
1. Introduction
Precision medicine or personalized cancer therapy involves the tailoring of antitumor treatments to the individual clinical features, tumor molecular profiles, and associated microenvironment of patients, with the aim of treating cancer more effectively and with less toxicity [Citation1,Citation2]. Although molecularly targeted agents and immunotherapeutics have revolutionized cancer treatment in the past decade, only a limited proportion of patients respond, while those who respond eventually acquire drug resistance and develop progressive disease. There is therefore a critical need to identify and validate robust biomarkers that can predict sensitivity or resistance to such therapies. The integration of tumor genomic profiling into clinical practice has provided key insights into the complex biology of cancer development and progression. The incorporation of such technologies has however also inadvertently introduced multiple challenges associated with data analyses, including the complexities associated with the identification of driver alterations, target classification with different levels of evidence, and the selection of rational treatments for patients at an individual level with appropriate drugs at the right time [Citation3,Citation4]. In this article, we will discuss the challenges of selecting, testing, and integrating biomarkers in the drug development process and strategies for managing the many complex decisions associated with genomically matched cancer therapies. Finally, we will discuss current and future approaches for integrating biomarkers in oncology drug development in the era of real-world evidence and immunotherapy.
2. Biomarkers in drug development
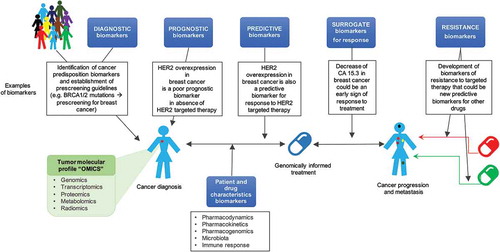
A cancer biomarker refers to a tumor characteristic or a response of the body in the presence of cancer that can be objectively measured and evaluated. This most commonly involves the assessment of a genomic alteration or a difference in protein expression levels [Citation5]. Personalized cancer therapy integrates diagnostic biomarkers (for predisposition and early detection of tumors in healthy patients), prognostic biomarkers (for evaluation of the possible natural course of the disease), predictive biomarkers (for prediction of response to a specific therapy), pharmacokinetic, pharmacodynamic, pharmacogenomic biomarkers (for evaluation of the interaction between the drug and patient), and surrogate biomarkers that could be used as intermediate end point biomarkers to determine early response or resistance to treatment ().
Figure 1. Types of biomarkers in the multistep drug development process.
It is now widely accepted that compounds which target driver genomic alterations involved in cancer initiation and progression can be successfully pursued as rational therapeutic strategies [Citation6]. Nevertheless, there are still only a limited number of effective targeted therapies associated with robust predictive biomarkers of response for the treatment of patients with solid tumors. One of the first predictive biomarkers established for treatment selection is the BCR-ABL fusion gene. The translocation between chromosomes 9 and 22 (the Philadelphia chromosome) was discovered in patients with chronic myelogenous leukemia in the 1960s, and subsequently shown to predict for response to imatinib, which was approved by the Food and Drug Administration (FDA) for this indication in 2001 [Citation7]. The development of modern and sophisticated molecular and pharmacological technologies has since contributed to an improvement in the time taken from driver genomic alteration discovery to matched drug FDA approval [Citation8] ().
Figure 2. Time between driver alteration discovery and targeted treatment approval (*year of FDA approval of the first indication for that drug).
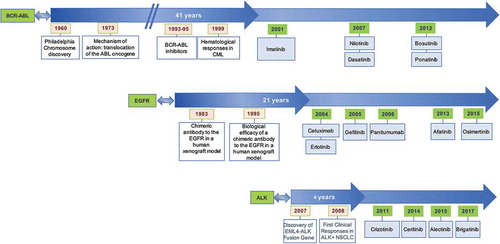
Other promising drugs which have good predictive biomarkers are currently being tested in clinical trials such as larotrectinib for cancers harboring NTRK gene fusions regardless of tumor histology [Citation9,Citation10]. There have also been successes in the development of second- and third-generation compounds, which improve on existing antitumor agents by effectively targeting resistance mechanisms to first-generation inhibitors. A recent example is osimertinib, which selectively targets EGFR T790M mutations in non-small cell lung cancers and doubled the progression-free survival from 8.5 to 17.2 months in first line of treatment when compared to other standard EGFR targeting tyrosine kinase inhibitors [Citation11]. Another success story of biomarker-driven targeted therapy is ceritinib in ALK rearranged non-squamous non-small cell lung cancer, with significant improvement of progression-free survival from 8.1 to 16.6 months when compared with standard chemotherapy [Citation12].
To continue improving on the status quo and to accelerate drug development processes, research efforts need to continue to be focused on the identification of predictive biomarkers of response, as well as intermediate end points that can provide early signals of efficacy or resistance to treatment [Citation13]. Prior to integrating such biomarkers in routine clinical practice, assays need to undergo preclinical validation with robust methodologies, followed by clinical qualification in prospective trials [Citation14,Citation15].
3. Challenges associated with tumor heterogeneity and clonal evolution
One of hallmarks of cancer is genomic instability and acquisition of abnormalities during the evolution of cancer. However, only a limited number of these alterations are bona fide cancer drivers, which confer selective growth advantages to the cancer cell; rather, the majority of these aberrations are more likely to represent passengers that do not affect the fitness of the malignant clone [Citation16]. Overall, it is now generally accepted that cancer cells harbor driver aberrations that are present in the majority of cancer cells, with the existence of subclonal alterations that wax and wane temporally and spatially with different cancer therapies [Citation17].
Although there are a number of sophisticated bioinformatic tools, which may aid in distinguishing cancer drivers from passenger alterations, intratumoral heterogeneity remains a major challenge for the identification and development of potential biomarkers. Also, while a molecular alteration is often established as a cancer driver based on data at a population level, these results are often not recapitulated at an individual patient level due to a number of other key factors, such as the tumor immune microenvironment [Citation18]. Tumors may also harbor distinct cancer cell clones with different molecular alterations, which may respond differentially to targeted therapies [Citation15]. There are now multiple well-known mechanisms of resistance to treatments targeting specific driver genomic alterations, such as mutations in the targeted gene (e.g. EGFR T790M and ALK C1156Y) or the activation of associated signaling pathways.
Potential strategies required for the development of robust biomarkers despite the challenges of evolving cancer alterations mandates translational efforts in the clinic to incorporate serial tumor samples in patients with different cancers. The serial collection of blood samples for the analysis of circulating tumor DNA (ctDNA), to support and add to tumor biopsy findings, will also aid in overcoming the logistical challenges associated with sequential tumor biopsies and may help to further evaluate the role of tumor heterogeneity [Citation19].
4. Challenges in the identification and testing of biomarkers
The initial companion diagnostic tests developed for matching patients rationally with molecularly targeted agents consisted mainly of single-gene assays. With the advances in molecular profiling technologies, our improved knowledge in tumor genomics, and the decrease in costs associated with next-generation sequencing (NGS) assays, there has been an exponential increase in the number of genes tested within multiplex panels using sophisticated platforms. This wider tumor genomic profiling has in turn led to further gains in our understanding of tumor biology and the dissection of resistance pathways related to a range of modern therapies. This has led to the development of rational combination therapies designed to overcome monotherapy drug resistance, e.g. regimens involving MEK and BRAF inhibitors [Citation20]. NGS is now a commonly utilized research tool integrated in different clinical settings, including molecular screening efforts set up to personalize antitumor therapies through the enrolment of patients onto genomically guided clinical trials.
It is clear that NGS offers undisputable benefits, but carries several challenges. The high-resolution data achieved with NGS allow the identification of a large number of genomic alterations, with a considerable increase in computational analysis and bioinformatics support that are now needed for the production and interpretation of these data. Although the large amount of data could potentially be used for the discovery of novel biological mechanisms underlying cancer progression and drug resistance, ultimately only a small fraction of the sequenced data is useful in the clinic for personalized treatment selection based on our current knowledge and availability of genomically matched therapies.
Although comprehensive molecular testing integrating the different ‘omics,’ such as proteomics and metabolomics, could help identify new predictive biomarkers of response and resistance, the lack of data reproducibility between multiple platforms and the associated challenges of analytical validation has limited their implementation in routine clinical practice. Currently, necessary efforts such as the mandatory validation of biomarkers in CLIA (clinical-laboratory improvement amendments)-certified laboratories prior to their use in the clinic to assure accuracy and reproducibility of laboratory procedures has served to ensure high-quality biomarker testing. In the future, the establishment of large knowledge bases of NGS data, increased collaborations between clinicians, scientists, clinical centers, and regulatory entities, as well as the redefinition of validation criteria may prove useful in improving biomarker development. Apart from these factors, tissue availability and tumor quality are other important challenges that need to be addressed for the application of multiple molecular and genomic analyses using current technologies.
5. Challenges in the implementation of biomarkers in clinical drug development
The next generation of clinical trials will need to take into account co-alterations that may drive resistance to single agent targeted therapies and to use such knowledge to rationally combine different anticancer drugs. However, the design of such drug regimens will require a deep understanding of the tumor biology involved, as well as preclinical and clinical pharmacology of drugs to avoid overlapping toxicities [Citation21].
Actionability of an alteration can be based on their diagnostic or prognostic value; however, the common accepted definition of an actionable alteration in precision oncology is based on the predictive response (sensitivity or resistance) to a currently genomically matched treatment that is FDA approved or available in clinical trials [Citation4]. Although some alterations may occur within an actionable gene, the alteration itself may not ultimately lead to functional change of the protein, and treating physicians should be aware of these challenges. Absence of genomic decision support for physicians can lead to the suboptimal treatment of patients by not offering genotype-matched treatments in the setting of a known pathogenic actionable genomic alteration or by targeting a non-actionable variant.
An example of knowledge base to support genomically informed matched clinical trials is the Institute for Personalized Cancer Therapy (IPCT) at the University of Texas MD Anderson Cancer Center, available through the Personalized Cancer Therapy website (https://personalizedcancertherapy.org or https://pct.mdanderson.org). This is a continuous effort to identify biomarkers by assessing the clinical significance and actionability of genomic alterations, reviewing the levels of evidence for drug effectiveness in tumors harboring for each alteration, and the subsequent identification of genomically matched targeted therapies for patients [Citation22]. Some alterations (especially tumor suppressor genes) may be considered non-actionable based on the availability of current treatments [Citation23]. For example, cancer cells with deleterious somatic or germline alterations in BRCA1 and BRCA2 genes are associated with defective DNA damage response. Targeting poly(ADP-ribose) polymerase (PARP), a key functional pathway of DNA repair, results in genomic instability in BRCA1/2-mutated cancers and eventual cancer cell death. Several PARP inhibitors (Olaparib, Rucaparib, and Niraparib) are now FDA-approved in different ovarian and breast cancers [Citation24–Citation26].
Immune checkpoint inhibitors, such as anti-cytotoxic T-lymphocyte associated antigen 4 (anti-CTLA-4), anti-programmed cell death (anti-PD-1), and anti-programmed cell death-ligand (anti-PD-L1) monoclonal antibodies, have significantly impacted cancer survival in several aggressive diseases. Pembrolizumab (anti-PD1 antibody) is now FDA-approved for tumors with microsatellite instability or pathogenic alterations in mismatch repair genes, highlighting the connection between the tumor genotype and different immune phenotypes [Citation27].
Despite rapid clinical advances and the regulatory approval of immunotherapies for multiple indications, the biological mechanisms that underlie antitumor immunity and sensitivity to these agents are still poorly understood. Moreover, the majority of patients with different cancers still do not respond to immunotherapies, highlighting the urgent need for the development of robust predictive biomarkers of response to guide the appropriate selection of patients. Although various molecular abnormalities, such as MSI status, PD-L1 expression levels [Citation28], and tumor mutational burden [Citation29], have been associated with enhanced antitumor responses to immune checkpoint inhibitors in selected populations of patients, no other biomarkers have been established as robust predictive markers of response.
There is a critical need of identification and development of biomarkers that could help select the best treatment for the patient and the right time in clinical care. Oncologists and scientists should be aware that the process of validation and implementation in the clinic of biomarkers is long and complicated, with several challenges such as tumor heterogeneity, actionability of alterations, and differences in techniques used to measure them, and they should work on developing strategies to overcome these limitations.
6. Expert opinion
The challenges of biomarkers in drug discovery and development may be considered at three different levels: (1) identifying the right target of the drug as biomarker; (2) validation of the biomarker test in question, and (3) development of matched biomarker-driven treatments (). Moving forward, it is clear that these biomarker challenges will be contingent on continuous technological improvements. For example, as DNA sequencing improves and becomes more cost-effective, whole genome sequencing could be routinely used to identify rare but highly penetrant germline alterations that could potentially lead to molecular screening programs and early detection of relevant tumors.
Figure 3. Summary of challenges of biomarkers in drug discovery and development.
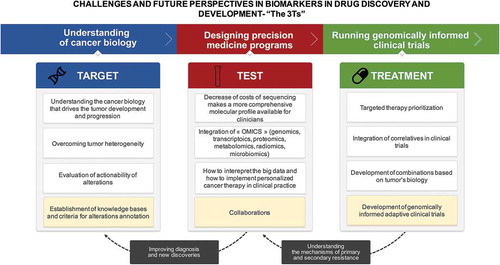
Using complex computational analyses and bioinformatics algorithms, we will also be able to integrate different biomarkers that are not limited to genomic profiles, but also include transcriptome, protein expression, and methylation. Furthermore, other patient characteristics may be integrated into prognostic and predictive signatures, such as radiomics, which integrates medical imaging data.
As a range of validated molecular profiling platforms make their way to routine clinical practice, additional challenges include the costs of implementing and validating newer tools in diagnostic laboratories, the bioinformatic efforts required to integrate and validate data, and ultimately the regulatory burden associated with accelerating the implementation of new technologies. The development of NGS protocols for use in small tissue samples or liquid biopsies (e.g. ctDNA) will provide invaluable opportunities to investigate genomic changes early in the malignant process and to detect tumor progression before traditional radiologic changes.
The establishment of knowledge bases and mutual sharing of data, as well as regular consensus meetings to discuss the reporting of biomarkers results will likely be a vital component of precision medicine research efforts and a key cog in informing the next generation of clinical trials. There is already a shift from the classical paradigm of Phase I clinical trials of identifying the maximum tolerated dose to ones that routinely integrate correlative studies in early clinical studies to improve our understanding of the underlying tumor biology and different putative mechanisms of resistance. Also, there is an urgent need to improve strategies to tailor both dose and schedule of drugs based on the pharmacodynamic effects, rather than just drug-related toxicities.
In the coming years, targeting key driver pathways and the immune response should involve target and therapy prioritization based on available levels of evidence, as well as the development of adaptive combination clinical trials to minimize the treatment of patients with ineffective and potentially toxic drugs. Ultimately, while the existing armamentarium of treatment modalities, such as targeted therapies, immunotherapies, and chemotherapies, has impacted patient benefit, it has brought new challenges in the identification and validation of predictive biomarkers for response to treatments, which will require the collaboration and sharing of knowledge between oncologists, immunologists, and molecular biologists to optimize their application in cancer medicine.
Declaration of Interest
F Meric-Bernstam has received research funding from Novartis, AstraZeneca, Taiho, Genentech, Calithera, Debiopharma, Bayer Healthcare, PUMA, Aileron, Jounce, CytoMx, eFFECTOR Therapeutics, Zymeworks, Curis and Pfizer Inc. F Meric-Bernstam acted as a paid consultant for Dialecta and Sumitomo Dainippon Pharma as well an advisory committee/board member for Inflection Biosciences, Clearlight Diagnostics, Pieris, Darwin Health and GRAIL. TA Yap has received research funding from AstraZeneca, Vertex Pharmaceuticals and Clearbridge Biomedics. He is also an advisory board member for AstraZeneca, Pfizer Inc, EMD Serono, Clovis, Bristol-Myers Squibb, Ignyta, Roche, Janssen Pharmaceuticals, Atrin and Aduro and has received travel support from AstraZeneca, Bristol-Myers Squibb, Merck Sharp Dohme, Vertex Pharmaceuticals, GlaxoSmithKline, EMD Serono and Tesaro. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Massard C, Michiels S, Ferté C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7(6):586–595.
- Meric-Bernstam F, Mills GB. Overcoming implementation challenges of personalized cancer therapy. Nat Rev Clin Oncol. 2012;9(9):542–548.
- Meric-Bernstam F, Johnson A, Holla V, et al. A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst. 2015;107(7):1–9.
- Johnson A, Zeng J, Bailey AM, et al. The right drugs at the right time for the right patient: the MD Anderson precision oncology decision support platform. Drug Discov Today. 2015;20(12):1433–1438.
- Atkinson AJ, Colburn WA, DeGruttola VG, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95.
- Dancey JE, Bedard PL, Onetto N, et al. The genetic basis for cancer treatment decisions. Cell. 2012;148(3):409–420.
- Druker B, Talpaz M, Piesta D, et al. Journal efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037.
- Shaw AT, Kim D-W, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394.
- Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–714.
- Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–739.
- Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med. 2017;378(2):113–125.
- Soria J-C, Tan DSW, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. 2017;389(10072):917–929.
- Smith AD, Roda D, Yap TA. Strategies for modern biomarker and drug development in oncology. J Hematol Oncol. 2014;7(70):1–16.
- FDA Biomarker Qualification Program. Washington, DC: FDA/Center for drug evaluation and research, 2018; 2005 [cited 2018 May 8. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram.
- Yap TA, Workman P. Exploiting the cancer genome: strategies for the discovery and clinical development of targeted molecular therapeutics. Annu Rev Pharmacol Toxicol. 2012;52(1):549–573.
- Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501.
- Greaves M, Maley C. Clonal evolution in cancer. Nature. 2012;481(7381):306–313.
- Andre F, Mardis E, Salm M, et al. Prioritizing targets for precision cancer medicine. Ann Oncol. 2014;25(12):2295–2303.
- Volik S, Alcaide M, Morin RD, et al. Cell-free DNA (cfDNA): clinical significance and utility in cancer shaped by emerging technologies. Mol Cancer Res. 2016;14(10):898–908.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2014;372(1):30–39.
- Yap TA, Omlin A, De Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31(12):1592–1605.
- Kurnit KC, Bailey AM, Zeng J, et al. Personalized cancer therapy: a publicly available precision oncology resource. Cancer Res. 2017;77(21):e123–e126.
- Chen B, Butte AJ. Leveraging big data to transform target selection and drug discovery. Clin Pharmacol Ther. 2016;99(3):285–297.
- Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164.
- Robson M, Im S-A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–533.
- Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961.
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520.
- Gettinger S, Rizvi NA, Chow LQ, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(25):2980–2987.
- Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in Melanoma. N Engl J Med. 2014;371(23):2189–2199.