
ABSTRACT
Introduction
Tuberculosis requires lengthy multi-drug therapy. Mycobacterium tuberculosis occupies different tissue compartments during infection, making drug access and susceptibility patterns variable. Antibiotic combinations are needed to ensure each compartment of infection is reached with effective drug treatment. Despite drug combinations’ role in treating tuberculosis, the design of such combinations has been tackled relatively late in the drug development process, limiting the number of drug combinations tested. In recent years, there has been significant progress using in vitro, in vivo, and computational methodologies to interrogate combination drug effects.
Areas covered
This review discusses the advances in these methodologies and how they may be used in conjunction with new successful clinical trials of novel drug combinations to design optimized combination therapies for tuberculosis. Literature searches for approaches and experimental models used to evaluate drug combination effects were undertaken.
Expert opinion
We are entering an era richer in combination drug effect and pharmacokinetic/pharmacodynamic data, genetic tools, and outcome measurement types. Application of computational modeling approaches that integrate these data and produce predictive models of clinical outcomes may enable the field to generate novel, effective multidrug therapies using existing and new drug combination backbones.
1. Introduction
Since the introduction of antibiotics, tuberculosis (TB) treatment has required months or years to achieve effective disease resolution. Culturable Mycobacterium tuberculosis (Mtb), the causative agent of TB, from patients decreases rapidly during the first few weeks of treatment [Citation1]. However, historically, short-duration therapies (e.g. up to a few months) resulted in patients with disease at the end of treatment and a high percentage of disease relapse [Citation1,Citation2], indicating that there are Mtb that can survive drug treatment to regrow and cause disease once drug treatment ends. Some patients are successfully treated with short treatment times, while others are not. There are currently no diagnostic tests to indicate when enough therapy for a relapse-free cure has been given to an individual patient. Thus, treatment times must be lengthy for all patients to ensure the best possible outcome for most patients. Why are such prolonged treatment regimens required to achieve the durable curing of TB? Decades of research into this question suggest that the answer is partly due to the immune response to TB infection and the resulting disease pathology and partly due to the ability of Mtb to alter its metabolism and physiology to survive changing environmental cues [Citation3].
Clinical trials have demonstrated that TB treatment must include multiple drugs given for several months to achieve an effective and durable cure. The current standard of care treatment for drug-sensitive (DS) TB is 6 months and consists of two phases of drug treatment: four drugs (isoniazid (H), rifampicin (R), pyrazinamide (Z), and ethambutol (E)) given for 2 months followed by two drugs (HR) for 4 months. This treatment has been used for over 40 years, with an estimated treatment effectiveness of 85% [Citation2,Citation4,Citation5]. Furthermore, the estimated number of TB cases resistant to currently used drugs (including drug-resistant (DR) and multidrug-resistant (MDR)) remains high, at more than 500,000 annually [Citation4]. Treatment for DR TB has historically required more than four drugs given for up to 24 months due to lower drug sterilizing abilities and toxicity, was without standardization, and has left many patients without positive outcomes (<50% curing) [Citation6]. Given the development of novel antibiotics, new clinical trial designs, and positive clinical trial results [Citation7,Citation8], there is the promise to develop more effective therapies for TB. Treatment guidelines now recommend a standardized and shorter treatment (BPaL) for most DR TB patients. Therefore, there is reason to be optimistic about the discovery of other effective treatment regimens for TB using new drugs and new design principles.
This review will discuss the lessons learned, the current state of primarily empirically determined drug treatment regimens, and developments in the systematic and rational exploration of combination drug space to identify the next generation of TB multidrug regimens. We will cover a brief history of how the current standard of TB drug treatment was empirically determined (Section 2), why multidrug therapies are needed to target Mtb during treatment (Section 3), and current and future approaches for identifying the best new candidates for shortening treatment time and increasing treatment efficacy (Section 4).
2. Empirical establishment of combination therapy for Mtb
2.1. TB treatment from palliative care to combination antimicrobial therapy
Historically, TB treatment was necessarily empirical and iterative. Until the mid-twentieth century, TB treatment consisted of palliative care and the isolation of infected individuals to minimize the spread of disease [Citation1,Citation2,Citation9]. Drug treatment for TB had been long sought after but not proven effective until the discovery of streptomycin (S) and para-aminosalicylic acid (PAS) in 1944 [Citation2,Citation9]. These antibiotics were quickly found to be efficacious for treating TB in the clinic [Citation2]. Only a few years after the first uses, treatment with both S and PAS in a clinical trial demonstrated the increased efficacy of combination therapies while preventing the development of common drug resistance in monotherapies [Citation10]. The discovery of isonicotinic acid hydrazide (H, isoniazid) in 1952, which also displays activity against Mtb, quickly led to a three-drug combination therapy H+S+PAS that had predictably high cure (>90%) in patients with as low as 4% disease relapse [Citation2,Citation10].
2.2. The modern short-course therapy
These advances in treatment were significant but required continuous antibiotic therapies for 24 months. In the 1960s, ethambutol (E) was introduced and led to the replacement of PAS in the three-drug therapy (HSE) because it shortened treatment time to 18 months while also being better tolerated than PAS. Many antibiotics currently used as second-line treatments for drug-resistant TB were also developed during the 1960s [Citation2]. Rifamycins, antibiotics produced by soil bacteria, were discovered in 1957 and chemically modified to show more efficacy in vivo during the 1960s [Citation2,Citation9,Citation11]. A semisynthetic antibiotic, rifampicin (R), was developed during the 1960s [Citation2] and added to the TB combination regimen (HRSE) because of the dramatic improvement in treatment time from 18 to 9 months [Citation1]. Pyrazinamide (Z) was rediscovered in 1972, and in clinical trials, it was shown to provide sterilizing activity in the first few months of treatment that further reduced treatment time to 6 months when combined with isoniazid and rifampicin (HRZ) [Citation1]. Treatment outcome improvement was achieved through the incremental addition or substitution of drugs. The short-course therapy and treatment regimen was developed in the late 1970s based on these decades of clinical trials. Ethambutol was added shortly after the development of the short-course therapy regimen to combat the resistance to isoniazid and led to the standard of care today (HRZE). Further treatment improvement using substitutions was recently realized after more than 40 years of attempts with the shortening of drug therapy to 4 months by substituting isoniazid and rifampicin for moxifloxacin (M) and rifapentine (P) [Citation7]. The design principles leading to our best TB treatment options have been primarily driven by the iterative addition or substitution of drugs to clinically effective combinations. To understand how to develop the next generation of effective TB drug combinations, it is helpful to consider what is achieved by multidrug therapy and what population of Mtb requires lengthy treatment so that it can be specifically targeted to achieve shorter-duration therapies.
3. Combination therapy as a means to treat TB in multiple physical compartments
TB’s disease pathology helps us understand why Mtb infections can be challenging to treat and require multidrug therapies for extended periods. The pathology of pulmonary TB involves a coordinated immune response that includes multiple cell types that evolve during the progression and treatment of the disease. The complex and dynamic nature of the infection environment suggests that Mtb must survive a variety of stressors and possess the physiological flexibility to adjust to the changing surroundings. The pulmonary lesions of TB disease are organized, inflammatory, immune structures that consist of multiple cell types surrounding a core of either intracellular or extracellular Mtb [Citation12,Citation13]. Several lesion (granuloma) types have been described, each with characteristic immune cell composition, organization, degrees of necrosis of host cells, and eventually cavitation [Citation14,Citation15]. Necrotic and cavitating lesions are associated with high levels of extracellular Mtb and are more difficult to treat in humans [Citation16] as well as animals [Citation17].
3.1. Differential drug distribution
There is evidence in humans that lesion types have differential responses to drug treatment that may be influenced by the amount of drug that reaches Mtb. Recent positron emission tomography/computed tomography (PET/CT) studies monitoring lesion response to drug treatment within individual patients during 14 days of drug treatment observed that some lesions within a patient shrink while others enlarge [Citation18] suggesting there is heterogeneity of lesion response to drugs within individual patients. Studies of drug distributions in patients with MDR TB demonstrated that linezolid (L) penetrates evenly through caseous lesions. In contrast, other drugs like clofazimine (C) do not penetrate the necrotic core of lesions, confirming what had been observed in animal models () [Citation19–23]. Together, these studies support the idea that Mtb in different lesions will respond to drugs differently in part because the concentrations of drugs where Mtb are located within the lesions are variable. These results provide a plausible explanation for why treatment may take a long time and why therapy with multiple drugs and/or higher doses provides the shortest and most effective cure. Furthermore, the design of new drug combination therapies should ensure that more than one drug is present in every lesion type at levels that will kill Mtb.
Figure 1. Diagram of tissue drug distribution in Mtb-infected lungs. Diagram of lungs with cellular (solid arrow) and caseous (dashed area) lesions (a). Pyrazinamide penetrates both lesion types (b) [Citation22,Citation101]. Rifampicin penetrates both lesion types and accumulates in the caseous core (c) [Citation22,Citation112]. Bedaquiline penetrates cellular lesions but is partially excluded from the core of caseous lesions (d) [Citation101]. The relative drug concentration scale (from low to high) is shown in greyscale bar in the upper right.
![Figure 1. Diagram of tissue drug distribution in Mtb-infected lungs. Diagram of lungs with cellular (solid arrow) and caseous (dashed area) lesions (a). Pyrazinamide penetrates both lesion types (b) [Citation22,Citation101]. Rifampicin penetrates both lesion types and accumulates in the caseous core (c) [Citation22,Citation112]. Bedaquiline penetrates cellular lesions but is partially excluded from the core of caseous lesions (d) [Citation101]. The relative drug concentration scale (from low to high) is shown in greyscale bar in the upper right.](/cms/asset/ede40c5e-f208-4551-beb7-3a5b7db5d210/iedc_a_2157811_f0001_b.gif)
3.2. Mtb dormancy and persistence
The lesion microenvironment (e.g. lipid carbon sources, iron deprivation) and stressors (e.g. acidic pH, oxygen levels) can induce a non-replicative or dormant state [Citation17,Citation24–27]. Mtb adapted to these conditions respond with changes in transcriptomic, proteomic, and metabolic activities [Citation28,Citation29]. Many antibiotics target actively replicating cells and thus may provide limited inhibition when treating infections containing non-replicating Mtb. Support for this idea comes from in vitro experiments [Citation28,Citation30,Citation31], such as those demonstrating that some drugs in the standard of care regimen targeting the growth of new mycolic acid cell wall components (H and E) are more effective against actively growing Mtb [Citation1,Citation2,Citation32–37]. The other drugs in the regimen (R and Z) can kill both actively growing and non-replicating Mtb and may partially explain why including these two drugs in regimens were associated with treatment shortening during the development of the current standard of care regimen [Citation1,Citation2,Citation36,Citation37]. More recently, drugs such as bedaquiline and pretomanid that display activity toward both replicating and non-replicating Mtb have been used effectively to treat Mtb in vitro [Citation38,Citation39] and clinically [Citation8,Citation40,Citation41]. Furthermore, there are antibiotics that can selectively kill non-replicating Mtb [Citation30,Citation31,Citation34,Citation42–47]. Thus, there is hope that treatment-shortening regimens will be found by combining drugs that target both replicating and non-replicating Mtb.
As defined in the Consensus Statement from Balaban et al., tolerance describes cells that survive lethal or inhibitory concentrations of a drug without genetic resistance [Citation48]. Tolerance of a subpopulation (persistence) allows the survival of a subgroup of cells for extended periods without changing the minimum inhibitory concentration (MIC) for the whole population [Citation48]. This is particularly relevant for mycobacterial cells because, as a population, they are intrinsically heterogeneous in their drug response. Differential drug susceptibility at the single-cell level is observed in vitro in a drug-specific manner [Citation49], suggesting that there is always a subpopulation of Mtb cells that may tolerate a single-drug treatment and therefore could contribute to the challenge in rapidly clearing Mtb using antibiotic treatments. Persister cells may also be a stepping-stone to resistance [Citation50], as has been observed in other bacterial species [Citation51,Citation52]. Moreover, in these studies, the tolerance to one drug preceded resistance development to a second drug. It may be that Mtb persisters in TB patients could present a risk for developing resistance to multiple drugs in the current treatment regimen. Care must also be taken to ensure that drug combinations are designed, so they are not prone to cross-resistance [Citation50]. Targeting tolerant and dormant cells is a focus of new TB drug development [Citation30].
3.3. Multidrug therapies reduce treatment time and drug resistance development
Drug resistance is a major issue in treating diseases such as bacterial infections and cancer. The likelihood of developing resistance increases with the duration of drug exposure. Therefore, the months-long treatment time for TB is sufficient for resistance development to occur [Citation53,Citation54]. Drug resistance can occur when cells acquire a factor (for example, a mutation in the drug target or mutation that increases the expression of an efflux pump) that allows cells carrying this factor to survive in the presence of drug concentrations that will inhibit or kill cells without the factor [Citation48,Citation55,Citation56]. These factors are heritable, and once resistance develops, a drug is ineffective at clinically useful concentrations; the resistant population can survive and expand into the space previously occupied by the now-killed susceptible bacteria.
The use of multiple drugs that target different cellular processes is a strategy that can prevent the development of resistance experimentally and clinically. By targeting various essential cell processes, the likelihood for a single cell to be simultaneously resistant to multiple drugs is low. Historically, multidrug therapies for TB showed improved curing with fewer patients relapsing with drug-resistant Mtb compared with single-drug therapies [Citation1]. Using as a reference the clinically determined spontaneous streptomycin resistance rate of one in 105, one in 1015 Mtb bacilli would be triple drug-resistant [Citation57]. Thus, multidrug therapies with more than three drugs should be effective because there should be fewer than one spontaneously triple drug-resistant Mtb cell in a severely infected patient, using the severe case of cavitary disease bacillary burden as a reference [Citation2,Citation58]. Based on these calculations, the likelihood of multidrug therapy success is supported by the success of three- and four-drug regimens for treating TB [Citation1,Citation7,Citation8].
Decreased resistance development was clinically demonstrated during the early trials using S and PAS. In one early study, S monotherapy resulted in 20% of patients developing resistance [Citation2] and the randomized control trials from the Medical Research Council (MRC) in the United Kingdom found streptomycin resistance in 70% of cases [Citation2,Citation59,Citation60]. In contrast, the MRC studies found that streptomycin resistance could be detected in 41% of cases receiving both S and PAS and was found in as few as 9% of cases when PAS was administered every 3 days [Citation2,Citation59,Citation60]. These early studies demonstrated the necessity of combination therapy to prevent resistance development. Therefore, it is not surprising that the development of short-course therapy for TB treatment utilizes at least three drugs. Clinical studies have demonstrated that we require combination therapy to bet-hedge treatment against variation in access and susceptibility to Mtb that resides in different lesions. Combination therapies should therefore include drugs that coordinate to rapidly reduce bacterial burden and then sterilize persister cells.
4. New era of combination therapy design
The use of animal and in vitro models of TB has been key to the development and design of drug combinations, as well as to the optimization of combination therapies. In the following sections, we will discuss the models, their uses, and computational approaches that will aid in identifying new combinations and designing future TB treatment regimens.
4.1. Optimizing combinations based on backbones
Dozens of drugs are being considered for inclusion in new TB drug combinations [Citation61–63]. These include drugs that have already been approved for use in humans, like moxifloxacin or linezolid, and are being repurposed or considered for use in TB treatments. There is considerable optimism and excitement that treatment improvement can be achieved with combinations including drugs, such as bedaquiline, and pretomanid, from newly discovered drug classes. However, the number of possible drug combinations among TB antibiotics is immense. Therefore, one successful approach has been to develop new combination therapies by augmenting and modifying effective backbones. Isoniazid, rifampicin, and pyrazinamide (HRZ) form the backbone of the current four-drug standard of care (HRZE) [Citation1]. Over the last 15 years, the three-drug combination of pretomanid, moxifloxacin, and pyrazinamide was developed based partly on preclinical models’ success [Citation64–66] and evaluated in a clinical study [Citation67]. Early optimism about the potential clinical efficacy of this three-drug combination backbone led to the initiation of the ongoing SimpliciTB clinical trial [Citation68] to evaluate the addition of bedaquiline to pretomanid, moxifloxacin, and pyrazinamide for the treatment of both DS and DR TB. Modifying these backbones through the addition and substitution of drugs has identified regimens with proven or potential treatment improvement [Citation7,Citation68,Citation69].
The Nix-TB [Citation8] and ZeNix-TB [Citation41] clinical trials recently demonstrated dramatic treatment shortening of DR TB with the three-drug combination of bedaquiline, pretomanid, and linezolid (BPaL). BPaL was developed, in part, with preclinical studies that evaluated the addition of various drugs to backbones such as BPa [Citation65,Citation70–72]. Despite the treatment shortening and efficacy of BPaL, toxicity is associated with high and prolonged dosing of linezolid [Citation8,Citation73]. Therefore, there is interest in identifying drugs to substitute for linezolid (e.g. less toxic oxazolidinones or other drugs from other classes).
The process of addition and substitution of drugs has continued to be used to identify new treatment-improving drug combinations. However, clinical trial successes and failures highlight the importance of making the best choices for combinations to test in the clinic. The choice of drugs in the current standard-of-care treatment regimen was identified through decades of clinical evaluation of the addition or substitution of drugs [Citation1]. Clinical trials evaluating the substitution of rifapentine for rifampicin [Citation74,Citation75] and the substitution of moxifloxacin for either ethambutol or isoniazid [Citation76] were unsuccessful at identifying treatment-shortening regimens. However, the combination of rifapentine, moxifloxacin, pyrazinamide, and ethambutol was demonstrated to be treatment-shortening in a recent clinical trial [Citation7] and shares two drugs with the current standard of care. The combination was in part tested because of the potential seen in the earlier unsuccessful clinical studies, and consideration of moxifloxacin as a potential treatment-shortening drug for inclusion was also strongly supported through testing in preclinical mouse models [Citation65,Citation66,Citation77–85]. The ongoing TRUNCATE-TB trial [Citation69] further explores the potential for treatment shortening using substitution and addition of drugs to the current standard of care. Another recent example comes from the early optimism in the STAND clinical trial [Citation67] evaluating pretomanid, moxifloxacin, and pyrazinamide that led to the addition of bedaquiline and the initiation of the ongoing SimpliciTB clinical trial [Citation68] to evaluated BPaMZ for treatment of both drug-sensitive (DS) TB and MDR TB. These examples of recent trials evaluated seven drug combinations and identified between one and two treatment-improving regimens and have taken more than 12 years to complete. This underscores that choosing combinations for evaluation is essential to maximize the possibility of identifying treatment-improving regimens.
Clinical trials have demonstrated that adjustments (addition or substitution) of the drugs to a backbone combination can lead to improvements in treatment outcomes. In addition, recent trials have established paths for new regimens based on novel backbones and significant shifts on old backbones. Parallel work on novel backbones and refinement of existing backbones should be pursued to maximize the potential to identify treatment-improving drug combinations.
4.2. Preclinical animal models of TB disease
The drug combination space to be explored with new drugs is large and cannot be surveyed in the clinic. Animal models are important for evaluating drugs and drug combination design in modern TB regimen design and have been extensively reviewed [Citation86–88]. Consideration of the drugs highlighted in the clinical trials above (e.g. moxifloxacin, bedaquiline, pretomanid) as potentially treatment-shortening drugs for inclusion in regimens was strongly supported through testing in preclinical mouse models of TB disease. While systematic evaluation of drug combination efficiencies in animal models is not feasible, evaluation in animal models is essential before progression to clinical trials. We will briefly cover animal models and their uses for drug combination design.
Animals have been used to test new TB drugs’ efficacy and regimens since the introduction of streptomycin [Citation2]. Additionally, our understanding of disease pathology and the biology of Mtb and host response to infection have depended on animal studies [Citation86–90]. Mice are the most used animal models, but most strains lack many of the pathological hallmarks of human TB disease [Citation91]. Other animal models (e.g. guinea pigs, rabbits, non-human primates) have granulomas more like those in human disease, but they cannot be practically used for large-scale drug treatment efficacy studies.
Mouse models of TB are the most practical and economical for drug efficacy testing due to their ubiquity in basic research and cost, ease of genetic modification, ease of handling, and the need for relatively small amounts of drugs [Citation83,Citation92]. The most commonly used mouse strains (e.g. BALB/c, C57BL/6, and Swiss) form lesions where Mtb is intracellular and lacks the caseous, necrotic lesions or cavitations that occur in human disease. Despite these and other limitations, drug efficacy studies using these strains have produced a large amount of data, and these strains have demonstrated utility in evaluating new drugs and combinations. The treatment-shortening effects of pyrazinamide and rifampicin [Citation93] were shown in mice. The newly approved multi-drug resistant treatment of bedaquiline, pretomanid, and linezolid was also effective in mouse studies [Citation65,Citation66,Citation71,Citation94]. Because of these successful examples and the practical aspects outlined above, these mouse strains will continue to play a prominent role in preclinical drug efficacy testing.
More recently, the C3HeB/FeJ mouse strain has been used in drug efficacy studies. These animals form heterogeneous lesion types: intracellular (similar to those observed in BALB/c mice), ones with caseous, necrotic cores that are similar in pathology to human lesions, and intracellular lesions enriched with neutrophils. C3Heb/FeJ mice also have heterogeneous drug responses. Treatment-responsive and non-responsive mice can be observed with pyrazinamide, which is thought to be influenced by heterogeneous lesion types. Drug combination efficacy studies have also shown the utility of the C3HeB/FeJ strain for evaluating drug combinations that show clinical efficacy [Citation81,Citation94–98].
Rats are commonly used in toxicology and pharmacology studies to evaluate the pharmacokinetics of drugs but were considered too resistant to Mtb infection to be useful. More recent studies have shown that Wistar rats can be infected and show similar pathology to mice [Citation86,Citation88]. Furthermore, a DprE inhibitor was tested in a rat infection model because optimal drug exposure was not attainable in mice [Citation99]. The common use of rats in toxicology makes future development of this animal model for studying drug combination efficacy and pharmacokinetics promising.
Guinea pigs, rabbits, and non-human primates (NHP) form lesions that show caseation necrosis, while cavitation can also be observed in rabbits and NHP. These human-like pathologies make these animal models important for evaluating drug efficacy. Guinea pigs were the preferred animal model in the mid-1900s and were influential in developing early TB drug regimens. Recent studies of combination drug efficacy showed similar outcomes to mice and the caseous necrotic lesion contains abundant extracellular Mtb during drug treatments [Citation86,Citation88]. Even with these advantages, guinea pigs are likely to remain models for follow-up studies because they are larger, more expensive, and require more drugs during treatment because they clear drugs faster than mice. Rabbits form caseating necrotic lesions as well as lung cavities after infection with Mtb. In humans, longer TB treatment, disease relapse, and development of resistance are all associated with lung cavity formation. Additionally, drug distribution studies in the caseous lesions and pharmacokinetic studies have shown similarities with humans [Citation17,Citation20,Citation21,Citation92,Citation100–102]. Rabbit models of TB disease will continue to provide important insight into the drug treatment of refractory lesions, but due to their size, cost, and other limitations, they will not take the place of mice in evaluating drug combination efficacy early in the development process. Non-human primates infected with Mtb have the pathological hallmarks, including cavitation, as well as the symptoms and range of drug treatment outcomes that occur in human TB disease [Citation86,Citation88]. Cynomolgus macaques, and more recently marmosets, have been used to study TB disease and treatment efficacy [Citation86,Citation88,Citation103–107]. A study in marmosets showed improved treatment of cavitary lesions with the current four-drug standard of care compared to an older streptomycin-containing drug combination [Citation103]. Despite the necessary scarcity of drug combinations that can be tested in NHPs, experiments in the NHPs provide rich information that helps us understand drug combination efficacies in the context of immune effects that are similar to those in human disease.
4.3. Integrative computational modeling
Preclinical animal models will continue to serve an important role in drug efficacy testing. However, none of these models are amenable to evaluating the thousands of candidate drug combinations; therefore, another path to realizing the potential treatment-improving drug combinations is to prioritize drug combinations for animal testing and enable exploration of novel drug combinations. In vivo efficacy is an integration of drug dosing and the time resolution of drug distribution (pharmacokinetics, PK) and susceptibility of Mtb populations in different metabolic states (pharmacodynamics, PD). One scalable and economical way to integrate these data is through computational modeling. Simulations that employ mathematical and computational modeling are being used to determine drug dosing and predict clinical outcomes of tuberculosis mono- and combination therapies [Citation21,Citation108–116]. Modeling approaches utilizing drug PK and PD are predictive of drug distribution in both plasma and lungs as well as predicting potential drug–drug interactions, both of which may aid in choosing drugs to combine that minimize adverse side effects (reviewed in [Citation117,Citation118]). Utilization of PK/PD data to model drug combination effects and develop improved treatment regimens is a highly active area and has been reviewed elsewhere [Citation117,Citation119,Citation120]. These models can include patient clinical and demographic data, infection burden, blood, and serum measurements for biomarkers, as well as tissue and serum drug concentrations. Experimental measurements from human, non-human primate, small animal studies, and the Mtb response in in vitro hollow fiber models (see below) can also be used as variables in mechanistic models that are being used to design improved multidrug regimens. Treatment regimens with established clinical efficacy (HRZE and BPaL) or that are being evaluated (BPaMZ) contain drugs with differential drug distributions and effects on Mtb that are actively growing or non-replicating [Citation19–23] (). Therefore, by using information about Mtb sensitivity combined with the penetration of these drugs into compartments containing Mtb, we may be able to explain why these regimens are clinically effective. By incorporating PK/PD with models of granuloma structure and immune effects, computational simulations predict drug and combination drug response in vivo and the clinic [Citation121–126]. These simulations of granuloma formation use in vitro drug exposure measurements in addition to tissue pharmacokinetic/pharmacodynamic drug distribution and immune cell recruitment measurements from animal studies to model the dynamism of the infection environment. In recent years, integrated PK/PD models have been developed for many TB antibiotics, including isoniazid, rifampicin, rifapentine, pyrazinamide, fluoroquinolones, pretomanid, and drug combinations [Citation21,Citation109–115,Citation127]. The use of clinical modeling for predicting treatment outcomes of new drug combinations has been limited because the small number of drug combinations that have been trialed in human studies limits the ability to evaluate the predictive ability of the models. Clinical data are also complicated by other factors, such as other conditions and compliance, which may make the outcomes harder to interpret in these mathematical models. Yet, the predictive abilities of PK/PD and integrative models of single- and combination-drug responses in vivo make these models, especially those that incorporate the immune state, important tools moving forward because they allow large-scale exploration of combination regimens and optimization of dosing.
Figure 2. Diagram of tuberculosis lesion with drug penetration and Mtb cell type susceptibility. Mtb-infected lungs are shown on the left with different lesion types. Higher magnification of the lesions, highlighting the difference in lesion structure and Mtb cell growth within these lesions. These examples are high-level and generalized descriptions of major lesion types; there is a wide spectrum of lesion types that are not fully represented here but are reviewed in-depth [Citation14,Citation15]. The standard of care (HRZE) and two drug combinations showing efficacy in clinical trials (BPaMZ and BPaL) are listed on the right. *Hypoxia refers to the area around the periphery of the caseum; hypoxia levels are variable in many lesion types. Antibiotics in each combination are listed below, and the relative penetration into lesion is shown with arrows (the upward arrow shows that drug penetrates readily into lesion, and the downward arrow shows drug penetration into a lesion may be uneven or restricted). The Mtb cell type susceptibility for each drug are displayed next to each drug. BPaMZ = bedaquiline + pretomanid + moxifloxacin + pyrazinamide. BPaL = bedaquiline + pretomanid + linezolid.
![Figure 2. Diagram of tuberculosis lesion with drug penetration and Mtb cell type susceptibility. Mtb-infected lungs are shown on the left with different lesion types. Higher magnification of the lesions, highlighting the difference in lesion structure and Mtb cell growth within these lesions. These examples are high-level and generalized descriptions of major lesion types; there is a wide spectrum of lesion types that are not fully represented here but are reviewed in-depth [Citation14,Citation15]. The standard of care (HRZE) and two drug combinations showing efficacy in clinical trials (BPaMZ and BPaL) are listed on the right. *Hypoxia refers to the area around the periphery of the caseum; hypoxia levels are variable in many lesion types. Antibiotics in each combination are listed below, and the relative penetration into lesion is shown with arrows (the upward arrow shows that drug penetrates readily into lesion, and the downward arrow shows drug penetration into a lesion may be uneven or restricted). The Mtb cell type susceptibility for each drug are displayed next to each drug. BPaMZ = bedaquiline + pretomanid + moxifloxacin + pyrazinamide. BPaL = bedaquiline + pretomanid + linezolid.](/cms/asset/6dff0a64-5a23-4323-8273-52f007e71d1a/iedc_a_2157811_f0002_b.gif)
4.4. In vitro models of drug combination effects
In vitro models are important for understanding the response of Mtb to its environment and to drug challenges. These models also play a crucial role in prioritizing new drugs for the development and testing in animal infection models (see [Citation86,Citation88,Citation128,Citation129] for comprehensive reviews of models and their uses). However, in vitro models cannot capture the entire complexity of the infection environment. Until recently, there have been sparse data to articulate a clear path on how to best use Mtb drug response in vitro to evaluate in vivo drug efficacy. The increase in data availability of in vitro drug combination data has emphasized the need to develop in vitro models of microenvironments where persisters reside and to balance the simplicity of these models for systematic measurement that can be used for the prediction of in vivo outcomes.
Mtb encounters and must adapt to numerous factors and stressors during infection, including changes in nutrient availability, pH, oxygen availability, and other host-mediated stressors (). In vitro growth conditions have been developed that mimic many of these important microenvironmental factors and allow for their study in laboratory settings [Citation86,Citation88,Citation128,Citation129]. These include liquid media conditions that model single as well as multiple factors and include in vitro immune cell-based infection assays (see for selected models and the environmental factors they mimic).
Table 1. Selected microenvironmental factors encountered by Mtb during infection and in vitro models developed to mimic these factors.
Broth-based in vitro conditions have been important for understanding the environmental factors influencing Mtb growth and survival, including the effects of different lipids as carbon sources [Citation17,Citation33,Citation130,Citation131]. In vitro studies have helped us understand the key role of cholesterol for Mtb survival during infection [Citation132–136], including its use as a carbon source via beta-oxidation through its secondary metabolites as important precursors to virulence [Citation131,Citation135–137]. Mtb utilization of other lipids for growth and virulence, as well as the metabolic flux of carbon and important cofactors (e.g. Acetyl-CoA and propionyl-CoA), was described in in vitro conditions that used single lipid carbon sources such as short-chain fatty acids.
Mtb must also respond to changes in pH and oxygen levels during infection, and the details of how Mtb adapts to these changes have been studied using in vitro models. Mtb response to low pH has been well studied using growth media buffered to different pH levels that mimic the acidification of the phagosome [Citation138] as well as the more neutral pH of portions of the extracellular caseum [Citation17]. The importance of studying Mtb during pH adaptation for understanding drug response has been well documented for pyrazinamide. Pyrazinamide is generally inactive in standard laboratory media at neutral pH and is more potent at a more acidic pH [Citation139–141]. Mtb response to intracellular residency has also been studied using in vitro infection in both primary cells and cell culture systems [Citation128,Citation142,Citation143]. These studies have identified important roles for the host cells in affecting Mtb response to antibiotics and in the availability of drugs at the sites where Mtb bacilli are found [Citation144–147]. For example, both clofazimine and bedaquiline can be taken up by host cells that then serve a role in drug delivery to the intracellular Mtb. Furthermore, in vitro host cell infection models identified several potential treatment-improving drug combinations [Citation78,Citation148] that were confirmed in mouse studies [Citation78,Citation97,Citation148–150].
Oxygen availability for Mtb varies during infection, including in the caseating lesions, which have hypoxic compartments [Citation17,Citation96,Citation151]. Mtb can respond to low oxygen levels by slowing growth or entering a dormant or non-replicative state where many drugs that require active replication are not as effective [Citation17,Citation24,Citation34,Citation152–155]. Hypoxic growth conditions can be induced using environmental chambers that regulate gas concentrations but make monitoring drug response more challenging. When oxygen is low, Mtb can conduct anaerobic respiration and generate ATP by utilizing alternative electron acceptors, such as nitrate [Citation156]. This process has been shown experimentally during intracellular infection [Citation157] as well as modeled in vitro using fatty acids combined with nitrate. The reduction of nitrate during anaerobic respiration produces nitrite, which can be toxic for Mtb [Citation157]. Consequently, anaerobic respiration of nitrate in vitro can lead to an accumulation of nitrite that also induces a hypoxic non-replicative/dormant state that may be relevant to the in vivo infection environment. As the field has started to focus on developing treatment regimens that target persisters, models where multiple factors are combined in a single in vitro system to study Mtb cellular and drug response have been described [Citation128,Citation154,Citation158–160]. These multi-stress models are thought to more closely mimic the complex environments that Mtb encounters during infection.
Traditional in vitro methods for culturing and treating Mtb using broth and solid media represent controlled and consistent but static environments. Hollow fiber systems are an alternative in vitro approach in which Mtb are housed within chambers and can be exposed to dynamic media states [Citation161]. This can include changes to the culture media for monitoring growth dynamics to changing environmental cues, changes in drug concentration over time as well as multiple drug treatments [Citation116,Citation127,Citation162–164]. Hollow fiber systems allow for evaluating the PK/PD profile of a drug or combination that is more consistent with in vivo PK studies [Citation116,Citation127,Citation162,Citation163]. To date, this approach cannot be scaled to accommodate large-scale systematic studies of drugs or combinations. However, it is the only system that can monitor sequential drug treatments and dynamic changes in drug concentration more similar to the treatment environment during infection.
4.5. Measures of drug combination effects: dose responses and interactions
Systematic high-order (three or more) drug combination studies are not routinely conducted because the scale of the experiments and the size of the combination space make them logistically challenging. The checkerboard assay is a traditional method for measuring a single pairwise drug–drug interaction, wherein every drug–dose combination is measured. Checkerboard assays are resource-intensive and require, for example, one microtiter plate to measure all possible dose combinations of two drugs at 10 doses (10 × 10 = 100 drug–dose combinations). Three-dimensional checkerboard assays have been developed to study high-order drug combinations [Citation165] but require thousands of microtiter plates to measure a few high-order drug combinations. New methods have been developed to approximate essential elements of the checkerboard to enable efficient, systematic measurements of drug interactions (pairs or high-order) using in vitro models [Citation148,Citation166,Citation167]. Combinations that decrease relapse and shorten treatment time in mouse models of tuberculosis have been developed based on data from in vitro experiments [Citation78,Citation148].
The development of new methods to measure drug interactions has also highlighted the different outcome measurements from these assays. Traditionally, there is a tradeoff between the ease and the interpretation of a measurement, and these data may not always be directly comparable. Clinically, standardization in MIC determination using solid and liquid media has been set by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) [Citation168,Citation169] and can be used to measure drug combinations. However, the method has not yet been adapted to scaling for systematic drug combination studies. Colony-forming units (CFUs) are time-consuming (assayed ~4 weeks after drug treatment) and are not amenable to large-scale systematic studies. However, CFUs are considered the best measure of the ability of a drug to reduce Mtb viability. In about 2 weeks, cell viability can be estimated by adding resazurin to charcoal agar outgrowth plates instead of waiting until visible colonies are formed [Citation50,Citation153]. This method can be scaled in multiwell plates but cannot be performed in drug treatment plates for very high throughput studies. Optical density (OD) is one of the easiest measurements of an in vitro assay because it can be made directly and repeatedly on drug-treated broth-based cultures and can easily scale with the use of multiwell plates. CFU and OD are used to determine minimal inhibitory concentrations (MIC) for drugs and therefore are highly interpretable. However, its interpretation is limited because it cannot necessarily differentiate between treatment cidality and growth inhibition. OD is further limited to optically clear media and apparati. Liquid resazurin assays report on cellular metabolic activity and can be scaled in multiwell plates while also providing a quantitative readout of cell viability [Citation170–172]. Resazurin assays typically require separate assay plates, increasing the difficulty and scalability for systematic measurements. Genetically modified Mtb that can produce light (either autoluminescent or with the addition of substrate) or fluorescence provide other alternative measurements to OD for estimating cell growth [Citation131,Citation173–180] with luminescence, including the advantage of potentially providing a readout of cell viability. The genetic modifications have the limitation of potentially disrupting the Mtb physiology and requiring antibiotic selection, which may confound drug studies. Recently, drug sterilization of Mtb was estimated by measuring ribosomal RNA (rRNA) synthesis [Citation181] as a sensitive and orthogonal metric to CFUs. Whichever measurement type is made, identifying candidate drug combinations requires both an efficient approach and an interpretable measurement.
4.6. Systematic studies of drug combination effects
The expansion of methods for high-throughput measurement of drug combinations has ushered in a new era of measuring drug treatment response across a variety of in vitro growth conditions. A major obstacle in drug combination studies performed in vitro is understanding how different growth environments model in vivo niches. Decades of studies of pyrazinamide demonstrate increased efficacy in acid pH media. Other drugs with growth condition-specific activities support the continued use of multiple models for understanding Mtb drug response [Citation30,Citation141,Citation182,Citation183]. A systematic study of drug combination effects on Mtb in eight growth conditions found drug potency and drug interaction depended on the conditions in which Mtb were treated [Citation158]. The authors found that the outcome of mouse drug combination treatment could be predicted using in vitro data from subsets of growth conditions and metrics (e.g. interactions and potencies). These model-dependent results indicate that some in vitro formulations provide orthogonal information while others provide parallel information about drug combination effects on Mtb. These findings suggest that Mtb in these models are in different physiological states, perhaps analogous to Mtb in different lesion compartments, and that computational models that include combination drug response data from different relatively simple growth environments were predictive of relapse. A separate study using three growth conditions (log-phase, acid pH, and non-replicating) and testing multiple drug pairs monitored drug interaction and predicted bacterial load. Pretomanid and either moxifloxacin or bedaquiline were among the best pairs in multiple conditions showing a large bacterial burden decline with little correlation to drug interaction [Citation184]. Both studies concluded that in vitro metrics of inhibition (e.g. decreased growth and bacterial load) were more informative than drug interactions for prioritizing drug combinations. Future studies are required to quantify the utility of any in vitro models for directly understanding the in vivo combination drug response and to continue developing in vitro models for combination drug studies that may reflect Mtb in various dormancy states that enhance persistence [Citation23,Citation152–154,Citation178,Citation185]. Additionally, computational methods have been developed to aid drug discovery [Citation186] and predict drug combination effects [Citation187,Citation188]. These computational methods can complement and assist the methods for systematic, high-throughput measurements across multiple growth conditions to accelerate the identification of new effective drug combinations.
We have also entered an era where functional genomic approaches utilizing genetic tools, such as CRISPRi, are determining genetic pathways that, when perturbed, render Mtb vulnerable to drug treatments [Citation189,Citation190]. Genes in the target pathways of isoniazid and rifampicin were identified as vulnerable (inhA and rpoB, respectively), highlighting the ability of this approach to identify druggable targets and pathways. One advantage of the CRISPRi approach is the identification of new druggable pathways and the intersection of these pathways with environmental cues without having chemical compounds in hand. Intriguingly, the authors identified many genes that lack chemical inhibitors but that are more vulnerable than inhA and rpoB, indicating that future compound screens focusing on these pathways may be successful. Because gene expression can be attenuated, investigation of both essential and non-essential pathways and determination of conditional essentiality under differing growth conditions is possible. Identifying pathways vulnerable to perturbation using CRISPRi can identify new druggable pathways while also helping understand Mtb physiology and survival during infection. Chemical genetic approaches using genetic hypomorphs and pooled parallel sequencing have successfully paired effective compounds with their putative pathways of action [Citation191]. Many more hits were identified using this approach than when wild-type Mtb was used, and the target pathways identified included the synthesis of essential cell components: DNA, cell wall, tryptophan, folate, and RNA. The authors identified a mycobacterial efflux pump, EfpA [Citation192], and optimized compounds to inhibit wild-type Mtb. A follow-up study identified a structurally distinct compound that works synergistically and inhibits resistance development [Citation193]. Furthermore, a recent study determined that EfpA can induce drug tolerance to many antitubercular drugs [Citation194], suggesting that expression levels may play a role in how Mtb responds to drugs during treatment. Screens to identify mediators and inhibitors of multidrug tolerance, such as CinA [35,459,278], which cleaves NAD adducts from isoniazid, ethionamide, pretomanid, and delamanid, may extend the use of current antibiotics. Both CRISPRi and chemical genetics approaches have the potential to identify targets and compounds important for understanding drug treatment and improving future combination therapies.
The increasing availability of in vitro models and ease of measurement for drug and drug combination response to TB therapies has also created new opportunities to interpret these data. The development of experimental and computational approaches shows promise for predicting in vitro drug interactions using fewer experiments. A basis for these predictions is to take advantage of signatures of gene expression changes. These signatures predict drug interactions by using in vitro measurement in response to single-drug treatment. This enables predictions of thousands of drug combination responses from RNA sequencing data from tens of treatment groups [Citation188,Citation195]. These approaches are being used in interpreting why drugs increase efficacy while others antagonize the activity of each other and which Mtb pathways mediate these drug interactions in vitro. Additionally, experimental and computational approaches have been developed that predict drug combination treatment outcomes in preclinical and clinical studies using in vitro measurements [Citation78,Citation148–150,Citation158,Citation160,Citation181]. The insights from these treatment prediction modeling approaches can help us understand the physiological and environmental factors most relevant during drug treatment while using in vivo data to refine computational models.
5. Conclusion
The push to empirically develop treatment for TB in the twentieth century was a success and heralded randomized control trial design, which is now the gold standard for clinical trials [Citation59,Citation60]. We are entering a new era to leverage the incredible advancements in drug discovery for tuberculosis that have led to many new antibiotics and targets to be identified by focusing on how to design optimized combination therapies [Citation196]. So far, a thoughtful empirical approach to determining effective drug combinations has led to the establishment of several combination backbones with successful clinical trials. Yet, a significant number of novel combinations remains to be considered experimentally, especially involving new antibiotics. In recent years, the field has made key advances in developing animal models, in vitro models, and outcome metrics. These models were designed to better mimic the microenvironments that house the bacteria that are hardest to reach and kill with antibiotics. In the next phase of TB drug combination optimization, we expect that computational models will be used to integrate data of different types to improve methodology and our understanding of how to rationally design optimized combination therapies.
6. Expert opinion
The TB field has successfully developed new antimycobacterial drugs over the last 20 years ([Citation61–63]). Though novel multidrug regimens have been found to be superior to the standard of care and a new regimen was approved for the treatment of MDR TB, the development of optimized combinations is not at the pace at which new compounds are developed. Nonetheless, the recent success of two clinical trials with new combinations, including those incorporating antibiotics already in use, points to the potential of the combination space to yield treatment-shortening regimens. On the other hand, the failure of some clinical trials, despite the supporting in vitro and preclinical animal data, highlights that we must improve these models and refine the metrics used to evaluate treatment outcomes.
To shorten the duration of treatment, therapies must rapidly sterilize the entire Mtb population. The underlying heterogeneity of tuberculosis lesions and the bacterial population creates distinct subpopulations of Mtb that must each be penetrated by antibiotics and then must be susceptible to those drugs at the levels reached in the tissue. To achieve this, we must continue to foster a collaborative and innovative research environment to integrate higher-quality measurement across a broad range of novel combinations. These data types should take advantage of in vivo and in vitro models with predictive value against treatment-shortening outcomes in preclinical animal models and the clinic. The response of Mtb in vivo to drug treatment depends on the microenvironment to which the population has adapted; therefore, Mtb in one niche may be susceptible to a particular drug combination while Mtb in another niche are tolerant. Likewise, the response of Mtb to drug combination treatment in in vitro experiments depends on the growth environment. How should drug combinations be evaluated when shown to be effective or synergistic in one condition but not in another? We must move forward by taking advantage of the scale of in vitro studies that allow for efficient and systematic measurement of the combination space and use computational modeling to understand which of those data are predictive of the in vivo response. Furthermore, as we refine which outcomes in preclinical animal models are most predictive of treatment shortening (and relapse-less cure) in clinical trials, we should update the standards of which in vitro experimental data are most practically useful for drug combination design, e.g. which conditions and outcome metrics can be used to model treatment efficacy of Mtb in niches that are most refractory to antibiotic treatment (such as persisters).
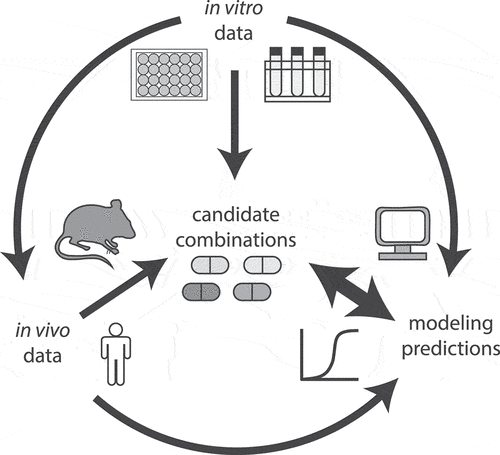
Our vision of an effective workflow for the design of combination therapy earlier in the drug development process is dependent on computational modeling to integrate disparate data on PD and PK in a lesion-specific manner (). We anticipate that computational analysis will also enable us to realize the full potential of data in a way that balances resources and experimental scale. For example, clinical and NHP data are the scarcest and are therefore only available for a few regimens. Smaller animal studies should be used to prioritize combinations for testing in NHP and in the clinic, and outcomes should be quantitatively compared so that we learn how to make these animal studies more predictive (e.g. using different PK measurements, lesion type, and outcome measures). On the other end of this scale are in vitro measures that can be acquired rapidly and are relatively high throughput. We should continue to improve the conditions and metrics used for in vitro measures to balance throughput and predictive value against in vivo efficacy. In turn, these optimized in vitro conditions may be used with genetic tools to identify vulnerable molecular pathways of Mtb in growth conditions that model where current therapies are least effective. Measuring and modeling the drug degradation in both in vitro and in vivo models, as well as understanding the tissue distribution of active or unbound forms of drugs, will play an important role in better understanding the most effective drugs to combine. In evaluating combinations, integrating data from in vivo (preclinical model) experiments and clinical trials on both treatment successes and failures will play an important role in learning what distinguishes an effective treatment combination from one that does not show efficacy. Finally, as we focus on evaluating different drug combinations for treatment efficacy, we must also consider the long-term potential of novel combinations with regard to their potential to promote phenotypic and genetic resistance.
Figure 3. Diagram of data flow for development of treatment improving candidate drug combinations. In vitro data of drug and combination effects can be directly used to inform candidate drug combination choice or can be used as input to design in vivo experiments and modeling efforts to predict optimal candidate combinations. Likewise, in vivo drug combination data (e.g. from preclinical animal models and human PK/PD studies) can be used directly to inform candidate combination choice or as input for modeling. Computational modeling that integrates multiple data sources, including effective in vitro combinations, drug tissue PK/PD, and preclinical drug combination efficacy can be leveraged to select candidate combination choice; as more combination data become available, the models can be refined and improved.
Article highlights
Tuberculosis combination therapy has primarily been empirically determined and based on three-drug combination backbones.
Combination therapies for long durations are required to sterilize multiple and diverse infection microenvironments.
Experimental advances allow systematic evaluation of new drug target pathways and drug combinations.
To shorten the duration of treatment, therapies must rapidly sterilize the entire Mycobacterium tuberculosis population.
Advances in modeling of drug therapies and treatment outcomes show potential to predict treatment outcomes and prioritize combinations for testing.
This box summarizes key points contained in the article.
Declaration of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
We thank Talia Greenstein, Eric Rubin, and Shumin Tan for their insightful feedback.
Additional information
Funding
References
- Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council Tuberculosis Units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10):S231–S279.
- Kerantzas CA, Jacobs WR. Origins of combination therapy for tuberculosis: lessons for future antimicrobial development and application. mBio. 2017;8(2):e01586–16.
- Connolly LE, Edelstein PH, Ramakrishnan L. Why is long-term therapy required to cure tuberculosis? PLoS Med. 2007;4(3):e120.
- World Health Organization. Global tuberculosis report 2020. Geneva: World Health Organization; 2020.
- Tiberi S, du Plessis N, Walzl G, et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect Dis. 2018;18(7):e183–e198.
- Seung KJ, Keshavjee S, Rich ML. Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis. Cold Spring Harb Perspect Med. 2015 Apr 27;5(9):a017863.
- Dorman SE, Nahid P, Kurbatova EV, et al. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N Engl J Med. 2021 May 6;384(18):1705–1718.
- Conradie F, Diacon AH, Ngubane N, et al., Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. 2020;382(10):893–902. .
- Riva MA. From milk to rifampicin and back again: history of failures and successes in the treatment for tuberculosis. J Antibiot (Tokyo). 2014;67(9):661–665.
- Iseman MD. Tuberculosis therapy: past, present and future. Eur Respir J Suppl. 2002 Jul;36(Supplement 36):87s–94s.
- Sensi P. History of the development of rifampin. Clin Infect Dis. 1983;5(Supplement_3):S402–S406.
- Furin J, Cox H, Pai M. Tuberculosis. Lancet. 2019;393(10181):1642–1656.
- Pai M, Behr MA, Dowdy D, et al. Tuberculosis. Nat Rev Dis Primers. 2016;2(1):16076.
- Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb). 2011 Nov;91(6):497–509.
- Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. 2014;26(6):601–609.
- Yegian D. Biology of tubercle bacilli in necrotic lesions. Am Rev Tuberc. 1952 Nov;66(5):629–631.
- Sarathy JP, Dartois V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev. 2020 Jun 17;33(3). DOI:10.1128/CMR.00159-19.
- Xie YL, de Jager VR, Chen RY, et al., Fourteen-day PET/CT imaging to monitor drug combination activity in treated individuals with tuberculosis. Sci Transl Med. 2021;13(579):eabd7618. .
- Strydom N, Gupta SV, Fox WS, et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 2019 Apr;16(4):e1002773.
- Ernest JP, Sarathy J, Wang N, et al. Lesion penetration and activity limit the utility of second-line injectable agents in pulmonary tuberculosis. Antimicrob Agents Chemother. 2021Jul12; 65(10). AAC0050621. DOI:10.1128/AAC.00506-21.
- Sarathy J, Blanc L, Alvarez-Cabrera N, et al. Fluoroquinolone Efficacy against tuberculosis is driven by penetration into lesions and activity against resident bacterial populations. Antimicrob Agents Chemother. 2019 May;63(5). DOI:10.1128/AAC.02516-18 .
- Prideaux B, Via LE, Zimmerman MD, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015;21(10):1223–1227.
- Sarathy JP, Zuccotto F, Hsinpin H, et al. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis. 2016;2(8):552–563.
- Wayne LG, Sohaskey CD. Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol. 2001;55(1):139–163.
- Lenaerts A, Barry CE 3rd, Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev. 2015 Mar;264(1):288–307.
- Hoff DR, Ryan GJ, Driver ER, et al. Location of intra- and extracellular M. tuberculosis populations in lungs of mice and Guinea pigs during disease progression and after drug treatment. PLoS One. 2011 Mar 21;6(3):e17550.
- Lenaerts AJ, Hoff D, Aly S, et al. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. 2007 Sep;51(9):3338–3345.
- Schrader SM, Vaubourgeix J, Nathan C. Biology of antimicrobial resistance and approaches to combat it. Sci Transl Med. 2020 Jun 24;12(549). DOI:10.1126/scitranslmed.aaz6992.
- Eoh H, Liu R, Lim J, et al. Central carbon metabolism remodeling as a mechanism to develop drug tolerance and drug resistance in Mycobacterium tuberculosis. Front Cell Infect Microbiol. 2022;12:958240.
- Iacobino A, Piccaro G, Giannoni F, et al. Fighting tuberculosis by drugs targeting nonreplicating Mycobacterium tuberculosis bacilli. Int J Mycobacteriol. 2017 Jul-Sep;6(3):213–221.
- Zhang M, Sala C, Dhar N, et al. In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014 Jun;58(6):3217–3223.
- Raghunandanan S, Jose L, Kumar RA. Dormant Mycobacterium tuberculosis converts isoniazid to the active drug in a Wayne’s model of dormancy. J Antibiot (Tokyo). 2018 Nov;71(11):939–949.
- Betts JC, Lukey PT, Robb LC, et al. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002 Feb;43(3):717–731.
- Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996 Jun;64(6):2062–2069.
- Karakousis PC, Williams EP, Bishai WR. Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J Antimicrob Chemother. 2008 Feb;61(2):323–331.
- Iacobino A, Piccaro G, Giannoni F, et al. Activity of drugs against dormant Mycobacterium tuberculosis. Int J Mycobacteriol. 2016 Dec;5(Suppl 1):S94–S95.
- Fattorini L, Piccaro G, Mustazzolu A, et al. Targeting dormant bacilli to fight tuberculosis. Mediterr J Hematol Infect Dis. 2013 Nov 19;5(1):e2013072.
- Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005 Jan 14;307(5707):223–227.
- Singh R, Manjunatha U, Boshoff HI, et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008 Nov 28;322(5906):1392–1395.
- Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med. 2009;360(23):2397–2405.
- Conradie F, Bagdasaryan TR, Borisov S, et al. Bedaquiline–pretomanid–linezolid regimens for drug-resistant tuberculosis. N Engl J Med. 2022;387(9):810–823.
- Bryk R, Gold B, Venugopal A, et al. Selective killing of nonreplicating mycobacteria. Cell Host Microbe. 2008 Mar 13;3(3):137–145.
- Darby CM, Nathan CF. Killing of non-replicating Mycobacterium tuberculosis by 8-hydroxyquinoline. J Antimicrob Chemother. 2010;65(7):1424–1427.
- Grant SS, Kawate T, Nag PP, et al. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem Biol. 2013 Oct 18;8(10):2224–2234.
- Gold B, Smith R, Nguyen Q, et al. Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. J Med Chem. 2016;59(13):6027–6044.
- Chatterjee D, Bonnett SA, Dennison D, et al. A class of hydrazones are active against non-replicating Mycobacterium tuberculosis. PLoS One. 2018;13(10). DOI:10.1371/journal.pone.0196348.
- Neyrolles O, Flint L, Korkegian A, et al. InhA inhibitors have activity against non-replicating Mycobacterium tuberculosis. PLoS One. 2020;15(11):e0239354.
- Balaban NQ, Helaine S, Lewis K, et al. Definitions and guidelines for research on antibiotic persistence. Nature Rev Microbiol. 2019;17(7):441–448.
- Aldridge BB, Fernandez-Suarez M, Heller D, et al. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science. 2012 Jan 6;335(6064):100–104.
- Schrader SM, Botella H, Jansen R, et al. Multiform antimicrobial resistance from a metabolic mutation. Sci Adv. 2021 Aug;7(35). doi:10.1126/sciadv.abh2037.
- Levin-Reisman I, Ronin I, Gefen O, et al. Antibiotic tolerance facilitates the evolution of resistance. Science. 2017 Feb 24;355(6327):826–830.
- Liu J, Gefen O, Ronin I, et al. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science. 2020 Jan 10;367(6474):200–204.
- McGrath M, Gey van Pittius NC, van Helden PD, et al. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2014 Feb;69(2):292–302.
- Ford CB, Shah RR, Maeda MK, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013 Jul;45(7):784–790.
- Coelho T, Machado D, Couto I, et al. Enhancement of antibiotic activity by efflux inhibitors against multidrug resistant Mycobacterium tuberculosis clinical isolates from Brazil. Front Microbiol. 2015;6:330.
- Laws M, Jin P, Rahman KM. Efflux pumps in Mycobacterium tuberculosis and their inhibition to tackle antimicrobial resistance. Trends Microbiol. 2022;30(1):57–68.
- Pyle MM. Relative numbers of resistant tubercle bacilli in sputa of patients before and during treatment with streptomycin. Proc Staff Meet Mayo Clin. 1947 Oct 15;22(21):465–473.
- Palaci M, Dietze R, Hadad DJ, et al. Cavitary disease and quantitative sputum bacillary load in cases of pulmonary tuberculosis. J Clin Microbiol. 2007 Dec;45(12):4064–4066.
- British Medical Journal. Streptomycin treatment of pulmonary tuberculosis: a medical research council investigation. Bmj. 1948;2(4582):769–782.
- British Medical Journal. Streptomycin in pulmonary tuberculosis. Bmj. 1948;2(4582):790–791.
- Bahuguna A, Rawat DS. An overview of new antitubercular drugs, drug candidates, and their targets. Med Res Rev. 2020 Jan;40(1):263–292.
- Campanico A, Moreira R, Lopes F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur J Med Chem. 2018 Apr 25;150:525–545. DOI:10.1016/j.ejmech.2018.03.020.
- Working Group on New TB Drugs. Compounds | Working Group for New TB Drugs. 2022. [November 22, 2022]. Available from: https://www.newtbdrugs.org/pipeline/compounds
- Nuermberger E, Tyagi S, Tasneen R, et al. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother. 2008;52(4):1522–1524.
- Tasneen R, Li SY, Peloquin CA, et al. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother. 2011 Dec;55(12):5485–5492.
- Tasneen R, Williams K, Amoabeng O, et al. Contribution of the nitroimidazoles PA-824 and TBA-354 to the activity of novel regimens in murine models of tuberculosis. Antimicrob Agents Chemother. 2015 Jan;59(1):129–135.
- Tweed CD, Wills GH, Crook AM, et al. A partially randomised trial of pretomanid, moxifloxacin and pyrazinamide for pulmonary TB. Int J Tuberc Lung Dis. 2021 Apr 1;25(4):305–314.
- Global Alliance for TB Drug Development. Trial to evaluate the efficacy, safety and tolerability of BPaMZ in Drug-Sensitive (DS-TB) adult patients and drug-resistant (DR-TB) adult patients: global alliance for TB drug development. 2020. [updated April 30]. Available from: https://ClinicalTrials.gov/show/NCT03338621
- University College L, National University Hospital S, Institute SCR. Two-month regimens using novel combinations to augment treatment effectiveness for drug-sensitive tuberculosis. 2018. [updated March 21]. https://ClinicalTrials.gov/show/NCT03474198
- Williams K, Minkowski A, Amoabeng O, et al. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother. 2012 Jun;56(6):3114–3120.
- Tasneen R, Betoudji F, Tyagi S, et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2016 Jan;60(1):270–277.
- Li SY, Tasneen R, Tyagi S, et al. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother. 2017 Sep;61(9). doi:10.1128/AAC.00913-17.
- Zhang X, Falagas ME, Vardakas KZ, et al. Systematic review and meta-analysis of the efficacy and safety of therapy with linezolid containing regimens in the treatment of multidrug-resistant and extensively drug-resistant tuberculosis. J Thorac Dis. 2015 Apr;7(4):603–615.
- Dorman SE, Goldberg S, Stout JE, et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J Infect Dis. 2012 Oct 1;206(7):1030–1040.
- Jindani A, Harrison TS, Nunn AJ, et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 2014;371(17):1599–1608.
- Gillespie SH, Crook AM, McHugh TD, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 2014 Oct 23;371(17):1577–1587.
- Andries K, Gevers T, Lounis N. Bactericidal potencies of new regimens are not predictive of their sterilizing potencies in a murine model of tuberculosis. Antimicrob Agents Chemother. 2010 Nov;54(11):4540–4544.
- Clemens DL, Lee BY, Silva A, et al. Artificial intelligence enabled parabolic response surface platform identifies ultra-rapid near-universal TB drug treatment regimens comprising approved drugs. PLoS One. 2019;14(5):e0215607.
- De Groote MA, Gilliland JC, Wells CL, et al. Comparative studies evaluating mouse models used for efficacy testing of experimental drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2011 Mar;55(3):1237–1247.
- Harper J, Skerry C, Davis SL, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis. 2012 Feb 15;205(4):595–602.
- Li SY, Irwin SM, Converse PJ, et al. Evaluation of moxifloxacin-containing regimens in pathologically distinct murine tuberculosis models. Antimicrob Agents Chemother. 2015 Jul;59(7):4026–4030.
- Mourik BC, Svensson RJ, de Knegt GJ, et al. Improving treatment outcome assessment in a mouse tuberculosis model. Sci Rep. 2018 Apr 9;8(1):5714.
- Nuermberger E. Using animal models to develop new treatments for tuberculosis. Semin Respir Crit Care Med. 2008 Oct;29(5):542–551.
- Nuermberger EL, Yoshimatsu T, Tyagi S, et al. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am J Respir Crit Care Med. 2004 Feb 1;169(3):421–426.
- Rosenthal IM, Zhang M, Almeida D, et al. Isoniazid or moxifloxacin in rifapentine-based regimens for experimental tuberculosis? Am J Respir Crit Care Med. 2008 Nov 1;178(9):989–993.
- Nuermberger EL, Jacobs Jr. WR, McShane H. Preclinical efficacy testing of new drug candidates. Microbiol Spectr. 2017;5(3). DOI:10.1128/microbiolspec.TBTB2-0034-2017
- Yang H-J, Wang D, Wen X, et al. One size fits all? Not in in vivo modeling of tuberculosis chemotherapeutics. Front Cell Infect Microbiol. 2021;11:613149.
- Gumbo T, Lenaerts AJ, Hanna D, et al. Nonclinical models for antituberculosis drug development: a landscape analysis. J Infect Dis. 2015;211(suppl_3):S83–S95.
- Dharmadhikari AS, Nardell EA. What animal models teach humans about tuberculosis. Am J Respir Cell Mol Biol. 2008 Nov;39(5):503–508.
- Singh AK, Gupta UD. Animal models of tuberculosis: lesson learnt. Indian J Med Res. 2018 May;147(5):456–463.
- Kramnik I, Beamer G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin Immunopathol. 2016 Mar;38(2):221–237.
- Franzblau SG, DeGroote MA, Cho SH, et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis. 2012;92(6):453–488.
- Grosset J. The sterilizing value of rifampicin and pyrazinamide in experimental short-course chemotherapy. Bull Int Union Tuberc. 1978 Mar;53(1):5–12.
- Xu J, Li SY, Almeida DV, et al. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob Agents Chemother. 2019 May;63(5). doi:10.1128/AAC.00021-19.
- Lanoix JP, Betoudji F, Nuermberger E. Sterilizing activity of pyrazinamide in combination with first-line drugs in a C3HeB/FeJ mouse model of tuberculosis. Antimicrob Agents Chemother. 2016 Feb;60(2):1091–1096.
- Lanoix JP, Lenaerts AJ, Nuermberger EL. Heterogeneous disease progression and treatment response in a C3HeB/FeJ mouse model of tuberculosis. Dis Model Mech. 2015 Jun;8(6):603–610.
- Lee BY, Clemens DL, Silva A, et al. Ultra-rapid near universal TB drug regimen identified via parabolic response surface platform cures mice of both conventional and high susceptibility. PLoS One. 2018;13(11):e0207469.
- Saini V, Ammerman NC, Chang YS, et al. Treatment-shortening effect of a novel regimen combining clofazimine and high-dose rifapentine in pathologically distinct mouse models of tuberculosis. Antimicrob Agents Chemother. 2019 Jun;63(6). doi:10.1128/AAC.00388-19.
- Chatterji M, Shandil R, Manjunatha MR, et al. 1,4-azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob Agents Chemother. 2014;58(9):5325–5331.
- Dartois V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol. 2014 Mar;12(3):159–167.
- Irwin SM, Prideaux B, Lyon ER, et al. Bedaquiline and pyrazinamide treatment responses are affected by pulmonary lesion heterogeneity in Mycobacterium tuberculosis infected C3HeB/FeJ mice. ACS Infect Dis. 2016 Apr 8;2(4):251–267.
- Liu Y, Tan S, Huang L, et al. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med. 2016 May 2;213(5):809–825.
- Via LE, England K, Weiner DM, et al. A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob Agents Chemother. 2015;59(7):4181–4189.
- Winchell CG, Mishra BB, Phuah JY, et al. Evaluation of IL-1 blockade as an adjunct to linezolid therapy for tuberculosis in mice and macaques. Front Immunol. 2020;11:11.
- Beites T, O’Brien K, Tiwari D, et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat Commun. 2019 Oct 31;10(1):4970.
- Kim S, Scanga CA, Miranda Silva C, et al. Pharmacokinetics of tedizolid, sutezolid, and sutezolid-M1 in non-human primates. Eur J Pharm Sci. 2020;151:105421.
- Coleman MT, Chen RY, Lee M, et al. PET/CT imaging reveals a therapeutic response to oxazolidinones in macaques and humans with tuberculosis. Sci Transl Med. 2014 Dec 3;6(265):265ra167.
- Rajoli RKR, Podany AT, Moss DM, et al. Modelling the long-acting administration of anti-tuberculosis agents using PBPK: a proof of concept study. Int J Tuberc Lung Dis. 2018 Aug 1;22(8):937–944.
- Fors J, Strydom N, Fox WS, et al. Mathematical model and tool to explore shorter multi-drug therapy options for active pulmonary tuberculosis. PLoS Comput Biol. 2020 Aug;16(8):e1008107.
- Pienaar E, Cilfone NA, Lin PL, et al. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J Theor Biol. 2015;367:166–179.
- Bigelow KM, Deitchman AN, S-Y L, et al. Pharmacodynamic correlates of linezolid activity and toxicity in murine models of tuberculosis. J Infect Dis. 2021;223(11):1855–1864.
- Rifat D, Prideaux B, Savic RM, et al. Pharmacokinetics of rifapentine and rifampin in a rabbit model of tuberculosis and correlation with clinical trial data. Sci Transl Med. 2018;10(435). DOI:10.1126/scitranslmed.aai7786.
- Lyons MA. Modeling and simulation of pretomanid pharmacodynamics in pulmonary tuberculosis patients. Antimicrob Agents Chemother. 2019 Sep 30;63(12). DOI:10.1128/AAC.00732-19.
- Lyons MA. Pretomanid dose selection for pulmonary tuberculosis: an application of multi‐objective optimization to dosage regimen design. CPT Pharmacometrics Syst Pharmacol. 2021;10(3):211–219.
- Gumbo T, Alffenaar JC. Pharmacokinetic/pharmacodynamic background and methods and scientific evidence base for dosing of second-line tuberculosis drugs. Clin Infect Dis. 2018 Nov 28;67(suppl_3):S267–S273.
- Gumbo T, Pasipanodya JG, Romero K, et al. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis. 2015 Aug 15;61 Suppl 1(suppl 1):S25–31.
- Humphries H, Almond L, Berg A, et al. Development of physiologically-based pharmacokinetic models for standard of care and newer tuberculosis drugs. CPT Pharmacometrics Syst Pharmacol. 2021 Nov;10(11):1382–1395.
- Riccardi N, Canetti D, Rodari P, et al. Tuberculosis and pharmacological interactions: a narrative review. Curr Res Pharmacol Drug Discov. 2021;2:100007.
- Alffenaar JC, Gumbo T, Dooley KE, et al. Integrating pharmacokinetics and pharmacodynamics in operational research to end tuberculosis. Clin Infect Dis. 2020 Apr 10;70(8):1774–1780.
- Garcia-Cremades M, Solans BP, Strydom N, et al. Emerging therapeutics, technologies, and drug development strategies to address patient nonadherence and improve tuberculosis treatment. Annu Rev Pharmacol Toxicol. 2022 Jan 6;62(1):197–210.
- Marino S, Kirschner DE. A multi-compartment hybrid computational model predicts key roles for dendritic cells in tuberculosis infection. Computation (Basel). 2016;4(4). DOI: 10.3390/computation4040039.
- Warsinske HC, Pienaar E, Linderman JJ, et al. Deletion of TGF-beta1 increases bacterial clearance by cytotoxic T cells in a tuberculosis granuloma model. Front Immunol. 2017;8:1843.
- Pienaar E, Sarathy J, Prideaux B, et al. Comparing efficacies of moxifloxacin, levofloxacin and gatifloxacin in tuberculosis granulomas using a multi-scale systems pharmacology approach. PLoS Comput Biol. 2017 Aug;13(8):e1005650.
- Marino S, Hult C, Wolberg P, et al. The role of dimensionality in understanding granuloma formation. Computation (Basel). 2018 Dec;6(4). doi:10.3390/computation6040058
- Hult C, Mattila JT, Gideon HP, et al. Neutrophil dynamics affect Mycobacterium tuberculosis granuloma outcomes and dissemination. Front Immunol. 2021;12:712457.
- Cicchese JM, Sambarey A, Kirschner D, et al. A multi-scale pipeline linking drug transcriptomics with pharmacokinetics predicts in vivo interactions of tuberculosis drugs. Sci Rep. 2021 Mar 11;11(1):5643.
- Gumbo T, Pasipanodya JG, Nuermberger E, et al. Correlations between the hollow fiber model of tuberculosis and therapeutic events in tuberculosis patients: learn and confirm. Clin Infect Dis. 2015 Aug 15;61 Suppl 1(suppl 1):S18–24.
- Parish T. In vitro drug discovery models for Mycobacterium tuberculosis relevant for host infection. Expert Opin Drug Discov. 2020 Mar;15(3):349–358.
- Dartois VA, Rubin EJ. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat Rev Microbiol. 2022 Apr 27;20(11):685–701.
- Russell DG, VanderVen BC, Lee W, et al. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe. 2010 Jul 22;8(1):68–76.
- VanderVen BC, Fahey RJ, Lee W, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 2015 Feb;11(2):e1004679.
- Griffin JE, Pandey AK, Gilmore SA, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol. 2012 Feb 24;19(2):218–227.
- Guerrini V, Prideaux B, Blanc L, et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 2018 Aug;14(8):e1007223.
- Lee W, VanderVen BC, Fahey RJ, et al. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem. 2013 Mar 8;288(10):6788–6800.
- Lovewell RR, Sassetti CM, VanderVen BC. Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr Opin Microbiol. 2016 Feb;29:30–36.
- Wilburn KM, Fieweger RA, VanderVen BC. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathog Dis. 2018 Mar 1;76(2). DOI:10.1093/femspd/fty021.
- Miner MD, Chang JC, Pandey AK, et al. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol. 2009 Jun;47(6):407–411.
- Baker JJ, Dechow SJ, Abramovitch RB. Acid fasting: modulation of Mycobacterium tuberculosis metabolism at acidic pH. Trends Microbiol. 2019 Nov;27(11):942–953.
- Tarshis MS, Weed WA Jr. Lack of significant in vitro sensitivity of Mycobacterium tuberculosis to pyrazinamide on three different solid media. Am Rev Tuberc. 1953 Mar;67(3):391–395.
- McDermott W, Tompsett R. Activation of pyrazinamide and nicotinamide in acidic environments in vitro. Am Rev Tuberc. 1954 Oct;70(4):748–754.
- Lamont EA, Dillon NA, Baughn AD. The bewildering antitubercular action of pyrazinamide. Microbiol Mol Biol Rev. 2020 May 20;84(2). DOI:10.1128/MMBR.00070-19.
- Daniel J, Maamar H, Deb C, et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011 Jun;7(6):e1002093.
- Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe. Semin Immunopathol. 2013 Sep;35(5):563–583.
- Giraud-Gatineau A, Coya JM, Maure A, et al. The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. Elife. 2020 May 4;9. DOI:10.7554/eLife.55692
- Cole ST. Inhibiting Mycobacterium tuberculosis within and without. Philos Trans R Soc Lond B Biol Sci. 2016 Nov 5;371(1707):20150506.
- Urtti A, Baik J, Rosania GR. Macrophages sequester clofazimine in an intracellular liquid crystal-like supramolecular organization. PLoS ONE. 2012;7(10). DOI:10.1371/journal.pone.0041410
- Greenwood DJ, Dos Santos MS, Huang S, et al. Subcellular antibiotic visualization reveals a dynamic drug reservoir in infected macrophages. Science. 2019 Jun 28;364(6447):1279–1282.
- Silva A, Lee B-Y, Clemens DL, et al. Output-driven feedback system control platform optimizes combinatorial therapy of tuberculosis using a macrophage cell culture model. Proc Nat Acad Sci. 2016;113(15):E2172–E2179.
- Horwitz MA, Clemens DL, Lee BY. AI‐enabled parabolic response surface approach identifies ultra short‐course near‐universal TB drug regimens. Adv Ther. 2019;3(4):1900086.
- Lee BY, Clemens DL, Silva A, et al. Drug regimens identified and optimized by output-driven platform markedly reduce tuberculosis treatment time. Nat Commun. 2017 Jan 24;8(1):14183.
- Lanoix JP, Ioerger T, Ormond A, et al. Selective inactivity of pyrazinamide against tuberculosis in C3HeB/FeJ mice is best explained by neutral pH of caseum. Antimicrob Agents Chemother. 2016 Feb;60(2):735–743.
- Gold B, Nathan C. Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol Spectr. 2017 Jan;5(1). DOI:10.1128/microbiolspec.TBTB2-0031-2016.
- Gold B, Roberts J, Ling Y, et al. Rapid, semiquantitative assay to discriminate among compounds with activity against replicating or nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015 Oct;59(10):6521–6538.
- Gold B, Warrier T, Nathan C. A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol Biol. 2015;1285:293–315. * Multi. * Multistress model amenable to systematic screening of compounds and combinations.
- Wayne LG. Dynamics of submerged growth of Mycobacterium tuberculosis under aerobic and microaerophilic conditions. Am Rev Respir Dis. 1976 Oct;114(4):807–811.
- Sohaskey CD. Nitrate enhances the survival of Mycobacterium tuberculosis during inhibition of respiration. J Bacteriol. 2008;190(8):2981–2986.
- Cunningham-Bussel A, Zhang T, Nathan CF. Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci U S A. 2013;110(45):E4256–E4265.
- Larkins-Ford J, Greenstein T, Van N, et al. Systematic measurement of combination-drug landscapes to predict in vivo treatment outcomes for tuberculosis. Cell Syst. 2021 Nov 17;12(11):1046–1063 e7.
- Deb C, Lee CM, Dubey VS, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One. 2009 Jun 29;4(6):e6077.
- Larkins-Ford J, Degefu YN, Van N, et al. Design principles to assemble drug combinations for effective tuberculosis therapy using interpretable pairwise drug response measurements. Cell Rep Med. 2022 Sep 20;3(9):100737.
- Gumbo T, Louie A, Deziel Mark R, et al. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190(9):1642–1651.
- Cavaleri M, Manolis E. Hollow fiber system model for tuberculosis: the European medicines agency experience. Clin Infect Dis. 2015;61(suppl 1):S1–S4.
- Pasipanodya JG, Nuermberger E, Romero K, et al. Systematic analysis of hollow fiber model of tuberculosis experiments. Clin Infect Dis. 2015 Aug 15;61 Suppl 1(suppl 1):S10–7.
- Mason AB, Dartois V. Drug sensitivity testing of Mycobacterium tuberculosis growing in a hollow fiber bioreactor. Methods Mol Biol. 2021;2314:715–731.
- Stein C, Makarewicz O, Bohnert JA, et al. Three dimensional checkerboard synergy analysis of colistin, meropenem, tigecycline against multidrug-resistant clinical Klebsiella pneumonia isolates. PLoS One. 2015;10(6):e0126479.
- Cokol M, Kuru N, Bicak E, et al. Efficient measurement and factorization of high-order drug interactions in Mycobacterium tuberculosis. Sci Adv. 2017;3(10):e1701881.
- Kehe J, Kulesa A, Ortiz A, et al. Massively parallel screening of synthetic microbial communities. Proc Nat Acad Sci. 2019;116(26):12804–12809.
- Schön T, Köser CU, Werngren J, et al. What is the role of the EUCAST reference method for MIC testing of the Mycobacterium tuberculosis complex? Clin Microbiol Infect. 2020;26(11):1453–1455.
- Schön T, Werngren J, Machado D, et al. Multicentre testing of the EUCAST broth microdilution reference method for MIC determination on Mycobacterium tuberculosis. Clin Microbiol Infect. 2021;27(2):288.e1–288.e4.
- Palomino JC, Martin A, Camacho M, et al. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002 Aug;46(8):2720–2722.
- Martin A, Camacho M, Portaels F, et al. Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob Agents Chemother. 2003;47(11):3616–3619.
- Taneja NK, Tyagi JS. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J Antimicrob Chemother. 2007;60(2):288–293.
- Queval CJ, Song OR, Delorme V, et al. A microscopic phenotypic assay for the quantification of intracellular mycobacteria adapted for high-throughput/high-content screening. J Vis Exp. 2014 Jan;17(83):e51114.
- Stanley SA, Barczak AK, Silvis MR, et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014 Feb;10(2):e1003946.
- Sorrentino F, Gonzalez Del Rio R, Zheng X, et al. Development of an intracellular screen for new compounds able to inhibit Mycobacterium tuberculosis growth in human macrophages. Antimicrob Agents Chemother. 2016 Jan;60(1):640–645.
- Rodrigues Felix C, Gupta R, Geden S, et al. Selective killing of dormant Mycobacterium tuberculosis by marine natural products. Antimicrob Agents Chemother. 2017 Aug;61(8). doi:10.1128/AAC.00743-17.
- Andreu N, Zelmer A, Fletcher T, et al. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One. 2010 May 24;5(5):e10777.
- Early JV, Mullen S, Parish T. A rapid, low pH, nutrient stress, assay to determine the bactericidal activity of compounds against non-replicating Mycobacterium tuberculosis. PLoS One. 2019;14(10):e0222970.
- Sharma S, Gelman E, Narayan C, et al. Simple and rapid method to determine antimycobacterial potency of compounds by using autoluminescent Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014 Oct;58(10):5801–5808.
- Zhang T, Li S-Y, Nuermberger EL. Autoluminescent Mycobacterium tuberculosis for rapid, real-time, non-invasive assessment of drug and vaccine efficacy. PLOS ONE. 2012;7(1):e29774.
- Walter ND, Born SEM, Robertson GT, et al. Mycobacterium tuberculosis precursor rRNA as a measure of treatment-shortening activity of drugs and regimens. Nat Commun. 2021;12(1). DOI:10.1038/s41467-021-22833-6
- Wayne LG, Sramek HA. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994 Sep;38(9):2054–2058.
- Pethe K, Sequeira PC, Agarwalla S, et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010 Aug 24;1(1):57.
- Drusano GL, Kim S, Almoslem M, et al. The funnel: a screening technique for identifying optimal two-drug combination chemotherapy regimens. Antimicrob Agents Chemother. 2021 Jan 20;65(2). doi:10.1128/AAC.02172-20.
- Sarathy JP, Liang HH, Weiner D, et al. An in vitro caseum binding assay that predicts drug penetration in tuberculosis lesions. J Vis Exp. 2017 May;8(123):55559.
- Macalino SJY, Billones JB, Organo VG, et al. In silico strategies in tuberculosis drug discovery. Molecules. 2020 Feb 4;25(3):665.
- Singh R, Ramachandran V, Shandil R, et al. In silico-based high-throughput screen for discovery of novel combinations for tuberculosis treatment. Antimicrob Agents Chemother. 2015;59(9):5664–5674.
- Srinivas V, Ruiz RA, Pan M, et al. Transcriptome signature of cell viability predicts drug response and drug interaction in Mycobacterium tuberculosis. Cell Rep Methods. 2021 Dec 20;1(8):None.
- Rock J, Kline KA. Tuberculosis drug discovery in the CRISPR era. PLoS Pathog. 2019 Sep;15(9):e1007975.
- Bosch B, DeJesus MA, Poulton NC, et al., Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell. 2021;184(17):4579–4592.e24. .
- Johnson EO, LaVerriere E, Office E, et al. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature. 2019 Jul;571(7763):72–78.
- Doran JL, Pang Y, Mdluli KE, et al. Mycobacterium tuberculosis efpA encodes an efflux protein of the QacA transporter family. Clin Diagn Lab Immunol. 1997 Jan;4(1):23–32.
- Johnson EO, Office E, Kawate T, et al. Large-scale chemical-genetic strategy enables the design of antimicrobial combination chemotherapy in mycobacteria. ACS Infect Dis. 2020 Jan 10;6(1):56–63.
- Rai D, Mehra S. The mycobacterial efflux pump EfpA can induce high drug tolerance to many antituberculosis drugs, including moxifloxacin, in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2021 Oct 18;65(11):e0026221.
- Ma S, Jaipalli S, Larkins-Ford J, et al. Transcriptomic signatures predict regulators of drug synergy and clinical regimen efficacy against tuberculosis. mBio. 2019 Nov 12;10(6). doi:10.1128/mBio.02627-19.
- Aldridge BB, Barros-Aguirre D, Barry CE 3rd, et al. The tuberculosis drug accelerator at year 10: what have we learned? Nat Med. 2021 Aug;27(8):1333–1337.