
Abstract
The study of biological processes has been aided greatly by the development of procedures to identify the large numbers of associated genes. However, the ability to study the identified genes experimentally is often impeded by the absence of technologies to perform such functional analyses. Here, a nonaxenic plant transformation system has been developed in Gossypium hirsutum (cotton) for the study of genes associated with root functions and root–organism interactions. The plant transformation system is compatible with modern high throughput plant transformation goals and the processing of large numbers of genes intended for the study of root function in G. hirsutum.
Introduction
The ability to generate large amounts of genomics data has reached an unprecedented state, in part, to advancements in nucleotide sequencing technology (Brenner et al. Citation2000). This ability to generate the data has been met with the capability to analyze and organize the data into meaningful expression maps using a variety of computer applications (Scheideler et al. Citation2002; Harris et al. Citation2004; Trapnell et al. Citation2009, Citation2010, Citation2012; Matsye et al. Citation2011; Risso et al. Citation2014). However, the ability to generate the data is often met with the daunting reality of what genes to focus in on from a large pool of hundreds to thousands of potential candidates. This situation is especially problematic in nonmodel systems. Therefore, it is important to consider this problem when faced with the need to genetically transform an organism at some level to test gene function (Wang et al. Citation2014).
For plants, the typical genetic engineering methodologies rely on the generation of germline transformants in processes that can take long periods of time even before seed stocks are accumulated (Barton et al. Citation1983; Bevan et al. Citation1983). Furthermore, bulking up seed stocks can present additional challenges. While an advantage of germline transformation is obtaining seeds having stably integrated gene cassettes, the method typically requires chemical selection on many different types of media during the course of the experiment. Thus, the procedures require great skill because contamination can occur at any step. It is also possible that the media can alter the growth or metabolic activities of the plant. Last, methods to accomplish the transformation in what may be considered nonmodel systems might not even exist.
It is not always necessary to generate germline transformants if only a specific organ type is the object of study. For example, the ability to generate genetically transformed roots was demonstrated by Tepfer (Citation1984), opening up the possibility to perform truly functional root genomics analyses. While the procedure was initially limited by the requirement for tissue culture, the subsequent development of plasmid vectors containing the enhanced green fluorescent protein (eGFP) reporter made possible the development of root cultures under nonaxenic conditions (Haseloff et al. Citation1997; Collier et al. Citation2005). While very useful, an inherent problem was that the early plasmid systems were not compatible with high throughput genomics analysis objectives because they lacked the capability to easily engineer genes into the vector. The development of the Gateway® system made that goal a reality (Curtis & Grossniklaus Citation2003). Later experiments adapted the Gateway® system, so that it could be used in agriculturally relevant plants (Klink et al. Citation2009; Matsye et al. Citation2012). Eventually, high throughput root genomics was demonstrated which ushered in a new era of functional root studies (Matthews et al. Citation2013). However, these plant systems are agricultural models.
Many agriculturally relevant plants that are not model systems, but are very important globally, still lack the requisite technology to easily study large numbers of genes. When the goal is to solve an urgent or emerging problem in a specific agriculturally relevant plant, it is imperative that a genetic transformation system be developed in that system. One such agriculturally relevant plant, Gossypium hirsutum, has several organisms (i.e. Meloidogyne incognita, Rotylenchulus reniformis and Fusarium oxysporum) whose interactions result in extensive agronomic loss. Developing a simplistic plant transformation strategy in G. hirsutum was the goal of this study in order to address that issue.
In this study, a Gateway®-based plasmid system is presented that is designed for its use in high throughput root genomics in G. hirsutum under nonaxenic conditions. The system is shown to be capable of expressing an eGFP reporter and a Glycine max NONEXPRESSOR OF PATHOGENESIS RELATED 1 (Gm-NPR1) transgene (Pant, Matsye, et al. Citation2014). A comparative analysis, including the efficiency of transformation in G. hirsutum, is presented showing the rate by which roots useful for experimentation are generated. The system presented here differs from other hairy root-based systems developed in G. hirsutum in that, by using eGFP as a reporter system, the experiments can be performed under nonaxenic conditions throughout the study (Triplett et al. Citation2008; Kim et al. Citation2011; Matsye et al. Citation2012; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). Furthermore, the platform is Gateway® compatible which simplifies the isolation and study of hundreds to thousands of genes identified in bioinformatics analyses (Matsye et al. Citation2012; Matthews et al. Citation2013; Pant, Matsye, et al. Citation2014; Klink & Thibaudeau Citation2014). This facet of the genetic engineering platform greatly lessens the amount of technical expertise that is required for successful plant transformation. Notably, this work is the compliment to Klink and Thibaudeau (Citation2014) for the analysis of G. hirsutum root interaction research.
Methods
pRAP15 plasmid
The pRAP15 vector was originally developed for Agrobacterium rhizogenes-mediated root genetic engineering experiments in G. max (Matsye et al. Citation2012; Matthews et al. Citation2013, Citation2014; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). The sequence of the pRAP15 vector used in the study was determined prior to the analysis (Eurofins MWG Operon, Huntsville, Alabama; Matsye et al. Citation2012). From that sequence information, the vector map for pRAP15 was made by taking its confirmed sequence information and entering it into PlasMapper Version 2.0 (Dong et al. Citation2004). The pRAP15 vector has a single Gateway® (Invitrogen®)-compatible attR1-ccdB-attR2 cassette. The attR1 and attR2 sites are LR bacteriophage λ-derived recombination sites, allowing for the integration of the gene of interest (GOI) by use of the LR clonase enzyme which functions as a recombinase (Invitrogen). The ccdB gene encodes a toxin, functioning as a selective agent for Escherichia coli selection (Bernard & Couturier Citation1991). The Gateway®-compatible attR1-ccdB-attR2 cassette was engineered into the pRAP15 vector between SpeI (5′) and XbaI (3′) sites. During the LR reaction, the ccdB gene is replaced in the pRAP15 by the GOI during the recombination reaction. Thus, the attR cassette is interrupted by the ccdB selectable marker gene that acts as an intron. Critical to this process functioning properly is the incorporation of a 5′→3′ CACC nucleotide sequence at the 5′ end of the forward PCR primer sequence prior to the PCR steps that generate the GOI amplicon. For cloning full length genes, this means that the CACC nucleotide sequence would precede the start codon (ATG). The LR clonase reaction engineers genes into pRAP15 and in the process releases the intron, resulting in a gene positioned in the correct orientation for overexpression. The ccdB gene provides a mechanism for selecting E. coli containing pRAP15 engineered with the GOI, eliminating E. coli containing nonengineered in pRAP15. The expression of the cassette is driven by the figwort mosaic virus sub-genomic transcript (FMV-sgt) promoter (Bhattacharyya et al. Citation2002). By using FMV-sgt as the promoter, the cassette is designed to drive the overexpression of full length genes and maintain high expression throughout plant–organism interactions (Klink et al. Citation2008, Citation2009; Matsye et al. Citation2012; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). The pRAP15 vector was engineered to have the tetracycline resistance gene for bacterial selection, found to be important for work with A. rhizogenes (Klink et al. Citation2008). The tetracycline resistance gene was engineered into a BstEII site that lies outside the left and right borders. The pRAP15 vector contains an eGFP gene driven by the rolD root promoter (White et al. Citation1985; Elmayan & Tepfer Citation1995; Haseloff et al. Citation1997). The eGFP gene product is a visual beacon for screening transformed roots in nonaxenic conditions (Collier et al. Citation2005). The inserted gene cassette is terminated by the cauliflower mosaic virus 35S terminator.
Gene cloning
The G. hirsutum root transformation method presented here was performed according to our published procedures using the pRAP15 vector with modifications (Matsye et al. Citation2012; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). The PCR primers used in the cloning experiments are provided in . RNA was extracted from G. hirsutum roots using the UltraClean® Plant RNA Isolation Kit (Mo Bio Laboratories®, Inc., Carlsbad, CA) and treated with DNase I to remove genomic DNA. The RNA was reversed transcribed into complimentary DNA (cDNA) using SuperScript First Strand Synthesis System for RT-PCR (Invitrogen®) with oligo d(T) as the primer according to protocol (Invitrogen®). PCR reactions containing no template and reactions using RNA processed in parallel, but with no Superscript® reverse transcriptase, also served as controls and produced no amplicon. These precautions proved no contaminating genomic DNA existed in the cDNA. Cloning the GOI and quantitative PCR (qPCR) was performed using cDNA synthesized from RNA (Pant, Matsye, et al. Citation2014). The GOI is Gm-NPR1-2 (Glyma09g02430), studied previously (Pant, Matsye, et al. Citation2014). To ensure the ligation of Gm-NPR1-2 into the pRAP15 occurs in the proper orientation, a 5′→3′ CACC nucleotide sequence was incorporated into the forward primer immediately upstream of the ATG start codon. PCR amplification of targeted genes was done using high fidelity Platinum® taq according to protocol (Invitrogen®). The cDNA used in the PCR reaction was dissociated for 10 min at 96°C, followed by PCR cycling and temperatures set for denaturation for 30 sec at 96°C, annealing for 60 sec at 55°C, and extension for 30 sec at 72°C (Matsye et al. Citation2012). Amplicons produced during the PCR reaction and representing Gm-NPR1-2 were gel purified in 1.0% agarose using the Qiagen® gel purification kit (Qiagen®, Valencia, CA, USA), ligated into the directional pENTR/D-TOPO® (Invitrogen®, Carlsbad, CA, USA) vector. The reaction contents were then mixed with chemically competent E. coli strain One Shot TOP10 (Invitrogen®) according to the manufacturer's instructions. Chemical selection of E. coli was performed on LB-kanamycin (50 μ g/ml) plates according to protocol (Invitrogen®; Pant, Matsye, et al. Citation2014). A representative colony was subsequently grown in 3 ml of LB-kanamycin (50 μ g/ml) overnight. The bacteria were pelleted and a plasmid prep (Qiagen®) was done. The presence of Gm-NPR1-2 was confirmed by PCR, and the amplicon was then sequenced (MWG Operon). After confirmation of the presence of Gm-NPR1-2, the pENTR/D-TOPO®-GOI-containing plasmid was used in an LR reaction according to the manufacturer's instructions (Invitrogen®). This procedure is designed to shuttle the Gm-NPR1-2 from the pENTR/D-TOPO® entry vector to the pRAP15 destination vector (Matsye et al. Citation2012). The resulting reaction contents were then transformed into chemically competent E. coli strain One Shot TOP10 according to the manufacturer's instructions (Invitrogen®). Chemical selection was on LB-tetracycline (5 μ g/ml) plates according to protocol (Invitrogen®). A representative colony was subsequently grown in 3 ml of LB-tetracycline (5 μ g/ml) overnight. The bacteria were pelleted, followed by a plasmid prep (Qiagen®). The presence of Gm-NPR1-2 was confirmed by PCR. The Gm-NPR1-2-engineered pRAP15 vector is then transformed into chemically competent A. rhizogenes strain 15834 (A. rhizogenes) using the freeze-thaw method on LB-tetracycline (5 μ g/ml; Hofgen & Willmitzer Citation1988; Triplett et al. Citation2008; Kim et al. Citation2011; Matsye et al. Citation2012). A PCR reaction using pRAP15 primers that amplify the 717 bp eGFP and the 690 bp A. rhizogenes root inducing (Ri) plasmid (EU186381) VirG gene (VirG) confirms that the A. rhizogenes contains both plasmids prior to plant transformation. The pRAP15 vector containing the 1773 bp NPR1-2 was confirmed (Pant, Matsye, et al. Citation2014).
Table 1. PCR primers. The primers used are A. rhizogenes VirG (Ar-VirG), G. hirsutum S21 (Gh-S21), G. max NPR1-2 (Gm-NPR1-2), and eGFP.
A. rhizogenes-mediated nonaxenic transformation
A modified version of the nonaxenic hairy root plant transformation procedure was developed here for use in G. hirsutum (Tepfer Citation1984; Klink et al. Citation2008, Citation2009; Matsye et al. Citation2012; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). Seeds of G. hirsutum were planted in prewetted sterilized sand, germinated, and grown for 14 days at ambient greenhouse temperatures (~26–29°C). The plants were cut at the hypocotyl with a freshly unwrapped, clean, and sterile scalpel in a Petri plate containing a small pool of the pRAP15-transformed A. rhizogenes. The procedure ensured that infection by A. rhizogenes occurred at the exact moment the plant was injured. The rootless plants (~25 plants per beaker) were placed in 400 ml beakers containing A. rhizogenes cultured in Murashige and Skoog (MS) media (Murashige & Skoog Citation1962), including vitamins (Duchefa Biochemie, the Netherlands) and 3.0% sucrose, pH 5.7 (MS media). Only the bottom 0.5 cm of the hypocotyl end of the plantlet was submerged in the MS media (please see Results section). No chemical selection was performed during the cocultivation. G. hirsutum underwent vacuum infiltration for 30 min. The vacuum then was released slowly, allowing the A. rhizogenes suspension to infiltrate the tissue. Cocultivation was performed overnight in MS media in the 400 ml beaker on a rotary shaker at 28°C without chemical selection. After an overnight cocultivation, the cut ends of G. hirsutum were placed individually 2–4 cm deep into fresh, coarse, nonsterilized, vermiculite (Palmetto Vermiculite Co., Woodruff, SC, USA) in 50-cell flat. The 50-cell flat containing the G. hirsutum plantlets was placed in a covered 32 quart Sterlite® Clearview Latch Box®. The plants then were covered and grown at a distance of 20 cm from standard fluorescent cool white 4100K, 32 watt bulbs emitting 2800 lumens (Sylvania®; Danvers, MA) for 14 days at ambient lab temperatures (~22°C). The plants then were uncovered and transferred to the greenhouse. Genetically engineered roots were identified by carefully dislodging the plant and root ball from its pot and inspecting them for the expression of eGFP using the Dark Reader Spot Lamp (Clare Chemical Research; Dolores, CO, USA). The remaining vermiculite was removed from these plants by washing the root ball in distilled, deionized water in a 1000 ml plastic beaker. The easily identified untransformed roots, evident by lacking fluorescence, were excised from the plants. The resulting plants are chimeras (having transformed roots and untransformed aerial stocks). The chimeras were planted in a sterilized 50-50 mixture of a Freestone fine sandy loam (46.25% sand, 46.50% silt, and 7.25% clay) and a sandy (93.00% sand, 5.75% silt, and 1.25% clay) soil and allowed to recover for two weeks. The nature of the hairy root system is that each transgenic root system functions as an independent transformant line (Tepfer Citation1984; Pant, Matsye, et al. Citation2014).
Quantitative real-time PCR (qPCR)
The qPCR experiments in G. hirsutum were performed according to Pant, Matsye, et al. (2Citation014). The qPCR primers used in the experiments are provided in . The ribosomal s21 primers were designed from the highly evolutionarily conserved Gossypium raimondii S21 (Gorai.009G233700.1) and served as a control. The transgene studied was Gm-NPR1-2 (Pant, Matsye, et al. Citation2014). The eGFP serves as the visual reporter (Haseloff et al. Citation1997). The G. hirsutum cDNA was made from RNA that was isolated from G. hirsutism roots. This cDNA was used as a template for qPCR experiments. The qPCR experiments used Taqman® 6-carboxyfluorescein (6-FAM) probes and Black Hole Quencher (BHQ1; MWG Operon). The qPCR differential expression tests were performed according to Livak and Schmittgen (Citation2001). The qPCR reaction conditions were prepared according to Pant, Matsye, et al. (2Citation014) and included a 20 μl Taqman Gene Expression Master Mix (Applied Biosystems, Foster City, CA), 0.9 µl of μM forward primer, 0.9 µl of 100 μM reverse primer, 2 µl of 2.5 µM 6-FAM (MWG Operon®) probe, and 9.0 µl of template DNA. The qPCR reactions were performed on an ABI 7300 (Applied Biosystems®). The qPCR conditions included a preincubation of 50°C for 2 min, followed by 95°C for 10 min. This step was followed by alternating 95°C for 15 sec followed by 60°C for 1 min for 40 cycles.
Results
Quality control measures for effective A. rhizogenes transformation
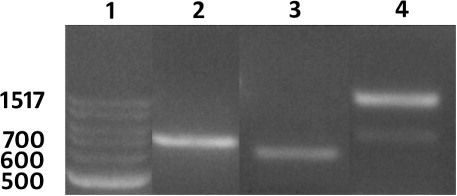
Prior to the plant transformation experiments, the sequence of the engineered pRAP15 transformation vector was determined and mapped (). The sequence information allowed for the accurate design of PCR primers that would be used later in quality control procedures during the various stages of the genetic engineering experiment. The sequenced pRAP15 plasmid was then transformed into A. rhizogenes with subsequent plasmid preps being performed on liquid cultures generated from independent colonies selected on tetracycline plates. The same procedure was used in experiments using pRAP15 that was genetically engineered to contain the Gm-NPR1-2 gene (Pant, Matsye, et al. Citation2014). PCR reactions using primers designed against two different regions of the genetically engineered pRAP15, including eGFP and a Gm-NPR1-2, show that A. rhizogenes is capable of being selected for by using the pRAP15 plasmid in freeze-thaw transformation procedures followed by selection on tetracycline (). Furthermore, the A. rhizogenes also are shown to contain the Ri plasmid as demonstrated using primers derived from the A. rhizogenes VirG (). These experiments indicate that the A. rhizogenes are competent to be used in experiments designed to genetically engineer G. hirsutum with a transgene under nonaxenic conditions because they harbor the eGFP visual reporter, the Gm-NPR1-2 transgene, and the Ri plasmid which is essential for root induction.
![Figure 1. pRAP15 vector. Legend: open reading frame (ORF), pink; ORF1, homologous to partitioning protein A (ParA); ORF2, no significantly homologous leader sequence 5′ fused to the Reporter gene (enhanced green fluorescent protein [eGFP]), green. ORF3, homologous to resolvase; ORF4, homologous to replicase A (RepA); ORF5, homologous to tetracycline resistance gene (TetR); ORF6, homologous to aminoglycoside adenyltransferase. Origin of replication (pBR322), black. Other gene (ccdB, attR1, attR2), magenta. Selectable marker (chloramphenicol [CAT]), orange. Terminator (cauliflower mosaic virus [CaMV] 35S), brown. Blue lettering, unique restriction sites.](/cms/asset/2ed2dfa6-cdc6-45a6-8e45-c20e46dec7f4/tjpi_a_1005181_f0001_c.jpg)
Quality control measures for efficient G. hirsutum transformation
The G. hirsutum transformation procedure is outlined here with many accompanying details provided in the Methods section (). The transformation procedure is initiated by pelleting down an A. rhizogenes culture, containing the engineered pRAP15-Gm-NPR1-2, grown in LB media. The bacterial pellet is washed with MS media, followed by resuspension in MS media (see Methods section). The G. hirsutum plants are removed from the sand in which they have been grown for 14 days. Subsequently, only the hypocotyl end of the plantlet is submerged in a Petri plate containing the transformed A. rhizogenes culture (). To provide an entry site for A. rhizogenes at the exact moment of plant injury, the plantlet is cut at the hypocotyl which removes the root (). The hypocotyl end of the G. hirsutum plants (now lacking the roots) are placed into a beaker containing A. rhizogenes (). The beaker, containing the plants, is placed in a vacuum chamber and infiltrated for 30 min. The vacuum is slowly released over a period of a couple of minutes and then covered with clear wrap which maintains humidity as the plants are rotated on a shaker overnight. The next day, the plants are removed from the beaker and planted into coarse vermiculite. The hypocotyl end of the plant is placed anywhere between 2 cm and 4 cm deep in the coarse vermiculite. The 50-cell flat is placed into an opaque humidity chamber, covered with its accompanying opaque lid and placed under fluorescent lights for 28 days (; ). Subsequently, the plants are brought to the greenhouse, removed from the humidity chamber, and grown for an additional 30 days on greenhouse benches to allow root growth. G. hirsutum transformation is demonstrated by both positive expression of the eGFP reporter, confirmed by a PCR reaction designed to amplify a fragment of the eGFP visual reporter gene (). To provide molecular proof of genetic transformation using the Gm-NPR1-2 transgene, qPCR experiments were done by isolating RNA from the G. hirsutum roots for cDNA template synthesis. These qPCR experiments demonstrated the presence of the Gm-NPR1-2 transgene only in G. hirsutum roots that underwent the plant genetic engineering procedure that included A. rhizogenes harboring the pRAP15 plasmid containing the Gm-NPR1-2 gene (). Thus, the qPCR experiments are monitoring presence against absence of the Gm=NPR1-2 transcript.

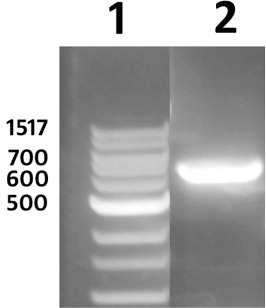
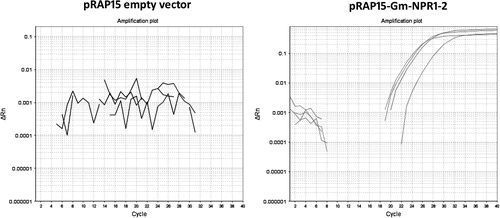
Table 2. A time line for production of composite G. max and G. hirsutum plants with transformed roots.
Specificity of the transgene analysis
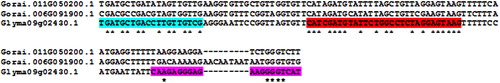
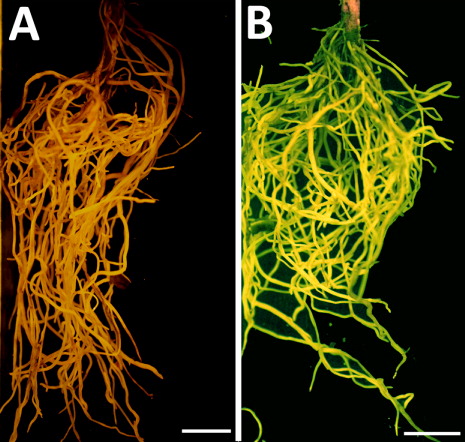
A genomics analysis employing the power of the G. raimondii genome was performed to determine the specificity of the qPCR analysis. The sequenced G. raimondii genome has two NPR1 homologs, Gorai.011G050200.1 and Gorai.006G091900.1. Assuming G. hirsutum has an NPR1 gene that would be similar to G. raimondii, the analysis determined that the G. raimondii NPR1 sequences differed from Gm-NPR1-2 in the region where the qPCR primers were designed (). This analysis indicated that the G. hirsutum NPR1 sequences, if present, would also likely be substantially different from the Gm-NPR1-2 transgene. After confirmation of the presence of the Gm-NPR1-2 transgene only in genetically engineered G. hirsutum roots that are also expressing the eGFP reporter (), the plantlets with genetically engineered roots were then planted into pots. After two additional weeks of acclimation, the plants are ready for experiments involving root pathogens (). To demonstrate this, the plants were removed to show the root fluorescence in the genetically engineered G. hirsutum ().

Relative rate of root regeneration
The procedure used here is very similar to a method employed to perform functional genomics analyses in G. max (Matsye et al. Citation2012; Pant, Matsye, et al. Citation2014). Since most of the steps use the same method and plasmids but differ in the plant species used, an analysis was done to compare the duration of the plant transformation steps for G. hirsutum and G. max (). The germination frequency (Step 1) was the same (~98%; ). The comparison shows that G. hirsutum takes longer than G. max (Step 2) to obtain plants that are ready for the plant genetic engineering procedure (7 vs. 14 days; ). The survival rate (~98%) after the plant transformation procedure (Step 3) was comparable (). Furthermore, the recovery time under fluorescent lights (Step 4) after the genetic engineering procedure (28 vs. 14 days) is nearly twice as long for G. hirsutum (). The recovery time in the greenhouse (Step 5; 30 vs. 14 days) is nearly twice as long for G. hirsutum (). However, the transformation efficiency (Step 6) for G. hirsutum whereby at least one genetically engineered root grows from a plant is highly efficient at each step (>90%; ). Subsequently, the time after which the G. hirsutum are ready for experiments (Step 7) is the same between G. hirsutum and G. max ().
Discussion
A major limitation to do functional genomics in many nonmodel plants is the inability to test gene function through the use of genetic engineering (Wahby et al. Citation2013). Since the original demonstration of the hairy root platform, a number of modifications have been made to simplify the procedure (Tepfer Citation1984; Collier et al. Citation2005; Klink et al. Citation2009). Based off of those technological improvements, an experimental platform is presented here that allows scientists to genetically engineer roots of the nonmodel G. hirsutum under nonaxenic conditions. The procedure is easy to perform, rapid, inexpensive, lacks tissue culture steps and can be used to process thousands of plants simultaneously. Much of the work presented here was supported by high school and undergraduate students. Thus, extensive training or scientific background is not required.
Platform details
In addition to its capability of being used under nonaxenic conditions, the platform presented here has the added feature of being compatible with the Gateway® technology (Klink et al. Citation2008; Matsye et al. Citation2012). The compatibility with the Gateway® technology allows for large numbers of genes to be directionally cloned first into the entry vector and then shuttled into pRAP15 overexpression vector. At that point, the gene can be expressed at high levels for biological investigation (Matsye et al. Citation2012; Matthews et al. Citation2013; Pant, Matsye, et al. Citation2014; Pant, Krishnavajhala, et al. Citation2014). Furthermore, as shown here, it is possible to isolate genes from one organism and express the gene in a heterologous system for experimental study. While complicating qPCR analyses, we have been able to compare roots that would normally lack the expression of a transgene completely to roots expressing the Gm-NPR1-2 transgene (absence/presence). This approach is similar to experiments that isolated genes from Arabidopsis thaliana and expressed them in G. max (Matthews et al. Citation2014). Thus, it is possible to engineer in and test traits that are not limited to G. hirsutum genes. In addition to overexpression, other vectors in the Gateway®-compatible pRAP series of plasmids are also capable of post-transcriptionally suppressing gene expression through RNA interference (RNAi; Klink et al. Citation2009).
Efficiency of plant transformation procedure
Prior to these studies, Triplett et al. (Citation2008) analyzed the capability that G. hirsutum has with regard to the hairy root platform. Triplett et al. (Citation2008) provided an important base line for the study presented here. While differences exist between the outcomes in the two analyses, each study is designed to accomplish a different goal. The results presented here show that 91% of the G. hirsutum plantlets that had gone through the transformation procedure had roots that were genetically engineered. This means that at least one genetically engineered root, as determined by the presence of the eGFP reporter, was observed on 91% of roots examined from the G. hirsutum plantlets. Therefore, with regard to efficiency, the level of transformation presented here is slightly higher than a tissue culture method presented by Triplett et al. (Citation2008; 3.8–56.2%). We suspect that many factors could be the cause for this difference, but the results demonstrate the robustness of the procedure presented here. Furthermore, depending on the type of research being done, the engineered G. hirsutum are ready for experimentation in as little as eight weeks. However, if large root systems are not required for the desired experiments, this time frame could be reduced by at least one week. Thus, the development of transgenic roots that are useful for experiments occurs nearly as rapidly as the system presented by Triplett et al. (Citation2008) with the added advantage that whole plants transformed root attached to a nonengineered aerial stock are generated without the need for tissue culture.
Compatibility for studies involving root-dwelling organisms
It is anticipated that the nonaxenic root transformation approach presented here would be useful for studies involving plant–organism interactions or other aspects of root biology in G. hirsutum (Klink & Thibaudeau Citation2014). Prior studies in other agriculturally relevant plant systems have already shown that this system is capable of producing tens of thousands of plants that can be easily maintained at one time for experimental study (Matthews et al. Citation2013; Pant, Matsye, et al. Citation2014). The same capability is expected to be true for G. hirsutum. In the system presented here, the G. hirsutum plants can be grown indefinitely under normal or predefined greenhouse conditions. Thus, long-term experiments can be performed which is compatible with their perennial habit. Furthermore, this long-term capability to culture the plants in the greenhouse is advantageous over tissue culture-reliant methods. The advantage is that the system presented here does not require the laborious and technically challenging step of changing tissue culture solutions at specific time points like what is required when performing the process under axenic conditions. Thus, analyzing the infection and reinfection capability of pathogens or symbionts over multiple generations is possible using the G. hirsutum hairy root system presented here. For certain reasons, another advantage is that germline transformants are not generated. Thus, the escape of the genes to wild relatives of these plants through pollen or other means is not possible or would be highly unlikely. In contrast, methods in other plant systems have been able to generate germline transformants from callus derived from tissues transformed through the hairy root method (Zdravković-Korać et al. Citation2004; Crane et al. Citation2006). Thus, the generation of germline transformed G. hirsutism directly from hairy roots is possible.
Acknowledgments
The authors thank Dr Robert L. Nichols and Cotton Incorporated for support. The research is further supported jointly between the College of Arts and Sciences (DBS) and the Mississippi Agricultural and Forestry Experimental Station (MAFES). The authors thank George Hopper, Reuben Moore, and Wes Burger (MAFES) whose support has made the research possible. Greenhouse space to support the research was provided by the Department of Biochemistry, Molecular Biology, Entomology and Plant Pathology (BMEP) and the Department of Plant and Soil Sciences (PSS) at Mississippi State University. The work of Anna Gaudin, Starkville High School, is acknowledged. Additional support was provided by Ashley Dowdy (DBS). A number of undergraduate students aided in this study, including Tineka Burkhead, Alison Antee, Annedrea McMillan, Hannah Miller, Erin Ball, Austin Martindale, Henry Pittman, Erin Curran, Courtney Gagliano, and Ashlee Gutierrez (DBS). The authors thank Dr Giselle Thibaudeau and Amanda Lawrence, Institute for Imaging and Analytical Technologies, Mississippi State University, for imaging expertise and technical suggestions during the course of research serving as the prerequisite to this work. VPK acknowledges Dr Ben Matthews and Dr Perry Cregan at the USDA-ARS (Beltsville, MD) for support.
Additional information
Funding
References
- Barton KA, Binns AN, Matzke AJ, Chilton M-D. 1983. Regeneration of intact tobacco plants containing full length copies of genetically engineered T-DNA, and transmission of T-DNA to R1 progeny. Cell. 32:1033–1043.
- Bernard P, Couturier M. 1991. The 41 Carboxy-terminal residues of the Mini-F plasmid ccdA protein are sufficient to antagonize the killer activity of the CcdB protein. Mol Gen Genet. 226:297–304.
- Bevan MW, Flavell RB, Chilton M-D. 1983. A chimeric antibiotic resistance gene as a selectable marker for plant cell transformation. Nature. 304:184–187.
- Bhattacharyya S, Dey N, Maiti IB. 2002. Analysis of cis-sequence of subgenomic transcript promoter from the Figwort mosaic virus and comparison of promoter activity with the cauliflower mosaic virus promoters in monocot and dicot cells. Virus Res. 90:47–62.
- Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, et al. 2000. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 18:630–634.
- Collier R, Fuchs B, Walter N, Kevin Lutke W, Taylor CG. 2005. Ex vitro composite plants: an inexpensive, rapid method for root biology. Plant J. 43:449–457.
- Crane C, Wright E, Dixon RA, Wang ZY. 2006. Transgenic Medicago truncatula plants obtained from Agrobacterium tumefaciens-transformed roots and Agrobacterium rhizogenes-transformed hairy roots. Planta. 223:1344–1354.
- Curtis MD, Grossniklaus U. 2003. A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 133:462–469.
- Dong X, Stothard P, Forsythe IJ, Wishart DS. 2004. PlasMapper: a web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 32(Web Server issue):W660–W664.
- Elmayan T, Tepfer M. 1995. Evaluation in tobacco of the organ specificity and strength of the rolD promoter, domain A of the 35S promoter and the 35S2 promoter. Transgenic Res. 4:388–396.
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32:D258–D261.
- Haseloff J, Siemering KR, Prasher DC, Hodge S. 1997. Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci USA. 94:2122–2127.
- Hofgen R, Willmitzer L. 1988. Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res. 16:9877
- Kim HJ, Murai N, Fang DD, Triplett BA. 2011. Functional analysis of Gossypium hirsutum cellulose synthase catalytic subunit 4 promoter in transgenic Arabidopsis and cotton tissues. Plant Sci. 180:323–332.
- Klink VP, Kim K-H, Martins VE, MacDonald MH, Beard HS, Alkharouf NW, Lee S-K, Park S-C, Matthews BF. 2009. A correlation between host-mediated expression of parasite genes as tandem inverted repeats and abrogation of the formation of female Heterodera glycines cysts during infection of Glycine max. Planta. 230:53–71.
- Klink VP, MacDonald MH, Martins VE, Park S-C, Kim K-H, Baek S-H, Matthews BF. 2008. MiniMax, a new diminutive Glycine max variety, with a rapid life cycle, embryogenic potential and transformation capabilities. Plant Cell Tiss Org Cult. 92:183–195.
- Klink VP, Thibaudeau G. 2014. Laser microdissection of ultrathin sections for studying plant-pathogen developmental processes at single cell resolution. J Plant Interact. 9:610–617.
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
- Matsye PD, Kumar R, Hosseini P, Jones CM, Tremblay A, Alkharouf NW, Matthews BF, Klink VP. 2011. Mapping cell fate decisions that occur during soybean defense responses. Plant Mol Biol. 77:513–528.
- Matsye PD, Lawrence GW, Youssef RM, Kim K-H, Matthews BF, Lawrence KS, Klink VP. 2012. The expression of a naturally occurring, truncated allele of an α-SNAP gene suppresses plant parasitic nematode infection. Plant Mol Biol. 80:131–155.
- Matthews BF, Beard H, Brewer E, Kabir S, MacDonald MH, Youssef RM. 2014. Arabidopsis genes, AtNPR1, AtTGA2 and AtPR-5, confer partial resistance to soybean cyst nematode (Heterodera glycines) when overexpressed in transgenic soybean roots. BMC Plant Biol. 14:96.
- Matthews BF, Beard H, MacDonald MH, Kabir S, Youssef RM, Hosseini P, Brewer E. 2013. Engineered resistance and hypersusceptibility through functional metabolic studies of 100 genes in soybean to its major pathogen, the soybean cyst nematode. Planta. 237:1337–1357.
- Murashige T, Skoog F. 1962. A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plantarum. 15:473–497.
- Pant SR, Krishnavajhala A, McNeece BT, Lawrence GW, Klink VP. 2014. The syntaxin 31-induced gene, LESION SIMULATING DISEASE1 (LSD1), functions in Glycine max defense to the root parasite Heterodera glycines. Plant Signal Behav.
- Pant SR, Matsye PD, McNeece BT, Sharma K, Krishnavajhala A, Lawrence GW, Klink VP. 2014. Syntaxin 31 functions in Glycine max resistance to the plant parasitic nematode Heterodera glycines. Plant Mol Biol. 85:107–121.
- Risso D, Ngai J, Speed TP, Dudoit S. 2014. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 32:896–902.
- Scheideler M, Schlaich NL, Fellenberg K, Beissbarth T, Hauser NC, Vingron M, Slusarenko AJ, Hoheisel JD. 2002. Monitoring the switch from housekeeping to pathogen defense metabolism in Arabidopsis thaliana using cDNA arrays. J Biol Chem. 277:10555–10561.
- Tepfer D. 1984. Transformation of several species of higher plants by Agrobacterium rhizogenes: sexual transmission of the transformed genotype and phenotype. Cell. 37:959–967.
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25:1105–1111.
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578.
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28:511–515.
- Triplett BA, Moss SC, Bland JM, Dowd MK. 2008. Induction of hairy root cultures from Gossypium hirsutum and Gossypium barbadense to produce gossypol and related compounds. In Vitro Cell Dev Biol Plant. 44:508–517.
- Wahby I, Caba JM, Ligero F. 2013. Agrobacterium infection of hemp (Cannabis sativa L.): establishment of hairy root cultures. J Plant Interact. 8:312–320.
- Wang X, Tang Q, Dong L, Dong Y, Su Y, Jia S, Wang Z. 2014. Construction of a standard reference plasmid containing seven target genes for the detection of transgenic cotton. Plasmid. 74:39–44.
- White FF, Taylor BH, Huffman GA, Gordon MP, Nester EW. 1985. Molecular and genetic analysis of the transferred DNA regions of the root-inducing plasmid of Agrobacterium rhizogenes. J Bacteriol. 164:33–44.
- Zdravković-Korać S, Muhovski Y, Druart P, Calić D, Radojević L. 2004. Agrobacterium rhizogenes-mediated DNA transfer to Aesculus hippocastanum L. and the regeneration of transformed plants. Plant Cell Rep. 22:698–704.