
ABSTRACT
Introduction
Hypocholesterolemia results from genetic – both monogenic and polygenic – and non-genetic causes and can sometimes be a source of clinical concern. We review etiologies and sequelae of hypocholesterolemia and therapeutics inspired from genetic hypocholesterolemia.
Areas covered
Monogenic hypocholesterolemia disorders caused by the complete absence of apolipoprotein (apo) B-containing lipoproteins (abetalipoproteinemia and homozygous hypobetalipoproteinemia) or an isolated absence of apo B-48 lipoproteinemia (chylomicron retention disease) lead to clinical sequelae. These include gastrointestinal disturbances and severe vitamin deficiencies that affect multiple body systems, i.e. neurological, musculoskeletal, ophthalmological, and hematological. Monogenic hypocholesterolemia disorders with reduced but not absent levels of apo B lipoproteins have a milder clinical presentation and patients are protected against atherosclerotic cardiovascular disease. Patients with heterozygous hypobetalipoproteinemia have somewhat increased risk of hepatic disease, while patients with PCSK9 deficiency, ANGPTL3 deficiency, and polygenic hypocholesterolemia typically have anunremarkable clinical presentation.
Expert opinion
In patients with severe monogenic hypocholesterolemia, early initiation of high-dose vitamin therapy and a low-fat diet are essential for optimal prognosis. The molecular basis of monogenic hypocholesterolemia has inspired novel therapeutics to help patients with the opposite phenotype – i.e. elevated apo B-containing lipoproteins. In particular, inhibitors of PCSK9 and ANGPTL3 show important clinical impact.
1. Introduction
Hypocholesterolemia, primarily due to depressed low-density lipoprotein (LDL) cholesterol levels, is often considered to be a benign condition that is protective against atherosclerotic cardiovascular disease (ASCVD). However, very low cholesterol states may sometimes be cause for alarm, especially when associated with underlying severe illness or when predisposing to certain debilitating neurological or other multisystemic conditions. Diagnosing the cause of hypocholesterolemia is important in order to commence appropriate treatment and monitoring [Citation1]. Here, we review the non-genetic and genetic causes of low LDL cholesterol states, the various underlying genetic and biochemical etiologies, and associations of the genetic disorders with ASCVD, diabetes, fatty liver disease and multisystemic disorders and vitamin deficiencies. We suggest an approach for treatment of patients at risk for multisystemic complications and outline the current state of therapeutics targeted to treat high LDL cholesterol that were inspired from understanding the pathways underlying the monogenic hypocholesterolemia disorders.
2. Non-genetic causes of hypocholesterolemia
The majority of cases of hypocholesterolemia are caused by secondary non-genetic factors () [Citation22]. Lifestyle factors, such as a vegan, vegetarian [Citation3] or low-fat diet, alcoholism [Citation2] and malnutrition, can contribute toward low cholesterol. Hypocholesterolemia may be a sign of an infectious disease [Citation15–17], underlying inflammatory state [Citation12], or severe illness [Citation4,Citation7–9,Citation23,Citation11,Citation13,Citation14,Citation18,Citation19,Citation24–26]. In some illnesses, the hypocholesterolemia may result from malabsorption [Citation19,Citation25]. In many critically ill patients, hypocholesterolemia is associated with a worse prognosis [Citation5] and is an independent predictor of mortality [Citation6,Citation10,Citation12]. In addition to lipid lowering therapies, other medications, such as interleukin-2 used for treating refractory metastatic cancer [Citation20,Citation27], and alternative therapies [Citation19] may contribute toward hypocholesterolemia.
Table 1. Non-genetic secondary causes of low LDL cholesterol.
3. Genetic causes of hypocholesterolemia
Numerous genetic etiologies can result in low LDL cholesterol (), including rare disorders causing a complete absence of apolipoprotein (apo) B-containing lipoproteins, an isolated absence of apo B-48 lipoproteins, and lastly disorders of reduced but not absent levels of apo B-containing lipoproteins. highlights the metabolic pathways of the main proteins involved in the monogenic hypocholesterolemia disorders.
Figure 1. Simplified lipid pathway demonstrating the role of the key proteins from each monogenic hypocholesterolemia disorder. The proteins involved in apo B lipoprotein production (apoB48, apoB100, MTP), secretion (SAR1B) and catabolism (LDLR) are shown in green. The proteins involved in modulating apoB lipoproteins (PCSK9 and ANGPTL3) are shown in red. Lipoproteins are labeled in blue, with smaller, denser lipoproteins corresponding with darker shades of blue. Abbreviations: ANGPTL3, angiopoietin-like 3; APOB, apolipoprotein B; HL, hepatic lipase; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LPL, lipoprotein lipase; LDLR, LDL receptor; LRP, low density lipoprotein receptor-related protein 1; MTP, microsomal triglyceride transfer protein; PCSK9, proprotein convertase subtilisin/kexin type 9; SAR1B, secretion associated Ras related GTPase 1B; TG, triglyceride; VLDL, very-low density lipoprotein.
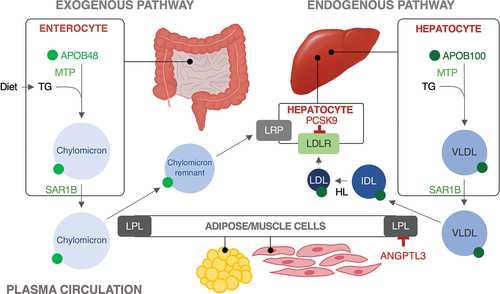
Table 2. Summary of the epidemiology, genetic, and clinical features of the primary causes of low LDL cholesterol.
Table 3. Treatment and clinical follow-up of patients with monogenic hypocholesterolemia: abetalipoproteinemia, homozygous familial hypobetalipoproteinemia, and chylomicron retention disease.
3.1. Complete absence of apo B-containing lipoproteins
3.1.1. Abetalipoproteinemia
Abetalipoproteinemia (ABL), or Bassen-Kornzweig syndrome [Citation21], is a rare autosomal recessive disorder reported in over 50 families [Citation28] with consanguinity in up to 75% of families [Citation29–34]. ABL is caused by extremely rare biallelic (i.e. homozygous or compound heterozygous) variants within the MTTP gene located on chromosome 4q23 [Citation35]. The gene codes for the 97 kDa microsomal triglyceride transfer protein (MTP) that forms a heterodimer with the 55 kDa protein disulfide isomerase (PDI) [Citation35,Citation36]. This heterodimer loads triglycerides (TG) onto apo B within enterocytes and hepatocytes [Citation35,Citation37,Citation38]. More than 75 variants [Citation29] within MTTP are causative for ABL [Citation39] that impair the formation and secretion of apo B-containing lipoproteins, specifically chylomicrons and very-low density lipoproteins (VLDL) within enterocytes and hepatocytes, respectively [Citation35,Citation37,Citation40]. The result is near absent or absent levels of chylomicrons, VLDL, LDL cholesterol (<0.1 mmol/L), apo B (<0.1 g/L), and TG (<0.2 mmol/L) in the blood [Citation35,Citation38]. Plasma cholesterol transport in ABL patients is largely carried out by high-density lipoprotein (HDL) particles [Citation26]. Impaired chylomicron production and secretion, which is the primary vehicle of absorption and transportation of fat-soluble vitamins, eventually results in severe deficiencies of fat-soluble vitamins.
The clinical presentation of ABL is heterogeneous [Citation41]. From infancy and early childhood, patients can exhibit diarrhea, oral fat intolerance, steatorrhea, and failure to thrive [Citation33,Citation37,Citation42,Citation43]. Nearly all patients exhibit fat malabsorption, acanthocytosis, and eventually severe deficiency of fat-soluble vitamins (A, D, E, and K) [Citation26,Citation38] that can cause multisystemic pathologies described in greater detail below. Hepatic sequelae are relatively common, including elevated serum transaminases, hepatomegaly, hepatic steatosis in about 15–30% of patients, as well as fibrosis and/or cirrhosis in <5% of patients [Citation28,Citation34,Citation43,Citation44]. There are infrequent reports of ileal adenocarcinoma and spinal cord glioblastoma in patients with ABL, although there is insufficient evidence for a clear convincing link of a higher cancer risk with ABL [Citation45,Citation46].
3.1.2. Homozygous Familial Hypobetalipoproteinemia
Familial hypobetalipoproteinemia (FHBL) is an autosomal co-dominant disorder caused by rare variants within APOB on chromosome 2p24.1, consisting largely of nonsense and frameshift variants, that result in early truncations [Citation47]. Individuals carrying a single variant within APOB have heterozygous familial hypobetalipoproteinemia (HetFHBL), while carriers of biallelic variants have homozygous familial hypobetalipoproteinemia (HoFHBL). Loss of function variants within APOB impair the formation and secretion of apo B lipoproteins, specifically VLDL and chylomicrons from hepatocytes and enterocytes, respectively [Citation37], resulting in absent or near absent levels of apo B-containing lipoproteins, in addition to very depressed levels of cholesterol and TG. Notably, truncated proteins larger than apo B-48 do not impede chylomicron secretion [Citation48]. The clinical presentation and sequelae in HoFHBL are essentially identical to ABL [Citation26]. A distinguishing feature is moderate hypocholesterolemia in parents of HoFHBL patients compared to no biochemical abnormalities in patients of ABL patients [Citation35].
3.2. Isolated absence of apo B-48 containing lipoproteins
3.2.1. Chylomicron retention disease
Chylomicron retention disease (CRD), or Anderson’s Disease [Citation49], is a very rare (approximately 55 cases described to date) [Citation50] autosomal recessive disorder caused by biallelic variants within SAR1B on chromosome 5p31.1, which encodes the secretion associated Ras related GTPase 1B (SAR1B) protein [Citation26]. SAR1B manages the formation of COPII vesicles required for proper trafficking of pre-chylomicron cargo within enterocytes from the endoplasmic reticulum to the Golgi; molecular defects within SAR1B impair chylomicron assembly and secretion, retaining pre-chylomicron transport vesicles in the cytoplasm [Citation50] and resulting in the absence of apo B-48 lipoproteins in circulation [Citation51]. Mutated SAR1B also impairs enterocyte production and secretion of HDL and apo A1 [Citation51,Citation52]. SAR1B has several functions, including roles in protection against inflammation, lipid homeostasis [Citation53], and neuronal development [Citation54], which may also affect the clinical presentation [Citation50]. Patients with CRD have 50% reduced levels of total, LDL, and HDL cholesterol, while TG levels are normal [Citation50]. Postprandial elevations in TG are blunted [Citation50,Citation51].
Patients with CRD can have similar but generally less severe clinical presentations to ABL and HoFHBL patients, including failure to thrive, malabsorption, diarrhea, steatorrhea, abdominal distention in infancy and early childhood and eventually fat-soluble vitamin deficiency that can lead to severe multi-systemic pathologies [Citation26,Citation51]. Hepatic steatosis and hepatomegaly [Citation51] may stem from reduced SAR1B-dependent VLDL secretion [Citation55]. Distinguishing features of CRD patients from ABL and HoFHBL patients include absence of acanthocytosis, milder neurological and ophthalmological complications [Citation50,Citation51], normal TG levels, low HDL cholesterol levels [Citation50], and more frequent elevations in creatine kinase [Citation50,Citation55].
3.3. Low but not absent apo B-containing lipoproteins
3.3.1. Heterozygous familial hypobetalipoproteinemia
Heterozygous familial HBL (HetFHBL) occurs in patients carrying one copy of a deleterious variant within APOB [Citation47]. Patients have depressed apo B (<30% of normal) and LDL cholesterol (<30% of normal) compared with noncarriers [Citation56], with LDL cholesterol levels as low as 8 mg/dL (0.2 mmol/L) [Citation57]. Unlike HoFHBL patients, severe malabsorption symptoms and flagrant vitamin deficiencies are largely absent in HetFHBL patients. However, neurological symptoms were described in an elderly 70-year-old HetFHBL patient, which may have resulted from the cumulative effects of life-long relatively low vitamin A and E levels [Citation58].
The main clinical concern in patients with HetFHBL is predisposition to hepatic disease, which fortunately affects only a minority of patients. Impaired VLDL secretion, stemming from reduced apo B production and secretion, translates to reduced hepatic TG transport and fat retention [Citation37]. HetFHBL patients have up to 2–3-fold higher hepatic fat content and approximately 2-fold [Citation59,Citation60] higher prevalence of hepatic steatosis than the general population [Citation58,Citation61–64]. Liver transaminases are frequently increased by 50% or more [Citation37,Citation60,Citation62]. There have been a few reported cases with more severe liver disease, including fibrosis [Citation58], cirrhosis [Citation65], and hepatocellular carcinoma [Citation37,Citation66]. Pelusi et al. has also demonstrated a link between nonalcoholic fatty liver disease and carrier status of APOB pathogenic variants [Citation67]. Despite the increased prevalence of hepatic steatosis, HetFHBL patients do not have a higher risk of insulin resistance, with a similar prevalence of diabetes as the general population [Citation48,Citation68].
As a result of the life-long reductions in apo B-containing lipoproteins, HetFHBL are protected from atherosclerosis, with an estimated 72% lower risk of coronary heart disease [Citation69]. HetFHBL patients exhibit lower arterial stiffness, a marker of vascular function associated with vessel wall protection [Citation60].
3.3.2. PCSK9 deficiency
PCSK9 deficiency is an autosomal semi-dominant disorder that results from loss-of-function variants within PCKS9 on chromosome 1p32.3. Loss of PCSK9 activity, which normally functions to direct the LDL receptor toward lysosomal degradation [Citation26], results in increased LDL uptake. Carriers of PCSK9 loss-of-function variants have low levels of total and LDL cholesterol and TG, with the extent of the lipid reduction dependent on zygosity, although the lipid reductions are less severe than in patients with ABL, HoFHBL, or CRD [Citation26]. The principle that PSCK9 loss-of-function variants offset genetically high levels of LDL cholesterol was observed in a familial hypercholesterolemia patient who had both a pathogenic LDLR variant and a PCSK9 variant, who had a near normal lipid profile and no complications [Citation70].
Patients with PCSK9 deficiency are generally healthy and have a reduced risk of ASCVD [Citation71], dementia [Citation72], fatty liver, and hepatic disease [Citation26,Citation56,Citation73,Citation74]. The PCSK9 p.Q152H variant protects against liver injury caused by endoplasmic reticulum storage disease [Citation75], and carriers of the p.R46L variant have a significantly lower risk of nonalcoholic fatty liver disease (OR = 0.42; 95% 0.22–0.81; p = 0.01), nonalcoholic steatohepatitis (OR = 0.48; 95% CI 0.26–0.87; p = 0.01), and fibrosis (OR = 0.55; 95% CI 0.32–0.94; p = 0.03) [Citation76]. However, despite the hepatoprotective effects apparent with PCSK9 deficiency, it did not appear to compensate for genetic susceptibility to nonalcoholic fatty liver disease in a HetFHBL patient [Citation77].
Both homozygotes and heterozygotes with PCSK9 deficiency are protected from ASCVD as a result of the lifelong reductions in LDL cholesterol levels, confirmed by Mendelian randomization and observational studies [Citation78–80]. The estimated degree of risk reduction for coronary heart disease, ranging from 17% to 88% across numerous studies, is proportional to the extent of reduction in LDL cholesterol levels [Citation71,Citation78,Citation79,Citation81]. Carriers have a significantly lower mean intima-medium thickness, indicating reduced atherosclerosis risk [Citation78]. Carriers of the PCSK9 p.R46L variant have a significantly lower age- and sex-adjusted risk for aortic valve stenosis (OR = 0.77; 95% CI 0.65–0.92) and myocardial infarction (OR = 0.76; 95% CI 0.64–0.89) [Citation82]. PCSK9 deficiency mediated reductions in LDL cholesterol by 10 mg/dL (0.26 mmol/L) significantly reduce risk of abdominal aortic aneurysm (OR = 0.76; 95% CI 0.68–0.86) and ischemic stroke (OR = 0.93; 95% CI 0.88–0.99), each controlled for sex and age [Citation71].
3.3.3. ANGPTL3 deficiency (familial combined hypolipidemia)
ANGPTL3 deficiency, also known as familial combined hypolipidemia, is an autosomal semi-dominant disorder [Citation83] resulting from rare loss of function variants in the ANGPTL3 gene located on chromosome 1q31.3 [Citation26]. In addition to reduced VLDL production, deficiency of ANGPTL3, which normally inhibits lipoprotein lipase and endothelial lipase activity [Citation84], results in enhanced hydrolysis of chylomicrons, VLDL and HDL particles [Citation85]. Carriers of biallelic variants have a near complete absence of circulating ANGPTL3, along with significant depressions in levels of all plasma lipids (i.e. LDL cholesterol, total cholesterol, apo B, TG, HDL cholesterol, and apo A1) [Citation83,Citation86]. Carriers of heterozygous variants have ANGPTL3 levels 42–50% lower than controls and have milder, yet significant, pan-hypolipidemia [Citation86,Citation87].
Patients with ANGPTL3 deficiency generally do not present with any adverse clinical findings [Citation26,Citation83]. They do not experience vitamin deficiencies [Citation26], are not at an elevated risk of hepatic steatosis, and appear to have normal serum transaminase levels [Citation60,Citation86]. Compared to heterozygous carriers and noncarriers, homozygotes demonstrate higher insulin sensitivity [Citation88].
Mounting evidence from Mendelian randomization studies suggest patients with ANGPTL3 deficiency are protected from ASCVD. In separate meta-analyses, Stitziel et al. and Dewey et al. found a 34% (OR = 0.66; 95% CI 0.44–0.98; p = 0.04) [Citation89] and 39% (OR = 0.61; 95% CI 0.45–0.81; p < 0.001) reduced risk of CAD in their respective cohorts of ANGPTL3 loss of function variant carriers [Citation87]. Individuals within the lowest tertile of circulating ANGPTL3 levels had a significantly reduced risk of myocardial infarction compared with the highest tertile (OR = 0.65; 95% CI 0.55–0.77; p = 2.2 × 10−7), which remained significant after adjustment for plasma LDL cholesterol and TG (OR = 0.71; p = 0.0001) [Citation89]. Carriers of ANGPTL3 loss-of-function variants also demonstrated a reduced risk of myocardial infarction that trended toward significance (OR = 0.66; 95% CI 0.39 to 1.06; p = 0.09) [Citation87]. In a family study, two out of three first-degree relatives had evidence of coronary atherosclerotic plaque while the three individuals with ANGPTL3 deficiency had none, including a middle-age individual with multiple risk factors for ASCVD [Citation89]. It is notable that patients with ANGPTL3 deficiency also have life-long depressions in HDL cholesterol, suggesting that protection from lower LDL cholesterol supersedes possible deleterious effects of concurrently low HDL cholesterol [Citation86].
3.3.4. LDLR gain-of-function variant
A novel large-effect gain-of-function variant within the LDLR gene on chromosome 19p13.2 discovered in an Icelandic family has become the latest identified monogenic cause of low LDL cholesterol. The LDL receptor is responsible for removal of LDL cholesterol in the blood. While LDLR gain-of-function variants have been reported in the past [Citation90–92], to date, only a single one, which is a 2.5 kb deletion spanning the 3’ UTR in 7 heterozygous members segregating in three generations of a single Icelandic family, has shown marked reductions in LDL cholesterol levels [Citation93]. This de novo variant, which caused a 1.79-fold increase in surface expression of the LDL receptor was unique to this family and segregated in an autosomal dominant manner. Carriers also had reductions in PCSK9 levels, which may have further contributed toward the depressed lipid profile. Mean levels of LDL cholesterol were 0.87 mmol/L among six heterozygous family members, which was within the 1st percentile for each carrier’s age and sex, and remained depressed over decades. Total and non-HDL cholesterol and apo B levels were also significantly lower compared to controls, while levels of HDL cholesterol and TG were normal. Noncarrier family members had normal lipid profiles. There were no detrimental effects among the carriers.
3.3.5. Polygenic hypocholesterolemia
While many hypocholesterolemia cases have a monogenic origin (16–52%), a considerable proportion of such patients have polygenic hypocholesterolemia (34–50%) [Citation94–96], which is the result of the cumulative effects of common variants or polymorphisms throughout the genome discovered from genome-wide association studies. Each common variant is individually associated with marginally depressed levels of LDL cholesterol, but cumulatively these small effects add up to produce a potentially large effect. Polygenic susceptibility has also been captured in some cases of monogenic hypocholesterolemia and may explain some of the heterogeneity among cases with a common monogenic basis [Citation94].
Polygenic hypocholesterolemia patients have LDL cholesterol and apo B levels depressed below the 5th percentile, although lipid reductions are attenuated compared with monogenic hypocholesterolemia [Citation95,Citation97]. The prevalence of hepatic disease is the same or lower. In a cohort study by Rimbert et al. of 38 patients with polygenic hypocholesterolemia and 40 patients with monogenic hypocholesterolemia, likelihood of hepatic steatosis was 87% lower in those with polygenic hypocholesterolemia [Citation95]. Non-alcoholic steatohepatitis, measured by elevations in ALT, was reported absent in the 34 patients with polygenic hypocholesterolemia evaluated while it was present in 47% (16/34) of patients with monogenic hypocholesterolemia [Citation95]. In contrast, a recent cohort study by Cefalù et al. found no difference in the prevalence of hepatic steatosis among 63 patients with polygenic hypocholesterolemia compared with 60 patients with monogenic hypocholesterolemia or 47 patients with hypocholesterolemia of unknown genetic origin [Citation97]. These discordant results cannot be explained by metabolic differences between the monogenic hypocholesterolemia cohorts in the two studies as they are both comprised mostly of carriers of loss-of-function APOB variants [Citation95,Citation97].
3.3.6. Congenital disorder of glycosylation (CDG)
Congenital disorders of glycosylation (CDG) are rare, autosomal recessive disorders causing defective glycosylation of proteins and lipids. CDG type I disorders are a newly recognized cause of hypocholesterolemia [Citation98]. Two prevalent CDG type I disorders that result in impaired N-glycosylation activity are asparagine-linked glycosylation protein 6 (ALG6)-deficiency and phosphomannomutase 2 (PMM2)-deficiency, caused by biallelic loss-of-function variants in ALG6 and PMM2, respectively [Citation99]. Patients present with levels of LDL cholesterol, total cholesterol and apo B below the 5th percentile. The proposed mechanism is increased levels of sterol regulatory element-binding protein 2 (SREBP2) elevates the expression and abundance of the LDL receptor, enhancing the uptake of LDL cholesterol. There is conflicting evidence that lower levels and impaired activity of PCSK9, from defective N-glycosylation, also contribute to the depressions in LDL cholesterol [Citation99,Citation100].
Clinical presentations vary among CDG type I patients, with a wide range of neurological, ophthalmological, gastrointestinal, musculoskeletal, and hepatic features. Neurological deficits present in nearly all patients, resulting in diagnosis commonly in infancy and early childhood [Citation100,Citation101]. These complications primarily result from impaired glycosylation rather than severe vitamin deficiencies. Heterozygous carriers have attenuated levels of LDL cholesterol and apo B without any other clinical abnormalities [Citation99].
4. Association with clinical phenotype and disease
We describe the association of the genetic disorders of low LDL cholesterol with ASCVD, diabetes, fatty liver disease, and multisystemic vitamin deficiencies.
4.1. Protection against ASCVD
Depressed levels of LDL cholesterol and apo B appear to be protective against ASCVD, a relationship supported by numerous Mendelian randomization and observational studies. Systematic evaluation of risk has not been completed for patients with ABL, CRD, LDLR gain-of-function, or polygenic hypocholesterolemia; however, the plethora of studies exploring risk in patients with FHBL, ANGPTL3 deficiency, and PCSK9 deficiency provide convincing evidence that genetically driven low levels of LDL cholesterol protect against atherosclerosis. Risk of coronary heart disease is 72% lower in FHBL patients [Citation69], 34–39% lower in patients with ANGPTL3 deficiency [Citation87,Citation89], and ranges from 17% to 88% lower in studies of patients with PCSK9 deficiency [Citation71,Citation78,Citation79,Citation81]. However, it is important to remember this genetically driven reduction in risk is not absolute as some patients with these variants develop ASCVD [Citation68,Citation69].
4.2. Fatty liver disease
Susceptibility to fatty liver disease in low LDL cholesterol states depends upon the molecular defect underlying the hypocholesterolemia. ABL and FHBL patients have defective VLDL secretion that leads to hepatic fat accumulation [Citation60], meaning some patients are predisposed to hepatic steatosis that sometimes develops into nonalcoholic steatohepatitis, fibrosis, and cirrhosis [Citation28]. Carriers of rare variants in APOB, resulting in ABL, are significantly more likely to develop liver disease and have higher aminotransferase levels [Citation102]. Patients with polygenic hypocholesterolemia also appear to have a similar risk of hepatic steatosis as monogenic hypocholesterolemia [Citation97]. There are also few reported cases of hepatic steatosis although not cirrhosis in patients with CRD [Citation50]. Patients with ANGTPL3 deficiency [Citation86], PCSK9 deficiency [Citation73], or LDLR gain-of-function [Citation93] do not appear to be at a higher risk for fatty liver; each of these patients have defects that increase catabolism of apo B lipoproteins rather than impaired VLDL secretion from the liver. In fact, individuals with PCSK9 deficiency may experience hepatoprotective effects that reduce risk of nonalcoholic fatty liver disease [Citation75,Citation76]. Of note, rare variants in other genes, such as CIDEB which encodes a structural protein on lipid droplets, also modulate the risk of fatty liver [Citation103].
4.3. Diabetes
Diabetes has sometimes been described in some patients with monogenic hypocholesterolemia [Citation104], prompting the question of whether these patients are genetically predisposed to diabetes. Despite the high prevalence of fatty liver disease in patients with FHBL, there is not any increased prevalence of insulin resistance [Citation105,Citation106] and diabetes appears to be absent [Citation48]. The prevalence of diabetes in patients with ANGPTL3 deficiency was 4.9%, relatively lower than the reference population at 9.2%, which may result from improved insulin sensitivity [Citation48]. Mendelian randomization studies suggest that patients with PCSK9 deficiency may be at a higher risk of developing type 2 diabetes [Citation107,Citation108]. However, associations of specific variants with diabetes are conflicted; Lotta et al. found that the loss-of-function p. R46L variant confers a 9% increased risk of type 2 diabetes (OR = 1.09; 95% CI 1.01–1.17; p = 0.03) [Citation109], while Bonnefond et al. found no significant associations [Citation110]. Overall, patients with FHBL or ANGPTL3 deficiency do not appear have a higher susceptibility to developing diabetes, while patients with PCSK9 deficiency may have a higher risk that needs to be further evaluated. There is no increased diabetes risk among patients who take PCSK9 inhibitor drugs. For other low cholesterol state phenotypes, i.e. ABL, CRD, polygenic hypocholesterolemia, and LDLR gain-of-function, a systematic evaluation of risk remains to be completed though diabetes does not appear to be a common feature among these patients.
4.4. Rare multisystem vitamin deficiencies
Rare multisystemic fat-soluble vitamin deficiencies affect patients with ABL, HoFHBL, and CRD, who each have impaired absorption and secretion of chylomicrons, the main carrier vehicle of fat-soluble vitamins (A, D, E, and K). These rare multisystemic vitamin deficiencies cause various neurological, ophthalmological, musculoskeletal, and coagulation defects. Deficiency of vitamin A underlies retinopathy and retinitis pigmentosa, which can result in loss of night and peripheral vision [Citation111,Citation112]. Complete blindness may eventually result if the severe vitamin deficiency is left untreated [Citation38,Citation42,Citation43]. Neuropathy, spinocerebellar ataxia, reduced proprioception, deficient deep tendon reflex, and dysarthria stem from vitamin E deficiency [Citation26,Citation38,Citation43,Citation113,Citation114]. Demyelination can affect neurons within both the central and peripheral nervous system [Citation38]. Beginning in the first or second decade of life, these neurological sequalae can lead to disability without treatment. Myopathy, rickets, osteopenia, osteomalacia, and osteoporosis [Citation26,Citation38,Citation43] arise largely from deficiency of vitamin D [Citation114], though vitamin K deficiency may also contribute to bone deterioration [Citation115,Citation116] in addition to coagulopathy with prolonged prothrombin time [Citation38], susceptibility to bruising, and hemorrhage [Citation114].
5. Approach to diagnosis and treatment
For patients with ABL, HoFHBL, and CRD, early diagnosis and treatment are essential for optimal prognosis [Citation112,Citation117]. Diagnosis can be based upon the presenting clinical features, the lipid profile, and genetic testing with next-generation DNA sequencing, when available. Prompt commencement of oral high dose vitamin therapy, consisting of 100–400 IU/kg/day vitamin A, 800–1200 IU/kg/day vitamin D, 100–300 IU/kg/day vitamin E, and 5–35 mg/week vitamin K, is essential to attenuate, halt, and potentially reverse the various severe multisystemic complications [Citation43]. Gastrointestinal manifestations, such as steatorrhea and malabsorption, experienced in early childhood or infancy can be resolved with a low-fat diet (<30% calories from fat) [Citation35,Citation68]. To promote normal growth, care should be taken to ensure that daily caloric intake is adequate and essential fatty acids may also be orally supplemented through 1–2 teaspoons (5–10 ml) of polyunsaturated fatty acid rich oil. Patients should be consistently followed-up on a 6–12-month basis for a complete evaluation. A detailed breakdown of the elements to be clinically evaluated and investigated in the laboratory are outlined in . Overall, a low-fat diet, high-dose vitamin therapy, essential fatty acid supplementation and consistent follow-up are key elements of the life-long treatment regimen of patients with ABL, HoFHBL, and CRD. Though other complications are largely absent, patients with HetFHBL should also be monitored for nonalcoholic fatty liver disease; diet and exercise may be recommended to improve liver transaminase levels [Citation113].
6. Translation to treatments for high cholesterol
Many of the monogenic hypocholesterolemia disorders have inspired novel therapies to treat elevated cholesterol in order to reduce ASCVD risk [Citation74,Citation118]. For instance, the small molecule inhibitor lomitapide targets MTP, mimicking the genetic defect in patients with ABL, while the antisense oligonucleotide mipomersen targets the messenger RNA of apo B-100, mimicking the genetic defect in FHBL patients with APOB truncations larger than apo B-48. Both are effective at lowering LDL cholesterol and other atherogenic lipids to reduce risk of ASCVD [Citation119,Citation120] on top of existing lipid-lowering interventions in patients with homozygous familial hypercholesterolemia [Citation121–124]. Gastrointestinal side-effects for lomitapide, injection site reactions and flu-like symptoms for mipomersen, and concerns of elevated transaminases, hepatic steatosis and nonalcoholic steatohepatitis for both treatments call for monitoring, potential dosage adjustments and if necessary, discontinuation of treatment [Citation120,Citation125]. While lomitapide is available in many places, marketing of mipomersen was suspended in 2017 due to side effects.
In contrast, for hypercholesterolemic patients with at least one functional LDLR allele [Citation126], the PCSK9 inhibitors, alirocumab, evolocumab [Citation127], and inclisiran [Citation128,Citation129], have proven to be transformational. They have a favorable safety profile with minimal injection-site reactions and are very efficacious at lowering LDL cholesterol. Alirocumab and evolocumab are monoclonal antibodies that target PCSK9 to achieve reductions in risk of cardiovascular events, such as myocardial infarction and stroke [Citation130,Citation131]. Inclisiran is a small interfering RNA that prevents translation of PCSK9 messenger RNA. Each PCSK9 inhibitor displays a similar LDL cholesterol-lowering effect [Citation126], but inclisiran may be advantageous for compliance as injections are required semiannually compared with every 2–4 weeks for the monoclonal antibodies [Citation128]. Finally, inspired by patients with ANGPTL3 deficiency, evinacumab is a monoclonal antibody that inhibits ANGPTL3 and lowers LDL cholesterol with remarkable efficacy in patients with severe and refractory familial hypercholesterolemia [Citation132,Citation133].
7. Conclusions
Hypocholesterolemia, i.e. low levels of LDL cholesterol, is a complex trait that can be caused by many different genetic causes, although most cases likely result from non-genetic lifestyle factors such as diet, underlying illness, or medications. Monogenic hypocholesterolemia varies in severity based upon the underlying biochemical and physiological defect. ABL and HoFHBL patients have the most severe phenotypes with a complete absence of apo B-containing lipoproteins, followed by CRD patients who have an absence of apo B-48 lipoproteins; patients with these disorders can have severe gastrointestinal and hepatic sequalae, in addition to multisystemic manifestations (i.e. neurological, musculoskeletal, hematological, and ophthalmological) caused by severe deficiencies of fat-soluble vitamins (i.e. A, D, E, and K). It is imperative to identify and begin treatment regimens for these patients early.
Patients with PCSK9 deficiency, ANGPTL3 deficiency, polygenic hypocholesterolemia, and LDLR gain-of-function variant appear to have no negative clinical manifestations. Depressed LDL cholesterol levels are predicted to protect against ASCVD, based on evidence from patients with FHBL, ANGPTL3 deficiency and PCSK9 deficiency. This has incentivized the development of various lipid-lowering therapies with these hypocholesterolemia disorders in mind intended for patients with high LDL cholesterol, with inhibitors of PCSK9 and ANGPTL3 showing the greatest potential for wide usage with a viable safety profile.
8. Expert opinion
With increased clinician experience, familiarity, and ongoing research involving patients with hypocholesterolemia, treatment guidelines are being updated with more effective regimens for optimal prognosis [Citation134]. But there remains room for further progress in understanding and adequately addressing low LDL cholesterol in patients. Early definitive diagnosis of the severe monogenic hypocholesterolemia disorders, as next-generation sequencing becomes more accessible and widely used, can pave the way for improved prognosis with early initiation of high-dose vitamin therapy and a low-fat diet. Barriers to effective care may include inadequate access to regular care and treatments due to a lack of financial coverage, as well as lack of access to genetic testing to confirm specific diagnoses.
Development of a PRS that captures more variation in the genome [Citation73] and ancestry-specific PRS can help us better understand and identify polygenic susceptibility to low LDL cholesterol in different populations. Testing and refinement of the component single nucleotide polymorphisms (SNPs) is equally as important since simply increasing the number of SNPs does not necessarily improve the ability to diagnose polygenic hypocholesterolemia [Citation97]. Hypocholesterolemia is often underinvestigated as some clinicians perceive that low cholesterol states are not associated with other diseases and are rather protective. Understanding how genetic status, LDL cholesterol, and apo B levels associate with the etiology of other diseases, such as breast cancer [Citation135] or PCSK9 and diabetes [Citation107–110], where the relationship is not clear can elucidate potential risks or benefits of the monogenic hypocholesterolemia inspired lipid-lowering therapies.
Treatment of the mirror-image condition, namely hypercholesterolemia, might benefit from future insights gained from studying natural human genetic variants. For example, a MTP inhibitor with similar effects as the p.G865V variant in MTTP, which blocks TG transfer but retains phospholipid transfer activity of MTP, may reduce risk of hepatic steatosis and gastrointestinal side-effects; this would be a significant improvement over the existing MTP inhibitor lomitapide [Citation136].
Also, new methods of delivery could help patients with hypercholesterolemia. For instance, development of novel gene editing technologies that target the regulators of synthesis, secretion, and metabolism of apo B lipoproteins could also pave the way for a more permanent solution in patients with elevated LDL cholesterol. In non-human primates, a CRISPR-Cas9 adenine base editor delivered to the liver precisely altered a single base pair to create a loss-of-function variant in PCSK9 resulted in sustained reductions of 90% and 60% over 8 months in levels of PCSK9 and LDL cholesterol, respectively [Citation137]. Vaccines also hold promise for generating long-lasting endogenous antibodies against PCSK9 [Citation138] or ANGPTL3 [Citation139,Citation140] to help achieve sustained reductions in lipid levels.
Finally, ongoing research might uncover potential new causal genes, for instance loci associated with reduced LDL cholesterol and also reduced hepatic fat content. Targeting such a putative gene product would circumvent some of the adverse liver effects seen with earlier generation treatments such as lomitapide and mipomersen. However, all findings of putative novel genes for low cholesterol [Citation141] will require calm circumspection and will need to be replicated and validated before they can be considered on a par with proven known causal genes.
In the next five to ten years, therapeutics for atherosclerosis prevention based on the hypocholesterolemia disorders, namely PCSK9 deficiency and ANGPTL3 deficiency, will reach the forefront of lipid management due to their efficacy and favorable safety profile, just as statins are currently. In addition to the mAb that already exist, which are currently already on the market, it is likely that at least some of the advanced therapeutics that are more immediately feasible, such as vaccines against PCSK9, will be at advanced stages of development or in clinical trials, for the management of high levels of LDL cholesterol.
Article highlights
Fat malabsorption in abetalipoproteinemia, hypobetalipoproteinemia, and chylomicron retention disease causes severe symptoms from infancy or childhood, such as diarrhea and failure to thrive, and eventually fat-soluble vitamin deficiency and hepatic disease
Fat-soluble vitamin deficiency results in progressively worsening neurological, hematological, musculoskeletal, and ophthalmological sequalae over a patient’s lifetime
Early initiation of a low-fat diet and high-dose fat-soluble vitamin therapy is essential in abetalipoproteinemia, hypobetalipoproteinemia, and chylomicron retention disease to preserve long-term neurological and ophthalmological function
PCSK9 deficiency and ANGPTL3 deficiency are protective against atherosclerotic cardiovascular disease
PCSK9 inhibitors (alirocumab, evolocumab and inclisiran) and ANGPTL3 inhibitors (evinacumab) are effective therapeutic agents to lower LDL cholesterol levels
Declaration of interest
RA Hegele is supported by the Jacob J. Wolfe Distinguished Medical Research Chair, the Edith Schulich Vinet Research Chair, and the Martha G. Blackburn Chair in Cardiovascular Research. He holds operating grants from the Canadian Institutes of Health Research (Foundation award), the Heart and Stroke Foundation of Ontario (G-21-0031455) and the Academic Medical Association of Southwestern Ontario (INN21–011). RA Hegele reports consulting fees from Acasti, Aegerion, Akcea/Ionis, Amgen, HLS Therapeutics, Novartis, Pfizer, Regeneron, Sanofi, and Ultragenyx.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet. 2009;10:109–121.
- Tabara Y, Ueshima H, Takashima N, et al. Mendelian randomization analysis in three Japanese populations supports a causal role of alcohol consumption in lowering low-density lipid cholesterol levels and particle numbers. Atherosclerosis. 2016;254:242–248.
- Wang F, Zheng J, Yang B, et al. Effects of vegetarian diets on blood lipids: a systematic review and meta‐analysis of randomized controlled trials. J Am Heart Assoc. 2015;4:e002408.
- Hartman C, Tamary H, Tamir A, et al. Hypocholesterolemia in children and adolescents with β-thalassemia intermedia. J Pediatr. 2002;141:543–547.
- Giovannini I, Boldrini G, Chiarla C, et al. Pathophysiologic correlates of hypocholesterolemia in critically ill surgical patients. Intensive care Med. 1999;25:748–751.
- Obialo CI, Okonofua EC, Nzerue MC, et al. Role of hypoalbuminemia and hypocholesterolemia as copredictors of mortality in acute renal failure. Kidney Int. 1999;56:1058–1063.
- Shalev H, Kapelushnik J, Moser A, et al. Hypocholesterolemia in chronic anemias with increased erythropoietic activity. Am J Hematol. 2007;82:199–202.
- Zwickl H, Hackner K, Köfeler H, et al. Reduced LDL-cholesterol and reduced total cholesterol as potential indicators of early cancer in male treatment-naïve cancer patients with pre-cachexia and cachexia. Front Oncol. 2020;10:1262.
- Ginsberg H, Grabowski GA, Gibson JC, et al. Reduced plasma concentrations of total, low density lipoprotein and high density lipoprotein cholesterol in patients with Gaucher type I disease. Clin Genet. 1984;26:109–116.
- Iseki K, Yamazato M, Tozawa M, et al. Hypocholesterolemia is a significant predictor of death in a cohort of chronic hemodialysis patients. Kidney Int. 2002;61:1887–1893.
- Rizos CV. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc Med J. 2011;5:76–84.
- Bonville DA, Parker TS, Levine DM, et al. The relationships of hypocholesterolemia to cytokine concentrations and mortality in critically ill patients with systemic inflammatory response syndrome. Surg Infect. 2004;5:39–49.
- Yalcinkaya A, Unal S, Oztas Y. Altered HDL particle in sickle cell disease: decreased cholesterol content is associated with hemolysis, whereas decreased apolipoprotein A1 is linked to inflammation. Lipids Health Dis. 2019;18:225.
- Zorca S, Freeman L, Hildesheim M, et al. Lipid levels in sickle-cell disease associated with haemolytic severity, vascular dysfunction and pulmonary hypertension. Br J Haematol. 2010;149:436–445.
- Flores MS, Obregón-Cárdenas A, Tamez E, et al. Hypocholesterolemia in patients with an amebic liver abscess. Gut Liver. 2014;8:415–420.
- Bima AI, Hooper AJ, van Bockxmeer FM, et al. Hypobetalipoproteinaemia secondary to chronic hepatitis C virus infection in a patient with familial hypercholesterolaemia. Ann Clin Biochem. 2009;46:420–422.
- Shor-Posner G, Basit A, Lu Y, et al. Hypocholesterolemia is associated with immune dysfunction in early human immunodeficiency virus-1 infection. Am j med. 1993;94:515–519.
- Lal CS, Kumar A, Kumar S, et al. Hypocholesterolemia and increased triglyceride in pediatric visceral leishmaniasis. Clin Chim Acta. 2007;382:151–153.
- Moutzouri E, Elisaf M, Liberopoulos N. Hypocholesterolemia. Curr Vasc Pharmacol. 2011;9:200–212.
- Wilson DE, Birchfield GR, Hejazi JS, et al. Hypocholesterolemia in patients treated with recombinant interleukin-2: appearance of remnant-like lipoproteins. J Clin Oncol. 1989;7:1573–1577.
- Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950;5:381–387.
- Gutiérrez-Cirlos C, Ordóñez-Sánchez ML, Tusié-Luna MT, et al. Familial hypobetalipoproteinemia in a hospital survey: genetics, metabolism and non-alcoholic fatty liver disease. Ann Hepatol. 2011;10:155–164.
- Nowak JK, Szczepanik M, Wojsyk-Banaszak I, et al. Cystic fibrosis dyslipidaemia: a cross-sectional study. J Cyst Fibros. 2019;18:566–571.
- Ettinger WH, Harris T, Verdery RB, et al. Evidence for inflammation as a cause of hypocholesterolemia in older people. J Am Geriatr Soc. 1995;43:264–266.
- Nakamura T, Takebe K, Yamada N, et al. Bile acid malabsorption as a cause of hypocholesterolemia seen in patients with chronic pancreatitis. Int J Pancreatol off J Int Assoc Pancreatol. 1994;16:165–169.
- Hegele RA, Borén J, Ginsberg HN, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European atherosclerosis society task force consensus statement. Lancet Diabetes Endocrinol. 2020;8:50–67.
- Kwong LK, Ridinger DN, Bandhauer M, et al. Acute dyslipoproteinemia induced by interleukin-2: lecithin: cholesteryl acyltransferase, lipoprotein lipase, and hepatic lipase deficiencies. J Clin Endocrinol Metab. 1997;82:1572–1581.
- Di Filippo M, Moulin P, Roy P, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol. 2014;61:891–902.
- Rodríguez Gutiérrez PG, González García JR, De León YA C, et al. A novel p.Gly417Valfs*12 mutation in the MTTP gene causing abetalipoproteinemia: presentation of the first patient in Mexico and analysis of the previously reported cases. J Clin Lab Anal. 2021;35:e23672.
- Barakizou H, Gannouni S, Messaoui K, et al. Abetalipoproteinemia: a novel mutation of microsomal triglyceride transfer protein (MTP) gene in a young Tunisian patient. Egypt J Med Hum Genet. 2016;17:251–254.
- Chardon L, Sassolas A, Dingeon B, et al. Identification of two novel mutations and long-term follow-up in abetalipoproteinemia: a report of four cases. Eur J Pediatr. 2009;168:983–989.
- Najah M, Youssef SM, Yahia HM, et al. Molecular characterization of Tunisian families with abetalipoproteinemia and identification of a novel mutation in MTTP gene. Diagn Pathol. 2013;8:54.
- Sivamurukan P, Boddu D, Pulimood A, et al. An unusual presentation of hemorrhagic disease in an infant: a probable case of abetalipoproteinemia. J Pediatr Hematol Oncol. 2021;43:e429.
- Walsh MT, Iqbal J, Josekutty J, et al. Novel abetalipoproteinemia missense mutation highlights the importance of the n-terminal β-barrel in microsomal triglyceride transfer protein function. Circ Cardiovasc Genet. 2015;8:677–687.
- Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis. 2014;37:333–339.
- Wang J, Hegele RA. Microsomal triglyceride transfer protein (MTP) gene mutations in Canadian subjects with abetalipoproteinemia. Hum Mutat. 2000;15:294–295.
- Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol. 2014;25:161–168.
- Zamel R, Khan R, Pollex RL, et al. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19.
- Burnett JR, Bell DA, Hooper AJ, et al. Clinical utility gene card for: abetalipoproteinaemia. Eur J Hum Genet. 2012;20:1–3.
- Wetterau JR, Aggerbeck LP, Laplaud PM, et al. Structural properties of the microsomal triglyceride-transfer protein complex. Biochemistry. 1991;30:4406–4412.
- Paquette M, Dufour R, Hegele RA, et al. A tale of 2 cousins: an atypical and a typical case of abetalipoproteinemia. J Clin Lipidol. 2016;10:1030–1034.
- Bredefeld C, Peretti N, Hussain MM, et al. New classification and management of abetalipoproteinemia and related disorders. Gastroenterology. 2020;160:1912–1916.
- Burnett JR, Hooper AJ, Ab HR, et al. ipoproteinemia. In: GeneReviews® [Internet] MP A, DB E, GM M, RA P, SE W, LJH B, KW G Amemiya A, editors. 2018 Oct 25[updated 2022 May 19]. Seattle (WA): University of Washington, Seattle; p. 1993–2023.
- Rashtian P, Najafi Sani M, Jalilian R. A male infant with abetalipoproteinemia: a case report from Iran. Middle East J Dig Dis. 2015;7:181–184.
- Al‐shali K, Wang J, Rosen F, et al. Ileal adenocarcinoma in a mild phenotype of abetalipoproteinemia. Clin Genet. 2003;63:135–138.
- Newman RP, Schaefer EJ, Thomas CB, et al. Abetalipoproteinemia and metastatic spinal cord glioblastoma. Arch Neurol. 1984;41:554–556.
- Burnett JR, Bell DA, Hooper AJ, et al. Clinical utility gene card for: familial hypobetalipoproteinaemia (APOB) – Update 2014. Eur J Hum Genet. 2015;23:890.
- Noto D, Arca M, Tarugi P, et al. Association between familial hypobetalipoproteinemia and the risk of diabetes. Is this the other side of the cholesterol–diabetes connection? A systematic review of literature. Acta Diabetol. 2017;54:111–122.
- Anderson CM, Townley RR, Freeman M, et al. Unusual causes of steatorrhoea in infancy and childhood. Med J Aust. 1961;48:617–622.
- Peretti N, Sassolas A, Roy CC, et al. Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J Rare Dis. 2010;5:24.
- Levy E, Poinsot P, Spahis S. Chylomicron retention disease: genetics, biochemistry, and clinical spectrum. Curr Opin Lipidol. 2019;30:134–139.
- Sané AT, Seidman E, Peretti N, et al. Understanding chylomicron retention disease through Sar1b GTPase gene disruption. Arterioscler Thromb Vasc Biol. 2017;37:2243–2251.
- Sané A, Ahmarani L, Delvin E, et al. SAR1B GTPase is necessary to protect intestinal cells from disorders of lipid homeostasis, oxidative stress, and inflammation. J Lipid Res. 2019;60:1755–1764.
- Li X, Yan M, Guo Z, et al. Inhibition of Sar1b, the gene implicated in chylomicron retention disease, impairs migration and morphogenesis of developing cortical neurons. Neuroscience. 2020;449:228–240.
- Peretti N. Lessons from chylomicron retention disease: a potential new approach for the treatment of hypercholesterolemia? Expert Opin Orphan Drugs. 2018;6:163–165.
- Tarugi P, Averna M, Di Leo E, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195:e19–27.
- Surakka I, Hornsby WE, Farhat L, et al. A novel variant in APOB gene causes extremely low LDL-C without known adverse effects. J Am Coll Cardiol: Case Reports. 2020;2:775–779.
- Musialik J, Boguszewska-Chachulska A, Pojda-Wilczek D, et al. A rare mutation in the APOB gene associated with neurological manifestations in familial hypobetalipoproteinemia. Int J Mol Sci. 2020;21:1439.
- Di Costanzo A, Di Leo E, Noto D, et al. Clinical and biochemical characteristics of individuals with low cholesterol syndromes: a comparison between familial hypobetalipoproteinemia and familial combined hypolipidemia. J Clin Lipidol. 2017;11:1234–1242.
- Sankatsing RR, Fouchier SW, de Haan S, et al. Hepatic and cardiovascular consequences of familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2005;25:1979–1984.
- Castellano G, Garfia C, Gomez-Coronado D, et al. Diffuse fatty liver in familial heterozygous hypobetalipoproteinemia. J Clin Gastroenterol. 1997;25:379–382.
- Sen D, Dagdelen S, Erbas T. Hepatosteatosis with hypobetalipoproteinemia. J Natl Med Assoc. 2007;99:284–286.
- Tanoli T, Yue P, Yablonskiy D, et al. Fatty liver in familial hypobetalipoproteinemia. J Lipid Res. 2004;45:941–947.
- Tarugi P, Lonardo A, Ballarini G, et al. Fatty liver in heterozygous hypobetalipoproteinemia caused by a novel truncated form of apolipoprotein B. Gastroenterology. 1996;111:1125–1133.
- Bonnefont-Rousselot D, Condat B, Sassolas A, et al. Cryptogenic cirrhosis in a patient with familial hypocholesterolemia due to a new truncated form of apolipoprotein B. Eur J Gastroenterol Hepatol. 2009;21:104–108.
- Lonardo A, Tarugi P, Ballarini G, et al. Familial heterozygous hypobetalipoproteinemia, extrahepatic primary malignancy, and hepatocellular carcinoma. Dig Dis Sci. 1998;43:2489–2492.
- Pelusi S, Baselli G, Pietrelli A, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep. 2019;9:3682.
- Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia: liver disease and cardiovascular disease. Curr Opin Lipidol. 2020;31:49–55.
- Peloso GM, Nomura A, Khera AV, et al. Rare protein-truncating variants in APOB, lower low-density lipoprotein cholesterol, and protection against coronary heart disease. Circ Genomic Precis Med. 2019;12:e002376.
- Bayona A, Arrieta F, Rodríguez-Jiménez C, et al. Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: a case report. Medicine (Baltimore). 2020;99:e21754.
- Small AM, Huffman JE, Klarin D, et al. PCSK9 loss of function is protective against extra-coronary atherosclerotic cardiovascular disease in a large multi-ethnic cohort. PLoS ONE. 2020;15:e0239752.
- Paquette M, Saavedra YGL, Poirier J, et al. Loss-of-function PCSK9 mutations are not associated with Alzheimer disease. J Geriatr Psychiatry Neurol. 2018;31:90–96.
- Rimbert A, Smati S, Dijk W, et al. Genetic inhibition of PCSK9 and liver function. JAMA Cardiol. 2021;6:353–354.
- Tada H, Okada H, Nomura A, et al. A healthy family of familial hypobetalipoproteinemia caused by a protein-truncating variant in the PCSK9 gene. Intern Med. 2020;59:783–787.
- Lebeau PF, Wassef H, Byun JH, et al. The loss-of-function PCSK9Q152H variant increases ER chaperones GRP78 and GRP94 and protects against liver injury. J Clin Invest. 2021;131:e128650.
- Grimaudo S, Bartesaghi S, Rametta R, et al. PCSK9 rs11591147 R46L loss-of-function variant protects against liver damage in individuals with NAFLD. Liver Int. 2021;41:321–332.
- Di Filippo M, Vokaer B, Seidah NG. A case of hypocholesterolemia and steatosis in a carrier of a PCSK9 loss-of-function mutation and polymorphisms predisposing to nonalcoholic fatty liver disease. J Clin Lipidol. 2017;11:1101–1105.
- Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272.
- Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol. 2017;28:161–169.
- Hopewell JC, Malik R, Valdés-Márquez E, et al. Differential effects of PCSK9 variants on risk of coronary disease and ischaemic stroke. Eur Heart J. 2018;39:354–359.
- Kent ST, Rosenson RS, Avery CL, et al. PCSK9 loss-of-function variants, low-density lipoprotein cholesterol, and risk of coronary heart disease and stroke: data from 9 studies of blacks and whites. Circ Cardiovasc Genet. 2017;10:e001632.
- Langsted A, Nordestgaard BG, Benn M, et al. PCSK9 R46L Loss-of-function mutation reduces lipoprotein(a), LDL cholesterol, and risk of aortic valve stenosis. J Clin Endocrinol Metab. 2016;101:3281–3287.
- Minicocci I, Montali A, Robciuc MR, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97:E1266–1275.
- Su X. ANGPLT3 in cardio-metabolic disorders. Mol Biol Rep. 2021;48:2729–2739.
- Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–2227.
- Minicocci I, Santini S, Cantisani V, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54:3481–3490.
- Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211–221.
- Robciuc MR, Maranghi M, Lahikainen A, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler Thromb Vasc Biol. 2013;33:1706–1713.
- Stitziel NO, Khera AV, Wang X, et al. ANGPTL3 Deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–2063.
- Gretarsdottir S, Helgason H, Helgadottir A, et al. A splice region variant in LDLR lowers non-high density lipoprotein cholesterol and protects against coronary artery disease. PLoS Genet. 2015;11:e1005379.
- Natarajan P, Peloso GM, Zekavat SM, et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat Commun. 2018;9:3391.
- van Zyl T, Jerling JC, Conradie KR, et al. Common and rare single nucleotide polymorphisms in the LDLR gene are present in a black south African population and associate with low-density lipoprotein cholesterol levels. J Hum Genet. 2014;59:88–94.
- Björnsson E, Thorleifsson G, Helgadóttir A, et al. Association of genetically predicted lipid levels with the extent of coronary atherosclerosis in Icelandic adults. JAMA Cardiol. 2020;5:13–20.
- Blanco-Vaca F, Martin-Campos JM, Beteta-Vicente Á, et al. Molecular analysis of APOB, SAR1B, ANGPTL3, and MTTP in patients with primary hypocholesterolemia in a clinical laboratory setting: evidence supporting polygenicity in mutation-negative patients. Atherosclerosis. 2019;283:52–60.
- Rimbert A, Vanhoye X, Coulibaly D, et al. Phenotypic differences between polygenic and monogenic hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2020;41:e64–71.
- Balder JW, Rimbert A, Zhang X, et al. Genetics, lifestyle, and low-density lipoprotein cholesterol in young and apparently healthy women. Circulation. 2018;137(8):820–831.
- Cefalù AB, Spina R, Noto D, et al. Comparison of two polygenic risk scores to identify non-monogenic primary hypocholesterolemias in a large cohort of Italian hypocholesterolemic subjects. J Clin Lipidol. 2022;16:530–537.
- van den Boogert MAW, Rader DJ, Holleboom AG. New insights into the role of glycosylation in lipoprotein metabolism. Curr Opin Lipidol. 2017;28:502–506.
- van den Boogert MAW, Larsen LE, Ali L, et al. N-glycosylation defects in humans lower low-density lipoprotein cholesterol through increased low-density lipoprotein receptor expression. Circulation. 2019;140:280–292.
- Chong M, Yoon G, Susan-Resiga D, et al. Hypolipidaemia among patients with PMM2-CDG is associated with low circulating PCSK9 levels: a case report followed by observational and experimental studies. J Med Genet. 2020;57:11–17.
- Al Teneiji A, Bruun TUJ, Sidky S, et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol Genet Metab. 2017;120:235–242.
- Bjornsson E, Gunnarsdottir K, Halldorsson GH, et al. Lifelong reduction in LDL (low-density lipoprotein) cholesterol due to a gain-of-function mutation in LDLR. Circ Genomic Precis Med. 2021;14:e003029.
- Verweij N, Haas ME, Nielsen JB, et al. Germline mutations in CIDEB and protection against liver disease. N Engl J Med. 2022;387:332–344.
- Pulai JI, Latour MA, Kwok PY, et al. Diabetes mellitus in a new kindred with familial hypobetalipoproteinemia and an apolipoprotein B truncation (apoB-55). Atherosclerosis. 1998;136:289–295.
- Della Corte C, Fintini D, Giordano U, et al. Fatty liver and insulin resistance in children with hypobetalipoproteinemia: the importance of aetiology. Clin Endocrinol (Oxf). 2013;79:49–54.
- Lonardo A, Lombardini S, Scaglioni F, et al. Hepatic steatosis and insulin resistance: does etiology make a difference? J Hepatol. 2006;44:190–196.
- Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375:2144–2153.
- Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105.
- Lotta LA, Sharp SJ, Burgess S, et al. Association between low-density lipoprotein cholesterol–lowering genetic variants and risk of type 2 diabetes: a meta-analysis. JAMA. 2016;316:1383.
- Bonnefond A, Yengo L, Le May C, et al. The loss-of-function PCSK9 p.R46L genetic variant does not alter glucose homeostasis. Diabetologia. 2015;58:2051–2055.
- Khan AO, Basamh O, Alkatan HM. Ophthalmic diagnosis and optical coherence tomography of abetalipoproteinemia, a treatable form of pediatric retinal dystrophy. J Aapos. 2019;23:176–177.
- Le R, Zhao L, Hegele RA. Forty year follow-up of three patients with complete absence of apolipoprotein B-containing lipoproteins. J Clin Lipidol. 2022;16:155–159.
- Lam MCW, Singham J, Hegele RA, et al. Familial hypobetalipoproteinemia-induced nonalcoholic steatohepatitis. Case Rep Gastroenterol. 2012;6:429–437.
- Vlasschaert C, McIntyre AD, Thomson LA, et al. Abetalipoproteinemia due to a novel splicing variant in MTTP in 3 siblings. J Investig Med High Impact Case Rep. 2021;9:232470962110224.
- Fusaro M, Mereu M, Aghi A, et al. Vitamin K and bone. Clin Cases Miner Bone Metab. 2017;14:200–206.
- Palermo A, Tuccinardi D, D’Onofrio L, et al. Vitamin K and osteoporosis: myth or reality? Metabolism. 2017;70:57–71.
- Duell PB, Bakhtiani PA, Connor SL. Long-term 40-year prognosis in abetalipoproteinemia is optimized by early diagnosis and treatment. J Clin Lipidol. 2014;8:336.
- Handhle A, Viljoen A, Ramachandran R, et al. Low cholesterol syndrome and drug development. Curr Opin Cardiol. 2020;35:423–427.
- Hegele RA, Tsimikas S. Lipid-lowering agents: targets beyond PCSK9. Circ Res. 2019;124:386–404.
- Blom DJ, Cuchel M, Ager M, et al. Target achievement and cardiovascular event rates with lomitapide in homozygous familial hypercholesterolaemia. Orphanet J Rare Dis. 2018;13:96.
- Berberich AJ, Hegele RA. Lomitapide for the treatment of hypercholesterolemia. Expert Opin Pharmacother. 2017;18:1261–1268.
- Fogacci F, Ferri N, Toth PP, et al. Efficacy and safety of mipomersen: a systematic review and meta-analysis of randomized clinical trials. Drugs. 2019;79:751–766.
- Nohara A, Otsubo Y, Yanagi K, et al. Safety and efficacy of Lomitapide in Japanese patients with homozygous familial hypercholesterolemia (HoFH): results from the AEGR-733-301 long-term extension study. J Atheroscler Thromb. 2019;26:368–377.
- Underberg JA, Cannon CP, Larrey D, et al. Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: five-year data from the lomitapide observational worldwide evaluation registry (LOWER). J Clin Lipidol. 2020;14:807–817.
- Aljenedil S, Alothman L, Bélanger AM, et al. Lomitapide for treatment of homozygous familial hypercholesterolemia: the Québec experience. Atherosclerosis. 2020;310:54–63.
- Brandts J, Dharmayat KI, Vallejo-Vaz AJ, et al. A meta-analysis of medications directed against PCSK9 in familial hypercholesterolemia. Atherosclerosis. 2021;325:46–56.
- Rakipovski G, Hovingh GK, Nyberg M. Proprotein convertase subtilisin/kexin type 9 inhibition as the next statin? Curr Opin Lipidol. 2020;31:340–346.
- Raal FJ, Kallend D, Ray KK, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med. 2020;382:1520–1530.
- Ray KK, Landmesser U, Leiter LA, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. 2017;376:1430–1440.
- Rosenson RS, Hegele RA, Fazio S, et al. The evolving future of PCSK9 inhibitors. J Am Coll Cardiol. 2018;72:314–329.
- Rosenson RS, Hegele RA, Koenig W. Cholesterol-Lowering Agents: pCSK9 inhibitors today and tomorrow. Circ Res. 2019;124:364–385.
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711–720.
- Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383:2307–2319.
- Bredefeld C, Hussain MM, Averna M, et al. Guidance for the diagnosis and treatment of hypolipidemia disorders. J Clin Lipidol. 2022;16:797–812.
- Lima Pessoa E, Costa Vilella dos Reis M, Sayuri Yamamoto T, et al. Familial heterozygous hypobetalipoproteinemia and breast cancer risk: a systematic review and suggestions for further research. Breast J. 2019;25:763–765.
- Wilson MH, Rajan S, Danoff A, et al. A point mutation decouples the lipid transfer activities of microsomal triglyceride transfer protein. PLoS Genet. 2020;16:e1008941.
- Musunuru K, Chadwick AC, Mizoguchi T, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434.
- Momtazi-Borojeni AA, Jaafari MR, Afshar M, et al. PCSK9 immunization using nanoliposomes: preventive efficacy against hypercholesterolemia and atherosclerosis. Arch Med Sci. 2021;17:1365–1377.
- Fowler A, Sampson M, Remaley AT, et al. A VLP-based vaccine targeting ANGPTL3 lowers plasma triglycerides in mice. Vaccine. 2021;39:5780–5786.
- Fukami H, Morinaga J, Nakagami H, et al. Vaccine targeting ANGPTL3 ameliorates dyslipidemia and associated diseases in mouse models of obese dyslipidemia and familial hypercholesterolemia. Cell Rep Med. 2021;2:100446.
- Zhang YY, Fu ZY, Wei J, et al. A LIMA1 variant promotes low plasma LDL cholesterol and decreases intestinal cholesterol absorption. Science. 2018;360:1087–1092.