been shown to be higher in females than males, the difference observed is not considered to be clinically significant [Citation55] and no difference in endpoints has been observed [Citation3]. No dose adjustments are recommended based solely on these parameters [Citation42,Citation44].
Apixaban is metabolized by O-demethylation, hydroxylation, and sulfation [Citation3,Citation42,Citation54] and is primarily mediated by the cytochrome P450 enzymes CYP3A4 and CYP3A5, although others including CYP1A2, CYP2C8, CYP2C9, CYP2C19, and CYP2J2 are also involved [Citation3,Citation42,Citation54,Citation57]. Apixaban is also a substrate for P-glycoprotein (P-gp) and breast cancer resistance protein (BRCP) [Citation58]. Therefore, drugs which are strong inducers or inhibitors of both CYP3A4 and P-gp such as phenytoin, rifampicin, carbamazepine, and ketoconazole demonstrate clinically significant interactions with apixaban, with numerous studies confirming this [Citation3,Citation59,Citation60]. Clarithromycin slightly increases apixaban exposure, but this can be addressed through monitoring for signs and symptoms of bleeding or anemia; especially, in elderly patients and in those with renal impairment. Amiodarone, which inhibits both CYP3A4 and P-glycoprotein, may increase apixaban and rivaroxaban exposure, and therefore increase the risk of bleeding but no dose adjustment is required [Citation61,Citation62]. The FDA in the United States have an online tool which can help to identify interactions resulting from inducer/inhibitor drugs and those which inhibit transporter systems [Citation63]. Following administration, unchanged apixaban is the major component present in the plasma and no other active metabolites are found [Citation3,Citation42].
The presence of mild or moderate hepatic impairment has little or no impact on the pharmacokinetics of apixaban and plasma protein binding is similar to that seen in healthy individuals [Citation3]. Therefore, no dose adjustments are recommended in patients with Child-Pugh A or B [Citation42,Citation44]. Apixaban should be avoided in the event of severe hepatic impairment (Child-Pugh C), primarily due to a lack of experience although these patients will likely have some abnormalities in their coagulation [Citation3,Citation42,Citation44].
Apixaban has multiple routes of elimination including metabolism (as outlined above), biliary, renal, and direct intestinal excretion [Citation3,Citation42,Citation64] with total plasma clearance being 3.3 L/hr and half-life approximately 12 hours [Citation3,Citation42]. Following repeated twice daily dosing of apixaban, steady state is reached within three days [Citation45]. Compared to the other available DOACs, apixaban is the least dependent on renal elimination with approximately 27% of the drug being cleared by this route and the rest by other means [Citation3,Citation39,Citation42,Citation65]. Consequently, the impact of renal impairment is less than with other DOACs and appears not to impact on Cmax or the relationship between plasma concentration and anti – factor Xa activity, although apixaban exposure is increased in the presence of renal impairment [Citation56,Citation66,Citation67].
Numerous studies support the use of apixaban in patients with impaired renal function with some suggesting that it has a lower rate of bleeding in this population when compared to warfarin [Citation3,Citation68–72]. Although unlicensed in some jurisdictions for use in dialysis patients, there is some evidence to support its use in this patient cohort [Citation70]. Despite this, uncertainty remains around the optimal dosing of apixaban in patients with severely impaired renal function with some evidence to suggest that a reduced dose may be more appropriate [Citation73]. In the UK and Europe, the manufacturers recommend that the dose of apixaban be reduced in patients with AF who have a creatinine clearance between 15-29 mL/min or who meet two of three criteria (aged ≥80 years, body weight ≤60 Kg or serum creatinine ≥ 133micromol/L) but make no such dose reduction recommendation for patients with VTE [Citation42,Citation43]. In these jurisdictions, apixaban is not licensed in patients with a creatinine clearance <15 mL/min [Citation42,Citation43]. However, further large-scale studies are required to identify the safest regimens in patients with renal dysfunction. A patient-level meta-analysis (COMBINE-AF) of randomized controlled trials concluded that standard-dose DOACs are safer and more effective than warfarin down to a creatinine-clearance of at least 25 mL/min [Citation74].
1.3.2. Edoxaban
The dissolution of edoxaban is pH dependent, demonstrating high solubility in acidic environments and becoming practically insoluble in basic solutions [Citation75]. Consequently, absorption primarily occurs in the proximal small intestine with significantly less absorption occurring in the colon [Citation75]. Following administration, the absolute oral bioavailability of edoxaban is estimated to be approximately 60% with Cmax occurring within 1 to 2 hours of ingestion [Citation76,Citation77]. Although studies on the effect of fasted/fed states on the absorption of edoxaban have demonstrated a modest increase in both Cmax and total exposure as well as a reduced rate of absorption in the presence of food [Citation78,Citation79], this is felt not to be of any clinical significance [Citation79] and the manufacturers recommend administration with or without food [Citation24,Citation25,Citation80].
In patients with swallowing difficulties, edoxaban may be crushed and mixed with water or apple puree for oral administration or mixed with water and administered via a gastric tube [Citation24,Citation25]. Edoxaban is stable in water or apple puree for 4 hours [Citation81]. Pharmacokinetic studies of edoxaban have demonstrated that manipulation and administration of the dosage form in this manner have little impact on Cmax or exposure although it may shorten the time taken to achieve maximum plasma concentration [Citation81].
Edoxaban is widely distributed throughout the body with a with a large volume of distribution of approximately 107 L [Citation6] which is the largest of the DOACs [Citation82]. Edoxaban kinetics can be described by a two-compartment model [Citation83] and it is therefore tempting to conclude that obesity would negatively impact on therapeutic efficacy due to distribution into the peripheries and consequent reduction in exposure [Citation6]. However, given the complex mechanisms behind the pharmacokinetics of drugs, individual parameters such as volume of distribution may not necessarily correlate with clinical efficacy. The available data on the use of edoxaban in the management of VTE in obesity are limited and certainly less clear than with apixaban and rivaroxaban, both of which have evidence to support their use [Citation82,Citation84]. Unlike with VTE however, data supporting the use of edoxaban in the management of AF in obese patients are available, with analysis of the ENGAGE AF-TIMI 48 trial demonstrating similar clinical efficacy in extremes of body weight [Citation85]. For patients with low body weight, the rate of edoxaban clearance appears to be reduced [Citation86] and this is reflected in guidance from the manufacturers to reduce dose in those with a body weight ≤60 Kg regardless of indication in those in the UK and Europe [Citation24,Citation25] and for those with thromboembolism in the United States [Citation80].
The plasma protein binding of edoxaban is approximately 55% which suggests low potential for interactions resulting from displacement from plasma proteins [Citation6].
Although increasing age has been shown to correlate inversely with non-renal clearance, in the absence of any other factors such as low body weight, it is not expected to have any clinically significant impact on the pharmacokinetics of edoxaban [Citation76]. Other intrinsic factors such as sex and race also do not appear to impact on its pharmacokinetics [Citation6]
Edoxaban is metabolized by numerous pathways, primarily hydrolysis mediated by carboxylesterase 1, but also conjugation and oxidation by CYP3A4 and CYP3A5 [Citation6,Citation25,Citation87] although metabolism of the active drug accounts for less than 10% of the total clearance [Citation6]. Edoxaban is a substrate for P-gp and studies have demonstrated that coadministration with strong inhibitors increases edoxaban exposure [Citation76,Citation88]. Consequentially, the UK and European manufacturer recommends a dose reduction for all indications in patients taking certain P-gp inhibitors such as ciclosporin, dronedarone, erythromycin, and ketoconazole [Citation24,Citation25], although the American datasheet recommends this only when treating VTE [Citation80]. Following administration, unchanged edoxaban is the major component present in the plasma with three active metabolites also present, although exposure to these in healthy subjects is less than 10% [Citation24,Citation25] and they are unlikely to contribute to anticoagulant activity in any significant way [Citation6].
The presence of mild or moderate hepatic impairment does not appear to impact on the pharmacokinetics or pharmacodynamics of edoxaban which is in keeping with the limited role hepatic metabolism plays in edoxaban clearance (<10%) [Citation6,Citation25,Citation89] although different recommendations are made with regard to this in Europe and America [Citation6,Citation24,Citation25,Citation80,Citation89]. The use of edoxaban has not been studied in patients with severe hepatic impairment, but it is expected that these patients will likely have some disturbances in their coagulation [Citation25].
Edoxaban has multiple routes of elimination with renal clearance of the unchanged compound contributing to approximately half of the total clearance and the remainder being cleared through metabolism (as outlined above) and biliary excretion [Citation6,Citation87]. Total plasma clearance is estimated to be 22 L/hr and its half-life is between 10 and 14 hours with steady state plasma concentrations being achieved after 3 days of regular dosing [Citation6,Citation24,Citation25,Citation80]. Given the significant role of the kidneys in the elimination of edoxaban, it is unsurprising that pharmacokinetic studies have demonstrated that exposure increases as renal function declines [Citation90–92] with a corresponding increase in anti-factor Xa activity [Citation93]. In keeping with these findings, the dose of edoxaban should be reduced in the presence of renal impairment, defined as a creatinine clearance of 15–50 mL/min [Citation24,Citation25,Citation80] with numerous studies demonstrating the safety and efficacy of this approach to dosing in both AF and VTE [Citation94–96]. It has been suggested that in patients with very good levels of renal function (creatinine clearance >95 mL/min), the efficacy of edoxaban decreases, likely as a result of lower plasma concentrations and thus anti-factor Xa activity, although interpretation of these findings is complicated [Citation97]. In the UK and Europe, the manufacturers do not make any recommendation to avoid edoxaban or attenuate the dose in this patient cohort [Citation24,Citation25], but the American datasheet does recommend avoiding edoxaban in patients with AF who have a creatinine clearance >95 mL/min as a result of findings from the ENGAGE AF-TIMI 48 study [Citation80].
1.3.3. Rivaroxaban
Rivaroxaban is primarily absorbed in the stomach with studies demonstrating a significant drop in both exposure and Cmax when the drug was released directly into the small intestine [Citation98]. The oral bioavailability of rivaroxaban is the highest among the DOACs, being approximately 80–100% under the right conditions, with Cmax being achieved 2–4 hours following oral administration [Citation7,Citation99]. At the lowest dose of 10 mg, the presence or absence of food appears to make no difference to bioavailability, however this is not the case for doses of 15 mg and 20 mg. When these higher doses are administered in a fasted state, both absorption rate and bioavailability drop to as low as 66%, although this can be increased to > 80% when taken with food [Citation7,Citation99,Citation100]. This is irrespective of the type of food (i.e. high carbohydrate or high fat) [Citation7]. Consequently, the manufacturers recommend taking doses of 10 mg or less with or without food but doses of 15 mg or 20 mg with food [Citation101–104].
In patients with swallowing difficulties, studies have demonstrated that exposure and Cmax of a 20 mg tablet crushed and administered in apple sauce were comparable to a whole tablet but when mixed with water and administered via a nasogastric tube Cmax was 18% lower [Citation7,Citation101,Citation102,Citation104]. Given these results, the administration of rivaroxaban, crushed and mixed with either water or apple sauce and administered orally or via a nasogastric tube, is considered appropriate [Citation105]. In light of the dose-proportional and predictable pharmacokinetics of rivaroxaban, these results are likely applicable to the lower doses [Citation102]. When prescribing rivaroxaban to a patient with swallowing difficulties or an enteral tube, such patients may have a reduced oral intake which might adversely impact on bioavailability.
The volume of distribution of rivaroxaban is approximately 55 L indicating that it is primarily distributed within blood plasma with only low-to-moderate affinity for tissue [Citation7,Citation99]. It is therefore expected that extremes of body weight would have only limited impact on the pharmacokinetics of rivaroxaban. A recent review of the effect of obesity on rivaroxaban found that both the pharmacokinetic and clinical profile of the drug was unchanged and that high body weight does not necessitate a change in dosing [Citation106]. Although small changes in Cmax and prothrombin time have been observed in patients weighing <50 Kg, this was not considered clinically relevant [Citation107] and the manufacturers explicitly state that no doses adjustment is needed based on body weight [Citation101,Citation102,Citation104].
The protein binding of rivaroxaban is high (approximately 95%) with albumin being the primary binding component [Citation7,Citation99]. Although this might suggest a higher potential for interactions, this does not appear to be the case with rivaroxaban having a favorable interaction profile [Citation99,Citation101–104].
Although a study examining the impact of age on the kinetics of rivaroxaban did demonstrate an increase in total exposure, factor Xa inhibition and prothrombin time in elderly patients, this was the result of reduced renal clearance rather than age itself [Citation108]. The same study concluded that gender had no impact on the pharmacokinetics or pharmacodynamics of rivaroxaban [Citation108] and no dose adjustments are currently recommended based on these parameters [Citation101,Citation102,Citation104].
Rivaroxaban undergoes metabolism by the cytochrome P450 enzymes CYP3A4 and CYP2J2 as well as non-CYP medicated hydrolysis with approximately 60% of the dose being subject to metabolic degradation [Citation7,Citation99]. Rivaroxaban is a substrate of P-gp and BRCP [Citation7]. The inhibition of cytochrome P450 enzymes and/or P-gp would therefore be expected to decrease rivaroxaban clearance, however it is only the strong inhibition of both CYP3A4 and P-gp simultaneously that appears to significantly increase rivaroxaban exposure [Citation109]. These interactions are predominantly limited to azole antifungals and protease inhibitors. Strong inducers of CYP3A4 such as rifampicin, phenytoin, and carbamazepine have been shown to significantly reduce rivaroxaban exposure and increase the potential for treatment failure [Citation110]. These findings are reflected in the manufacturers’ recommendations [Citation101,Citation102,Citation104]. Unchanged rivaroxaban is the predominant molecule found in plasma, accounting for approximately 90%, and no major active metabolites are present [Citation7].
The presence of mild hepatic impairment has limited impact on the pharmacokinetics of rivaroxaban, and it can be safely used in patients with Child-Pugh A [Citation101,Citation102,Citation104,Citation111,Citation112]. In patients with moderate hepatic impairment, both Cmax and exposure are increased 1.27-fold and 2.27-fold, respectively, and elimination is prolonged [Citation111] which is in keeping with the significant role that the liver plays in the clearance of rivaroxaban. Due to the increased exposure to rivaroxaban in the presence of moderate hepatic impairment, factor Xa inhibition is significantly higher than in healthy subjects resulting in greater prolongation of prothrombin time [Citation111]. Consequently, rivaroxaban is contraindicated in patients with Child Pugh B and C, or those with hepatic disease associated with coagulopathy and clinically relevant bleeding risk [Citation101,Citation102,Citation104,Citation112].
Rivaroxaban is excreted via a combination of metabolism, as already described, and renal clearance. Approximately 36% of the drug is eliminated unchanged in the urine with a further 30% eliminated by this route in the form of metabolites [Citation7,Citation99]. Renal elimination of the active metabolite is highly dependent on active renal secretion involving both P-gp and BRCP [Citation99]. Faecal clearance of both active drug and metabolites accounts for a further 28% [Citation7,Citation99]. Rivaroxaban exhibits a clearance of approximately 10 L/hr and has a terminal half-life of 11–13 hours [Citation99]. Studies examining the effects of renal impairment on rivaroxaban have demonstrated reduced clearance resulting in increased exposure and higher levels of factor Xa inhibition when compared to healthy subjects [Citation113]. Increases in rivaroxaban concentrations of 44%, 52%, 64% and 56% have been observed in patients with mild, moderate, and severe renal impairment and end-stage renal disease respectively [Citation114]. Despite this, rivaroxaban does undergo a significant amount of non-renal clearance allowing for its safe use down to quite low levels of renal function (≥15 mL/min). Recommendations for dosing in renal impairment are dependent on both the level of renal function and the indication and vary slightly between Europe and the United States [Citation101,Citation102,Citation104].
1.3.4. Dabigatran
Dabigatran is administered as the inactive prodrug, dabigatran etexilate, and requires conversion into its pharmacologically active form in vivo [Citation115]. It is primarily absorbed in the stomach and small intestine, where the acidic environment aids dissolution and absorption [Citation116,Citation117], which is further enhanced by the tartaric acid within the capsules providing an acidic micro-environment [Citation9,Citation65,Citation117]. Unsurprisingly, the use of proton pump inhibitors has been shown to reduce Cmax at steady state by approximately 28% although this is not considered to be clinically significant, and they can be safely co-administered [Citation9,Citation10,Citation28,Citation29]. The oral bioavailability of dabigatran etexilate is low, at approximately 6%, but once absorbed it is rapidly and almost completely converted into the active form by nonspecific esterase enzymes via two intermediaries [Citation9,Citation65,Citation117,Citation118]. The presence of food does not impact the extent of absorption but can increase the time taken to achieve Cmax [Citation9,Citation117,Citation118]. The manufacturers recommend taking the capsules with or without food [Citation10,Citation28,Citation29]. Following oral administration, Cmax is achieved in approximately 1.5 hours [Citation117,Citation119].
Unlike the other DOACs, dabigatran capsules should not be crushed or otherwise manipulated to aid in administration to patients with swallowing difficulties or enteral tubes [Citation10,Citation28,Citation29]. Bioavailability is increased by 75% after a single dose and 37% after multiple doses if the capsule contents are taken without the shell [Citation10,Citation28,Citation29].
The volume of distribution of dabigatran is approximately 60–70 L which indicates moderate distribution of the drug into tissue [Citation117] and suggests that extremes of body weight have the potential to impact on the pharmacokinetics of the drug. A study examining the effects of obesity (weight >120 Kg) on peak plasma concentration demonstrated that a significant proportion of patients (20%) had peak plasma concentrations below the usual range and below the median trough level reported in the literature [Citation120]. In separate studies, dabigatran trough concentrations have also been found to be approximately 20% lower in patients weighing >100 Kg when compared to those weighing 50–100 Kg [Citation10,Citation29]. Despite this, it is not possible to draw solid conclusions on the impact of extreme body weight on the clinical efficacy of dabigatran based on pharmacokinetic studies alone. Studies examining the clinical significance of obesity on the efficacy of dabigatran in the management of AF have observed no difference in the rates of stroke or systemic embolism in obese patients, although case reports of treatment failure associated with low plasma levels have been reported in patients with a BMI >40 Kg/m2 where the data is less robust [Citation121–123]. Regarding the management of VTE, there is a lack of data to support to use of dabigatran in patients weighing >120 Kg [Citation84]. In patients weighing <50 Kg, the manufacturers concede that this may have a minor impact in the pharmacokinetics of the drug but that it has not been sufficiently studied to make any specific dose recommendations [Citation10,Citation29].
The plasma protein binding of dabigatran is approximately 35% which suggests that interactions involving displacement from plasma proteins are unlikely [Citation117].
Pharmacokinetic studies of dabigatran have demonstrated that exposure of dabigatran is significantly higher in elderly subjects, most likely as the result of the reduction in renal function associated with advancing age [Citation9]. Additionally, both Cmax and exposure are higher in female than male patients [Citation9,Citation117,Citation118]. Although no dose adjustments are recommended based on gender [Citation10,Citation28,Citation29], in the UK and Europe dose reductions are recommended in elderly patients [Citation10,Citation29].
Dabigatran is not a substrate for cytochrome P450 enzymes and is therefore not metabolized by this family of isoenzymes, nor does it impact on the metabolism of other drugs via these processes [Citation9,Citation117,Citation124]. Aside from the initial metabolism of dabigatran etexilate into the pharmacologically active dabigatran by nonspecific esterase enzymes, the only other metabolic process that the molecule undergoes is glucuronidation [Citation9,Citation115,Citation124]. This process results in the production of four pharmacologically active dabigatran acylglucuronides, which account for between 10% and 24% of total plasma dabigatran [Citation9,Citation10,Citation28,Citation29,Citation119,Citation124,Citation125]. Overall, hepatic metabolism plays only a minor role in the clearance of dabigatran, accounting for approximately 20%, with most of the absorbed dose being subject to renal clearance [Citation65,Citation115]. Although dabigatran etexilate is a substrate of P-gp, dabigatran is not, suggesting that any interactions related to this transporter are limited to absorption [Citation117]. Because of the role of P-gp in the efflux of dabigatran etexilate into the intestinal lumen, co-administration of P-gp inhibitors results in increased exposure. Concomitant use of strong inhibitors such as ketoconazole, dronedarone, itraconazole, cyclosporin and glecaprevir/pibrentasvir result in a significant increase in dabigatran exposure and is contraindicated in the UK and Europe [Citation10,Citation29] although these recommendations differ from those in the United States [Citation28]. Weaker P-gp inhibitors, such as verapamil can also increase exposure and some dose reductions are recommended by UK and European manufacturers [Citation10,Citation29]. Conversely, strong inducers of P-gp, such as rifampicin, carbamazepine, and phenytoin are known to reduce dabigatran exposure [Citation10,Citation28,Citation29].
The presence of moderate hepatic impairment, defined as Child-Pugh B, does not appear to have any significant impact on pharmacokinetics or pharmacodynamics of dabigatran [Citation126]. Furthermore, the study authors concluded that moderate impairment does not impact negatively on the safety profile of dabigatran and that it could be used in these patients without dose adjustment [Citation126]. Despite this, and although recommendations differ between locations, in Europe and the UK dabigatran is contraindicated in patients with hepatic impairment or liver disease that is expected to have any impact on survival [Citation10,Citation29].
Dabigatran is highly dependent on renal elimination with approximately 80% of plasma dabigatran being eliminated unchanged via the kidneys [Citation9,Citation65,Citation117,Citation119]. The remaining 20% undergoes hepatic glucuronidation (as outlined above) with the resultant metabolites being almost exclusively excreted in the bile with only traces found in the urine [Citation65,Citation124]. Total plasma clearance is estimated to be between 71 and 144 L/hr and half-life is between 12 and 14 hours with steady state plasma concentrations being achieved after 2–3 days of regular dosing in healthy patients [Citation9,Citation117]. Given the significant role of renal clearance in the elimination of dabigatran, it is unsurprising that clearance is prolonged and both Cmax and exposure increase in proportion with the degree of renal impairment present [Citation127]. The effect is more pronounced on exposure than Cmax with a 1.5-, 3.2-, and 6.3-fold increase in exposure observed with mild, moderate and severe renal impairment respectively following a single dose [Citation127]. This increase in exposure is associated with an increase in several measures of clotting time such as activated partial thromboplastin time [Citation127]. An additional study examining the impact of renal impairment on dabigatran kinetics demonstrated an inverse relationship between renal function and trough plasma levels [Citation128]. Analysis of data from the RE-LY trial has shown that bleeding risk corresponds positively with trough dabigatran levels with the risk of bleeds doubling when trough levels were above 210 ng/mL [Citation128]. Both the UK and European manufacturers contraindicate the use of dabigatran in patients with a creatinine clearance <30 mL/min and recommend dose reductions in certain circumstances in patients with a creatinine clearance between 30 mL/min and 50 mL/min [Citation10,Citation29]. These recommendations differ from those in America, although dose reductions are recommended there due to impaired renal function [Citation28].
2. Practicalities of DOAC use in specific populations
2.1. Estimating renal function when dosing DOACs
When calculating renal function for the purpose of dosing, the Cockcroft-Gault formula (estimated creatinine clearance) should be used in preference to estimated glomerular filtration rate [Citation129–133]. This is a consequence of the fact that during clinical trials, estimates of renal function were determined using Cockcroft-Gault with subsequent dosing recommendations being based on this, and subsequent analysis of data has shown that by using other estimates of renal function there is a failure to dose correctly in a substantial proportion of patients [Citation133,Citation134]. In addition to using the correct formula for estimating renal function, it is important to use actual body weight in the creatinine clearance calculation as was done in the phase three clinical trials [Citation131,Citation133]. These points are universal to all DOACs and doing otherwise risks incorrect dosing [Citation131–133].
2.2. The use of DOACs in obesity and morbid obesity
Historically, the use of DOACs in obese patients has been discouraged. The 2016 International Society of Thrombosis and Haemostasis (ISTH) guidance recommended against using DOACs in patients with a BMI >40 Kg/m2 or those weighing >120 Kg due to the lack of clinical data and pharmacokinetic/pharmacodynamic studies suggesting decreased exposure in these patients [Citation135]. However, evidence and clinical experience has evolved and a more recent review by the ISTH has concluded that there are similar outcomes in terms of VTE for apixaban and rivaroxaban when compared to warfarin and there is sufficient evidence to recommend the use of both agents at standard doses for the prevention and management of VTE in patients with a BMI >40 Kg/m2 or weighing >120 Kg [Citation84]. This is supported by a recent study demonstrating that anti-Xa levels among obese patients are not substantially different from patients with normal BMI and weight [Citation136]. At present, they recommend against the use of dabigatran, edoxaban, and betrixaban due to a lack of convincing data [Citation84]. A recent retrospective study conducted through the VENUS network demonstrated that DOACs were associated with lower major bleeding than warfarin in obese patients treated for VTE [Citation137]
As with the management of VTE, there is evidence to support the use of DOACs for AF in obese patients. A recent review concluded that, when compared to warfarin, DOACs were associated with a better efficacy and safety profile across all categories of BMI, including those who were morbidly obese [Citation138], supporting data from the studies already mentioned [Citation48,Citation85,Citation122]. Despite this, the European Society of Cardiology advises that once a patient’s BMI exceeds 40 Kg/m2, data on the safety and efficacy of DOACs is less robust, and that in those with BMI >50 Kg/m2, therapeutic drug monitoring or switching to a vitamin K antagonist may be a reasonable approach [Citation123].
3. Conclusion
Although there are similarities in some of the pharmacokinetic properties, there is much more diversity with respect to this in terms of absorption, distribution, metabolism, and elimination. These differences, far from being purely academic, have real-world consequences and can impact on the choice of DOAC for patients. Differences in renal clearance, distribution into body tissue, interaction potential and issues such as their suitability for dosage form manipulation should influence prescribing decisions. Ultimately, it is not the case that one DOAC is ‘better’ than another, but that one might be more suited to a particular patient and their current circumstances.
4. Expert opinion
The class of DOAC drugs have revolutionized the management of atrial fibrillation (and other thrombotic diseases) by enabling patients to be treated without routine, inconvenient, and costly monitoring of INR, as it the case when warfarin is used. Data from pharmacovigilance endeavors, and interrogation of routine healthcare data in registries and observational studies have afforded a more granular picture of the specific benefits and disadvantages of particular DOAC agents than is feasible in randomized controlled trials, which are by comparison small, and recruit a restricted cohort which may not fully represent the full cohort of patients treated in clinical practice. Together the available data suggest that the DOAC class of agents are broadly similar in many respects. This observation underlies initiatives to switch patients from one agent to another when patent expiry or other market forces affect the relative cost of drugs within the class. However, such initiatives must allow for inter-individual differences and patient-centered care, particularly in patients with hepatic or renal dysfunction where the routes of elimination of particular drugs may have important consequences for the elimination of these drugs, and therefore the anticoagulant effect of treatment. Increasing research into the ‘real-world’ safety and efficacy of DOACS is likely to expand their use into new populations, as has been the case with obesity which was initially a contra-indication for all DOACs, but which is now known to be compatible with prescribing of particular agents. Whilst great care should be taken in interpreting the results of non-randomized data in making treatment recommendations, the increasing availability of data is likely to expand the benefits of DOACs to a wider population.
Article highlights
Whilst the pharmacodynamic properties of direct oral anticoagulants (DOACs) are similar among the class, there are important differences which have implications for patient care.
The routine monitoring of DOACs is generally not considered necessary for most patients.
The pharmacokinetic properties of DOACs vary considerably between agents, and this can impact the suitability for individual patients.
There is potential for numerous drug interactions with DOACs which are generally related to their metabolism and a thorough understanding of these mechanisms is necessary for safe prescribing.
Accurate and appropriate estimation of renal function using the Cockcroft-Gault formula using actual body weight is required to correctly dose DOACs.
There is increasing evidence to support the use of DOACs in patients who are obese, and this is becoming more routine in clinical practice.
Declarations of interest
PE Penson owns four shares in AstraZeneca PLC and has received honoraria and/or travel reimbursement for events sponsored by AKCEA, Amgen, AMRYT, Link Medical, Napp, Sanofi; GYHL has been a consultant and speaker for BMS/Pfizer, Boehringer Ingelheim, Anthos and Daiichi-Sankyo; no fees are received personally. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One reviewer has received honoraria/grant funding from Atricure, Boston Scientific, Cheisi-U.S.A., CSL-Behring, and Medtronic. The remaining reviewers have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Chen A, Stecker E, Warden BA. Direct oral anticoagulant use: a practical guide to common clinical challenges. J Am Heart Assoc. 2020;9. doi: 10.1161/JAHA.120.017559
- DeHaas KA. The direct oral anticoagulants apixaban, rivaroxaban, and edoxaban. AmSoci Clin Lab Sci. 2017;30:2–6. doi: 10.29074/ascls.30.1.2
- Byon W, Garonzik S, Boyd RA, et al. Apixaban: a clinical pharmacokinetic and pharmacodynamic review. Clin Pharmacokinet. 2019;58(10):1265–1279. doi: 10.1007/s40262-019-00775-z
- Bonar R, Favaloro EJ, Mohammed S, et al. The effect of the direct factor Xa inhibitors apixaban and rivaroxaban on haemostasis tests: a comprehensive assessment using in vitro and ex vivo samples. Pathology. 2016;48(1):60–71. doi: 10.1016/j.pathol.2015.11.025
- Bounameaux H, Camm AJ. Edoxaban: an update on the new oral direct factor Xa inhibitor. Drugs. 2014;74(11):1209–1231. doi: 10.1007/s40265-014-0261-1
- Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non-vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55(6):641–655. doi: 10.1007/s40262-015-0342-7
- Mueck W, Stampfuss J, Kubitza D, et al. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet. 2014;53(1):1–16. doi: 10.1007/s40262-013-0100-7
- Graff J, von Hentig N, Misselwitz F, et al. Effects of the oral, direct factor Xa inhibitor rivaroxaban on platelet-induced thrombin generation and prothrombinase activity 1. J Clin Pharmacol. 2007;47(11):1398–1407. doi: 10.1177/0091270007302952
- Stangier J, Stähle H, Rathgen K, et al. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin Pharmacokinet. 2008;47(1):47–59. doi: 10.2165/00003088-200847010-00005
- Electronic Medicines Compendium. Pradaxa 150mg hard capsules [Internet]. 2022 [cited 2023 Jan 27]. Available from: https://www.medicines.org.uk/emc/product/4703/smpc.
- Eisert WG, Hauel N, Stangier J, et al. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol. 2010;30(10):1885–1889. doi: 10.1161/ATVBAHA.110.203604
- Mainbourg S, Cucherat M, Provencher S, et al. Twice- or once-daily dosing of direct oral anticoagulants, a systematic review and meta-analysis. Thromb Res. 2021 [cited 2023 Sep 12];197:24–32.
- Jakowenko N, Nguyen S, Ruegger M, et al. Apixaban and rivaroxaban anti-Xa level utilization and associated bleeding events within an academic health system. Thromb Res. 2020;196:276–282. doi: 10.1016/j.thromres.2020.09.002
- Rose DZ, Chang JY, Yi X, et al. Direct oral anticoagulant failures in atrial fibrillation with stroke: retrospective admission analysis and novel classification system. Neurohospitalist. 2023;13(3):256–265. doi: 10.1177/19418744231161390
- Rose DZ, Burgin WS. Direct oral anticoagulant failure in stroke/transient ischaemic attack: neurologic and pharmacokinetic considerations. Eur Heart J Case Rep. 2020 [cited 2023 Sep 12];4:1–2. doi: 10.1093/ehjcr/ytaa178
- Steffel J, Verhamme P, Potpara TS, et al. The 2018 European Heart rhythm Association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur Heart J. 2018;39(16):1330–1393. doi: 10.1093/eurheartj/ehy136
- Margetić S, C¨elap I, Huzjan AL, et al. DOAC dipstick testing can reliably exclude the presence of clinically relevant DOAC concentrations in Circulation. Thromb Haemost. 2022 [cited 2023 May 23];122:1542–1548. doi: 10.1055/a-1753-2748
- Merrelaar AE, Bögl MS, Buchtele N, et al. Performance of a qualitative point-of-care strip test to detect DOAC exposure at the emergency department: a cohort-type cross-sectional diagnostic accuracy study. Thromb Haemost. 2022 [cited 2023 May 23];122(10):1723–1731. doi: 10.1055/s-0042-1750327
- Willekens G, Studt J, Mendez A, et al. A universal anti‐Xa assay for rivaroxaban, apixaban, and edoxaban measurements: method validation, diagnostic accuracy and external validation. Br J Haematol. 2021;193(6):1203–1212. doi: 10.1111/bjh.17470
- Samama M, Contant G, Spiro TE, et al. Laboratory assessment of rivaroxaban: a review. Thromb J. 2013;11(1):11. doi: 10.1186/1477-9560-11-11
- Foulon-Pinto G, Lafuente C, Jourdi G, et al. Assessment of DOAC in GEriatrics (adage study): Rivaroxaban/apixaban concentrations and thrombin generation profiles in NVAF very elderly patients. Thromb Haemost. 2023 [cited 2023 May 23];123(4):402–414. doi: 10.1055/a-1981-1763
- Douxfils J, Adcock DM, Bates SM, et al. 2021 update of the International council for standardization in haematology recommendations for laboratory measurement of direct oral anticoagulants. Thromb Haemost. 2021;121(08):1008–1020. doi: 10.1055/a-1450-8178
- Wang X, Mondal S, Wang J, et al. Effect of activated charcoal on apixaban pharmacokinetics in healthy subjects. Am J Cardiovasc Drugs. 2014;14(2):147–154. doi: 10.1007/s40256-013-0055-y
- Daiichi Sankyo Europe GmbH. Lixiana summary of product characteristics [Internet]. 2020 [cited 2022 Dec 23]. Available from: https://www.ema.europa.eu/en/documents/product-information/lixiana-epar-product-information_en.pdf.
- Electronic Medicines Compendium. Lixiana 60mg film-coated tablets [Internet]. 2022 [cited 2022 Dec 23]. Available from: https://www.medicines.org.uk/emc/product/6905/smpc#gref.
- Ollier E, Hodin S, Lanoiselée J, et al. Effect of activated charcoal on rivaroxaban complex absorption. Clin Pharmacokinet. 2017;56(7):793–801. doi: 10.1007/s40262-016-0485-1
- Alikhan R, Rayment R, Keeling D, et al. The acute management of haemorrhage, surgery and overdose in patients receiving dabigatran. Emer Med J. 2014;31(2):163–168. doi: 10.1136/emermed-2012-201976
- Boehringer Ingelheim Pharmaceuticals Inc. Pradaxa prescribing infomation [Internet]. 2011 [cited 2023 Jan 27]. Ava: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022512s007lbl.pdf.
- Boehringer Ingelheim International GmbH. Pradaxa summary of product characteristics [Internet]. 2018 [cited 2023 Jan 27]. Available from: https://www.ema.europa.eu/en/documents/product-information/pradaxa-epar-product-information_en.pdf.
- Electronic Medicines Compendium. Ondexxya 200 mg powder for solution for infusion [Internet]. 2022 [cited 2022 Dec 12]. Available from: https://www.medicines.org.uk/emc/product/10933/smpc.
- Electronic Medicines Compendium. Praxbind 2.5 g/50 mL solution for injection/infusion [Internet]. 2022 [cited 2023 Feb 3]. Available from: https://www.medicines.org.uk/emc/product/5073.
- Alexion Europe SAS. Ondexxya summary of product characterisitcs [Internet]. 2021 [cited 2023 Feb 10]. Available from: https://www.ema.europa.eu/en/documents/product-information/ondexxya-epar-product-information_en.pdf.
- Alexion Pharmaceuticals Inc. Andexxa prescribing information [Internet]. 2022 [cited 2023 Feb 10]. Available from: https://www.fda.gov/media/113279/download.
- Benz AP, Xu L, Eikelboom JW, et al. Andexanet alfa for specific anticoagulation reversal in patients with acute bleeding during treatment with edoxaban. Thromb Haemost. 2022;122:998–1005.
- National Institute of Health. Trial of andexanet alfa in ICH patients receiving an oral FXa inhibitor [Internet]. 2023 [cited 2023 Feb 10]. Available from: https://clinicaltrials.gov/ct2/show/NCT03661528.
- Müller M, Eastline J, Nagler M, et al. Application of prothrombin complex concentrate for reversal of direct oral anticoagulants in clinical practice: indications, patient characteristics and clinical outcomes compared to reversal of vitamin K antagonists. Scand J Trauma Resusc Emerg Med. 2019 [cited 2023 Sep 12];27. PMC6480883/. doi: 10.1186/s13049-019-0625-3
- Amin S, Kasischke KA, Elsayed K, et al. No time to lose: cases of anticoagulant reversal before Thrombolysis in acute ischemic stroke patients. Cureus. 2022 [cited 2023 Sep 12];14. doi: 10.7759/cureus.21406
- Byon W, Nepal S, Schuster AE, et al. Regional gastrointestinal absorption of apixaban in healthy subjects. J Clin Pharmacol. 2018;58(7):965–971. doi: 10.1002/jcph.1097
- Vakkalagadda B, Frost C, Byon W, et al. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am J Cardiovasc Drugs. 2016;16(2):119–127. doi: 10.1007/s40256-015-0157-9
- Song Y, Chang M, Suzuki A, et al. Evaluation of crushed tablet for oral administration and the Effect of food on apixaban pharmacokinetics in healthy adults. Clin Ther. 2016;38(7):1674–1685.e1. doi: 10.1016/j.clinthera.2016.05.004
- Frost C, Wang J, Nepal S, et al. Apixaban, an oral, direct factor X a inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75:476–487.
- Electronic Medicines Compendium. Eliquis 5 mg film-coated tablets [Internet]. 2022 [cited 2022 Dec 12]. Available from: https://www.medicines.org.uk/emc/product/2878/smpc.
- Bristol-Myers Squibb/Pfizer EEIG. Eliquis summary of product characteristics [Internet]. 2021 [cited 2022 Dec 23]. Available from: https://www.ema.europa.eu/en/documents/product-information/eliquis-epar-product-information_en.pdf.
- Bristol-Myers Squibb Company PI. Eliquis (apixaban) prescribing information. [Internet]. 2021 [cited 2022 Dec 12]. Available from: https://packageinserts.bms.com/pi/pi_eliquis.pdf.
- Frost C, Nepal S, Wang J, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor X a inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76:776–786.
- Song Y, Wang X, Perlstein I, et al. Relative bioavailability of apixaban solution or crushed tablet formulations administered by mouth or nasogastric tube in healthy subjects. Clin Ther. 2015;37(8):1703–1712. doi: 10.1016/j.clinthera.2015.05.497
- Upreti VV, Wang J, Barrett YC, et al. Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br J Clin Pharmacol. 2013;76:908–916. doi: 10.1111/bcp.12114
- Choi Y, Kushnir M, Billett HH. Apixaban is safe and effective in morbidly obese patients: a retrospective analysis of 390 patients with BMI ≥40. Blood. 2017;130:1105. doi: 10.1182/blood.V130.Suppl_1.1105.1105
- Cohen A, Sah J, Lee T, et al. Effectiveness and safety of apixaban vs. Warfarin in venous thromboembolism patients with obesity and morbid obesity. J Clin Med. 2021;10(2):200. doi: 10.3390/jcm10020200
- Cohen AT, Pan S, Byon W, et al. Efficacy, safety, and exposure of apixaban in patients with high body weight or obesity and venous thromboembolism: insights from AMPLIFY. Adv Ther. 2021;38(6):3003–3018. doi: 10.1007/s12325-021-01716-8
- O’Kane CP, Avalon JCO, Lacoste JL, et al. Apixaban and rivaroxaban use for atrial fibrillation in patients with obesity and BMI ≥50 kg/m 2. Pharmacother J Human Pharmacol Drug Ther. 2022;42(2):112–118. doi: 10.1002/phar.2651
- Ballerie A, Nguyen Van R, Lacut K, et al. Apixaban and rivaroxaban in obese patients treated for venous thromboembolism: drug levels and clinical outcomes. Thromb Res. 2021;208:39–44. doi: 10.1016/j.thromres.2021.10.009
- Jamieson MJ, Byon W, Dettloff RW, et al. Apixaban use in obese patients: a review of the pharmacokinetic, Interventional, and observational study data. Am J Cardiovasc Drugs. 2022;22(6):615–631. doi: 10.1007/s40256-022-00524-x
- He K, Luettgen JM, Zhang D, et al. Preclinical pharmacokinetics and pharmacodynamics of apixaban, a potent and selective factor Xa inhibitor. Eur J Drug Metab Pharmacokinet. 2011;36(3):129–139. doi: 10.1007/s13318-011-0037-x
- Frost CE, Song Y, Shenker A, et al. Effects of age and sex on the single-dose pharmacokinetics and pharmacodynamics of apixaban. Clin Pharmacokinet. 2015;54(6):651–662. doi: 10.1007/s40262-014-0228-0
- Cirincione B, Kowalski K, Nielsen J, et al. Population pharmacokinetics of apixaban in subjects with nonvalvular atrial fibrillation. CPT Pharmacometrics Syst Pharmacol. 2018;7(11):728–738. doi: 10.1002/psp4.12347
- Wang L, Zhang D, Raghavan N, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–458. doi: 10.1124/dmd.109.029694
- Zhang D, He K, Herbst JJ, et al. Characterization of Efflux Transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41(4):827–835. doi: 10.1124/dmd.112.050260
- Fernandez S, Lenoir C, Samer C, et al. Drug interactions with apixaban: a systematic review of the literature and an analysis of VigiBase, the World Health Organization database of spontaneous safety reports. Pharmacol Res Perspect. 2020;8(5):8. doi: 10.1002/prp2.647
- Candeloro M, Eikelboom JW, Chan N, et al. Carbamazepine, phenytoin, and oral anticoagulants: drug‐drug interaction and clinical events in a retrospective cohort. Res Pract Thromb Haemost. 2022;6(2):6. doi: 10.1002/rth2.12650
- Shurrab M, Jackevicius CA, Austin PC, et al. Association between concurrent use of amiodarone and DOACs and risk of bleeding in patients with atrial fibrillation. Am J Cardiol. 2023 [cited 2023 Sep 12];186:58–65.
- Lin SY, Bin LY, Ho LT, et al. Impact of amiodarone on plasma concentration of direct oral anticoagulant in patients with atrial fibrillation. J Formos Med Assoc. 2023 [cited 2023 Sep 12];122(8):776–784. doi: 10.1016/j.jfma.2023.02.012
- U.S. Food and Drug Administration. FDA’s Examples of Drugs that Interact with CYP Enzymes and Transporter Systems. 2023.
- Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81. doi: 10.1124/dmd.108.023143
- Fawzy AM, Lip GYH. Pharmacokinetics and pharmacodynamics of oral anticoagulants used in atrial fibrillation. Expert Opin Drug Metab Toxicol. 2019;15(5):381–398. doi: 10.1080/17425255.2019.1604686
- Chang M, Yu Z, Shenker A, et al. Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J Clin Pharmacol. 2016;56(5):637–645. doi: 10.1002/jcph.633
- Byon W, Sweeney K, Frost C, et al. Population pharmacokinetics, pharmacodynamics, and exploratory exposure-response analyses of apixaban in subjects treated for venous thromboembolism. CPT Pharmacometrics Syst Pharmacol. 2017;6:340–349. doi: 10.1002/psp4.12184
- Siontis KC, Zhang X, Eckard A, et al. Outcomes associated with apixaban use in patients with end-stage kidney disease and atrial fibrillation in the United States. Circulation. 2018;138(15):1519–1529. doi: 10.1161/CIRCULATIONAHA.118.035418
- Stanifer JW, Pokorney SD, Chertow GM, et al. Apixaban versus warfarin in patients with atrial fibrillation and advanced chronic kidney disease. Circulation. 2020;141(17):1384–1392. doi: 10.1161/CIRCULATIONAHA.119.044059
- Chen C, Cao Y, Zheng Y, et al. Effect of rivaroxaban or apixaban in atrial fibrillation patients with stage 4–5 chronic kidney disease or on dialysis. Cardiovasc Drugs Ther. 2021;35(2):273–281. doi: 10.1007/s10557-021-07144-8
- Cohen AT, Sah J, Dhamane AD, et al. Effectiveness and safety of apixaban versus warfarin in venous thromboembolism patients with chronic kidney disease. Thromb Haemost. 2022;122(6):926–938. doi: 10.1055/s-0041-1740254
- Gurevitz C, Giladi E, Barsheshet A, et al. Comparison of low and Full dose apixaban versus warfarin in patients with atrial fibrillation and renal dysfunction (from a national registry). Am J Cardiol. 2021;159:87–93. doi: 10.1016/j.amjcard.2021.08.022
- Knueppel P, Bang SH, Troyer C, et al. Evaluation of standard versus reduced dose apixaban for the treatment of venous thromboembolism in patients with severe renal disease (ESRD-VTE). Thromb Res. 2022;220:91–96. doi: 10.1016/j.thromres.2022.10.014
- Harrington J, Carnicelli AP, Hua K, et al. Direct oral anticoagulants versus warfarin across the spectrum of kidney function: patient-level network meta-analyses from COMBINE AF. Circulation. 2023 [cited 2023 Sep 12];147(23):1748–1757. doi: 10.1161/CIRCULATIONAHA.122.062752
- Parasrampuria DA, Kanamaru T, Connor A, et al. Evaluation of regional gastrointestinal absorption of edoxaban using the enterion capsule. J Clin Pharmacol. 2015;55(11):1286–1292. doi: 10.1002/jcph.540
- Yin OQP, Tetsuya K, Miller R. Edoxaban population pharmacokinetics and exposure–response analysis in patients with non-valvular atrial fibrillation. Eur J Clin Pharmacol. 2014;70(11):1339–1351. doi: 10.1007/s00228-014-1736-4
- Matsushima N, Lee F, Sato T, et al. Bioavailability and safety of the factor Xa inhibitor edoxaban and the effects of quinidine in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(4):358–366. doi: 10.1002/cpdd.53
- Liu L, Li X, Liu Y, et al. Bioequivalence and pharmacokinetic study of 2 edoxaban tablets in healthy Chinese subjects. Clin Pharmacol Drug Dev. 2022;11(12):1440–1446. doi: 10.1002/cpdd.1156
- Mendell J, Tachibana M, Shi M, et al. Effects of food on the pharmacokinetics of edoxaban, an oral direct factor Xa inhibitor, in healthy volunteers. J Clin Pharmacol. 2011;51(5):687–694. doi: 10.1177/0091270010370974
- Daiichi Sankyo Co. L. Edoxaban (Savaysa) prescribing information [Internet]. 2015 [cited 2022 Dec 23]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206316lbl.pdf.
- Duchin K, Duggal A, Atiee GJ, et al. An open-label crossover study of the pharmacokinetics of the 60-mg edoxaban tablet crushed and administered either by a nasogastric tube or in apple puree in healthy adults. Clin Pharmacokinet. 2018;57(2):221–228. doi: 10.1007/s40262-017-0554-0
- Moll S, Crona DJ, Martin K. Direct oral anticoagulants in extremely obese patients: OK to use? Res Pract Thromb Haemost. 2019;3(2):152–155. doi: 10.1002/rth2.12178
- Yin OQP, Miller R. Population pharmacokinetics and dose–exposure proportionality of edoxaban in healthy volunteers. Clin Drug Investig. 2014;34(10):743–752. doi: 10.1007/s40261-014-0229-7
- Martin KA, Beyer‐Westendorf J, Davidson BL, et al. Use of direct oral anticoagulants in patients with obesity for treatment and prevention of venous thromboembolism: updated communication from the ISTH SSC subcommittee on control of anticoagulation. J Thromb Haemost. 2021;19(8):1874–1882. doi: 10.1111/jth.15358
- Boriani G, Ruff CT, Kuder JF, et al. Edoxaban versus warfarin in patients with atrial fibrillation at the extremes of body weight: an analysis from the ENGAGE AF-TIMI 48 trial. Thromb Haemost. 2021;121(2):140–149. doi: 10.1055/s-0040-1716540
- Krekels EHJ, Niebecker R, Karlsson MO, et al. Population pharmacokinetics of edoxaban in patients with non-valvular atrial fibrillation in the ENGAGE AF-TIMI 48 study, a phase III clinical trial. Clin Pharmacokinet. 2016;55(9):1079–1090. doi: 10.1007/s40262-016-0378-3
- Bathala MS, Masumoto H, Oguma T, et al. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in Humans. Drug Metab Dispos. 2012;40(12):2250–2255. doi: 10.1124/dmd.112.046888
- Mikkaichi T, Yoshigae Y, Masumoto H, et al. Edoxaban Transport via P-Glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42(4):520–528. doi: 10.1124/dmd.113.054866
- Mendell J, Johnson L, Chen S. An open-label, phase 1 study to evaluate the effects of hepatic impairment on edoxaban pharmacokinetics and pharmacodynamics. J Clin Pharmacol. 2015;55(12):1395–1405. doi: 10.1002/jcph.550
- Jönsson S, Simonsson USH, Miller R, et al. Population pharmacokinetics of edoxaban and its main metabolite in a dedicated renal impairment study. J Clin Pharmacol. 2015;55(11):1268–1279. doi: 10.1002/jcph.541
- Shimizu T, Tachibana M, Kimura T, et al. Population pharmacokinetics of edoxaban in Japanese atrial fibrillation patients with severe renal impairment. Clin Pharmacol Drug Dev. 2017;6(5):484–491. doi: 10.1002/cpdd.329
- Lip GYH, Agnelli G. Edoxaban: a focused review of its clinical pharmacology. Eur Heart J. 2014;35(28):1844–1855. doi: 10.1093/eurheartj/ehu181
- Ono R, Nishimura K, Takahashi H, et al. Impact of renal function on anti-factor Xa activity concentrations with edoxaban use in patients with non-valvular atrial fibrillation. Drugs R D. 2022;22(4):281–288. doi: 10.1007/s40268-022-00403-5
- Yu HT, Yang P-S, Kim T-H, et al. Impact of renal function on outcomes with edoxaban in real-world patients with atrial fibrillation. Stroke. 2018;49(10):2421–2429. doi: 10.1161/STROKEAHA.118.021387
- Fazio G, Dentamaro I, Gambacurta R, et al. Safety of edoxaban 30 mg in elderly patients with severe renal impairment. Clin Drug Investig. 2018;38(11):1023–1030. doi: 10.1007/s40261-018-0693-6
- Buller HR, Decousus H, Grosso MA, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369:1406–1415.
- Bohula EA, Giugliano RP, Ruff CT, et al. Impact of renal function on outcomes with edoxaban in the ENGAGE AF-TIMI 48 trial. Circulation. 2016;134(1):24–36. doi: 10.1161/CIRCULATIONAHA.116.022361
- Hakeam HA, Al-Sanea N. Effect of major gastrointestinal tract surgery on the absorption and efficacy of direct acting oral anticoagulants (DOACs). J Thromb Thrombolysis. 2017;43(3):343–351. doi: 10.1007/s11239-016-1465-x
- Bratsos S. Pharmacokinetic properties of rivaroxaban in healthy human subjects. Cureus. 2019. doi: 10.7759/cureus.5484
- Stampfuss J, Kubitza D, Becka M, et al. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51(7):549–561. doi: 10.5414/CP201812
- Janssen Pharmaceuticals. XARELTO (rivaroxaban) [Internet]. 2014 [cited 2023 Jan 13]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022406s015lbl.pdf.
- Electronic Medicines Compendium. Xarelto 15mg & 20mg film-coated tablets treatment initiation pack [Internet]. 2022 [cited 2023 Jan 13]. Available from: https://www.medicines.org.uk/emc/product/8419/smpc#gref.
- Electronic Medicines Compendium. Xarelto 10 mg film-coated tablets [Internet]. 2022 [cited 2023 Jan 13]. Available from: https://www.medicines.org.uk/emc/product/6402/smpc.
- Bayer AG Xarelto summary of product characteristics. 2018 [cited 2023 Jan 13]. Available from: https://www.ema.europa.eu/en/documents/product-information/xarelto-epar-product-information_en.pdf.
- Moore KT, Krook MA, Vaidyanathan S, et al. Rivaroxaban crushed tablet suspension characteristics and relative bioavailability in healthy adults when administered orally or via nasogastric tube. Clin Pharmacol Drug Dev. 2014;3(4):321–327. doi: 10.1002/cpdd.123
- Ashton V, Mudarris L, Moore KT. The pharmacology, efficacy, and safety of rivaroxaban in obese patient populations. Am J Cardiovasc Drugs. 2021;21(3):283–297. doi: 10.1007/s40256-020-00434-w
- Kubitza D, Becka M, Zuehlsdorf M, et al. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59-7939) in healthy subjects. J Clin Pharmacol. 2007;47(2):218–226. doi: 10.1177/0091270006296058
- Kubitza D, Becka M, Roth A, et al. The influence of age and gender on the pharmacokinetics and pharmacodynamics of rivaroxaban-an oral, direct factor Xa inhibitor. J Clin Pharmacol. 2013;53:249–255. doi: 10.1002/jcph.5
- Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76(3):455–466. doi: 10.1111/bcp.12075
- Ngo LT, Yang S, Tran QT, et al. Effects of carbamazepine and phenytoin on pharmacokinetics and pharmacodynamics of rivaroxaban. Pharmaceutics. 2020;12(11):1040. doi: 10.3390/pharmaceutics12111040
- Kubitza D, Roth A, Becka M, et al. Effect of hepatic impairment on the pharmacokinetics and pharmacodynamics of a single dose of rivaroxaban, an oral, direct factor Xa inhibitor. Br J Clin Pharmacol. 2013;76(1):89–98. doi: 10.1111/bcp.12054
- Qamar A, Vaduganathan M, Greenberger NJ, et al. Oral anticoagulation in patients with liver disease. J Am Coll Cardiol. 2018;71(19):2162–2175. doi: 10.1016/j.jacc.2018.03.023
- Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct factor Xa inhibitor. Br J Clin Pharmacol. 2010;70(5):703–712. doi: 10.1111/j.1365-2125.2010.03753.x
- Ashton V, Kerolus‐Georgi S, Moore KT. The pharmacology, efficacy, and safety of rivaroxaban in renally impaired patient populations. J Clin Pharmacol. 2021;61(8):1010–1026. doi: 10.1002/jcph.1838
- Moj D, Maas H, Schaeftlein A, et al. A comprehensive whole-body physiologically based pharmacokinetic model of dabigatran etexilate, dabigatran and dabigatran glucuronide in healthy adults and renally impaired patients. Clin Pharmacokinet. 2019;58(12):1577–1593. doi: 10.1007/s40262-019-00776-y
- Grainger B, Holloway R, Merriman E, et al. Evidence of impaired dabigatran absorption following laparoscopic Roux‐en‐Y gastric bypass surgery: the Auckland regional experience (2011–2018). Br J Haematol. 2020;191: doi: 10.1111/bjh.17004
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin App Thrombosis/Hemostasis. 2009;15:9S–16S. doi: 10.1177/1076029609343004
- Stangier J, Eriksson BI, Dahl OE, et al. Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement. J Clin Pharmacol. 2005;45(5):555–565. doi: 10.1177/0091270005274550
- Stangier J, Rathgen K, Stähle H, et al. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007;64(3):292–303. doi: 10.1111/j.1365-2125.2007.02899.x
- Piran S, Traquair H, Chan N, et al. Peak plasma concentration of direct oral anticoagulants in obese patients weighing over 120 kilograms: a retrospective study. Res Pract Thromb Haemost. 2018;2(4):684–688. doi: 10.1002/rth2.12146
- Jacobs M, Ezekowitz M, Nagarakanti R, et al. Body mass index from the RE-LY trial: further evidence of the obesity paradox. Eur Heart J. 2022;43(Supplement_2):43. doi: 10.1093/eurheartj/ehac544.629
- Coates J, Bitton E, Hendje A, et al. Clinical outcomes of dabigatran use in patients with non-valvular atrial fibrillation and weight >120 kg. Thromb Res. 2021;208:176–180. doi: 10.1016/j.thromres.2021.11.007
- Steffel J, Collins R, Antz M, et al. 2021 European Heart rhythm Association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. EP Europace. 2021;23:1612–1676. doi: 10.1093/europace/euab065
- Blech S, Ebner T, Ludwig-Schwellinger E, et al. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in Humans. Drug Metab Dispos. 2008;36(2):386–399. doi: 10.1124/dmd.107.019083
- Ebner T, Wagner K, Wienen W. Dabigatran acylglucuronide, the major human metabolite of dabigatran: In Vitro formation, stability, and pharmacological activity. Drug Metab Dispos. 2010;38(9):1567–1575. doi: 10.1124/dmd.110.033696
- Stangier J, Stähle H, Rathgen K, et al. Pharmacokinetics and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor, are not affected by moderate hepatic impairment. J Clin Pharmacol. 2008;48(12):1411–1419. doi: 10.1177/0091270008324179
- Stangier J, Rathgen K, Stähle H, et al. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate. Clin Pharmacokinet. 2010;49(4):259–268. doi: 10.2165/11318170-000000000-00000
- Skripka A, Sychev D, Bochkov P, et al. Factors affecting trough plasma dabigatran concentrations in patients with atrial fibrillation and chronic kidney disease. High Blood Pressure Cardiovasc Prev. 2020;27(2):151–156. doi: 10.1007/s40292-020-00373-2
- Medicines and Healthcare Products Regulatory Agency. Prescribing medicines in renal impairment: using the appropriate estimate of renal function to avoid the risk of adverse drug reactions [Internet]. 2019 [cited 2022 Dec 12]. Available from: https://www.gov.uk/drug-safety-update/prescribing-medicines-in-renal-impairment-using-the-appropriate-estimate-of-renal-function-to-avoid-the-risk-of-adverse-drug-reactions.
- MacCallum PK, Mathur R, Hull SA, et al. Patient safety and estimation of renal function in patients prescribed new oral anticoagulants for stroke prevention in atrial fibrillation: a cross-sectional study. BMJ Open. 2013;3(9):e003343. doi: 10.1136/bmjopen-2013-003343
- NHS North West Coast Strategic Networks. Consensus statement on how to calculate the creatinine clearance (CrCl) which is necessary when assessing the dose of direct-acting oral anticoagulants (DOACs) [Internet]. 2018 [cited 2022 Dec 12]. Available from: https://www.england.nhs.uk/north/wp-content/uploads/sites/5/2019/01/consensus-statement-on-CrCl-calculation-for-DOACs-final.pdf.
- Fanikos J, Burnett AE, Mahan CE, et al. Renal function and direct oral anticoagulant treatment for venous thromboembolism. Am J Med. 2017;130(10):1137–1143. doi: 10.1016/j.amjmed.2017.06.004
- Fanikos J, Burnett AE, Mahan CE, et al. Renal function considerations for stroke prevention in atrial fibrillation. Am J Med. 2017;130(9):1015–1023. doi: 10.1016/j.amjmed.2017.04.015
- Schwartz JB. Potential Effect of Substituting Estimated Glomerular Filtration Rate for Estimated Creatinine Clearance for Dosing of Direct Oral Anticoagulants. J Am Geriatr Soc. 2016;64(10):1996–2002. doi: 10.1111/jgs.14288
- Martin K, Beyer‐Westendorf J, Davidson BL, et al. Use of the direct oral anticoagulants in obese patients: guidance from the SSC of the ISTH. J Thromb Haemost. 2016;14(6):1308–1313. doi: 10.1111/jth.13323
- Harkness W, Pipitone O, Joss J, et al. Observed Apixaban Anti-Xa Levels in Obese Patients. Ann Pharmacother. 2022 [cited 2023 Sep 12];56(11):1215–1221. doi: 10.1177/10600280221077158
- Martin KA, Lancki N, Li C, et al. DOAC compared with warfarin for VTE in patients with obesity: a retrospective cohort study conducted through the VENUS network. J Thromb Thrombolysis. 2023 [cited 2023 Sep 12];55(4):685–690. doi: 10.1007/s11239-023-02774-1
- Barakat AF, Jain S, Masri A, et al. Outcomes of direct oral anticoagulants in atrial fibrillation patients across different body mass index categories. JACC Clin Electrophysiol. 2021;7(5):649–658. doi: 10.1016/j.jacep.2021.02.002