
ABSTRACT
Introduction
Fibrotic lung diseases represent a large subset of diseases with an unmet clinical need. Oligonucleotide therapies (ONT) are a promising therapeutic approach for the treatment of pulmonary disease as they can inhibit pathways that are otherwise difficult to target. Additionally, targeting the lung specifically with ONT is advantageous because it reduces the possibilities of systemic side effects and tolerability concerns.
Areas Covered
This review presents the chemical basis of designing various ONTs currently known to treat fibrotic lung diseases. Further, the authors have also discussed the delivery vehicle, routes of administration, physiological barriers of the lung, and toxicity concerns with ONTs.
Expert opinion
ONTs provide a promising therapeutic approach for the treatment of fibrotic diseases of the lung, particularly because ONTs directly delivered to the lung show little systemic side effects compared to current therapeutic strategies. Dry powder aerosolized inhalers may be a good strategy for getting ONTs into the lung in humans. However, as of now, no dry powder ONTs have been approved for use in the clinical setting, and this challenge must be overcome for future therapies. Various delivery methods that can aid in direct targeting may also improve the use of ONTs for lung fibrotic diseases.
1. Introduction
Pulmonary fibrosis is observed in several lung diseases such as interstitial lung diseases (ILDs), chronic asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF). Per the CDC in 2022, prior to the COVID-19 pandemic, chronic pulmonary diseases were the third leading cause of death in the US behind cardiovascular disease and neoplasms. Additionally, chronic lung disease affects over 500 million people worldwide, and is increasing in prevalence [Citation1]. Particularly, interstitial lung diseases (ILDs) which result in pulmonary fibrosis have significant unmet needs; in 2019, over 600,000 Americans were diagnosed with ILDs. The most extensively studied and aggressive form of ILD, idiopathic pulmonary fibrosis (IPF), has a poor prognosis and a median survival of 3–5 years if left untreated [Citation2]. Thus, fibrotic pulmonary diseases represent a significant global disease burden, and increased efforts should be made to understand the pathogenesis of these diseases and to find novel treatment strategies and drug delivery systems.
ILD is a group of diseases that cause fibrosis in the lungs, making it difficult to breathe and get oxygen into the bloodstream. Microscopic honeycombing, fibroblastic foci, loss of epithelial tissue, and architectural distortion are histological patterns of airway remodeling observed in ILD [Citation3]. Diseases that fall under this category include idiopathic pulmonary fibrosis (IPF), connective tissue disease-related ILDs associated with rheumatoid arthritis or scleroderma, idiopathic non-specific interstitial pneumonia, chronic pneumonitis, and sarcoidosis-related ILD. More recently, COVID-19 viral infection has been shown to produce lung fibrosis in about 2–6% of infected individuals, and pulmonary fibrosis may be a major long-term complication of COVID-19 infection [Citation4]. Currently, successful reversal and termination of lung fibrosis is achieved by lung transplant, however there is poor survival for those on the waitlist because usually patients are not referred for transplant until the disease is highly advanced [Citation5]. Current treatments for IPF include anti-fibrotic agents including nintedanib and pirfenidone, which have limited efficacy, particularly in severe subsets, as well as tolerability concerns [Citation6].
Due to the function of the lung, it naturally encounters many harmful substances such as air pollutants/allergens, which can increase risk for pulmonary disease. Several chronic lung disorders that result in fibrosis have a substantial genetic component leading to dysregulation of gene expression [Citation7]. Gene regulation through novel therapeutic approaches such as RNA interference (RNAi) poses as potential effective treatment. Oligonucleotide therapy (ONT) provides a benefit in that it allows researchers to pursue targets that otherwise would be difficult to drug. In addition, the lung is a prime candidate for ONT as there is direct access to the lung through inhaled therapy, and may have less systemic effects with lung delivery, enabling the pursuit of targets that otherwise present safety concerns [Citation8]. In addition, direct delivery to the lung can avoid enzymatic decay and allows rapid drug absorption as compared to oral therapeutics delivered to the gut. This review will outline the pathogenesis of the most prominent chronic pulmonary diseases, existing ONT therapies for lung-related pathologies (), and the current challenges/strategies for ONT delivery to the lung.
Table 1. Clinical trials of oligonucleotide therapies for pulmonary fibrotic disease.
1.1. Pathophysiology of IPF and other chronic lung diseases with fibrotic lung indication
Much remains to be elucidated about the mechanisms underlying IPF, but chronic dysregulation of type 2 alveolar epithelial cells (AEC2s) is thought to be a primary mechanism of this disease [Citation9]. AEC2s, found in alveoli, are surfactant producers that also serve as stem cells for AEC1s. These cells can contribute to the repair of AEC1s after inflammation or injury [Citation10]. It is thought that apoptosis of alveolar cells may be an initial response to injury, leading to activation of signaling pathways that secrete fibrogenic growth factors and cytokines that contribute to fibroblast migration and myofibroblast expansion/differentiation. These include secreted factors such as TGF-β, platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), TNF-α, angiotensin, several matrix metalloproteinases including MMP1, MMP7, MMP19, CC motif chemokine 2 (CCL2 or MCP-1), and stromal cell-derived factor 1 (CXCL12) [Citation11]. Activated myofibroblasts enhance abnormal ECM deposition in the context of IPF, though the mechanism is not fully understood. It is possible that α-integrins, which are receptors for cell adhesion to ECM proteins, play a role in this mechanism. It has been reported that TGF-β produced by AECs activates fibroblasts through αvβ6 integrin that mediates matrix stiffness and myofibroblast invasion [Citation12]. Additionally, endothelial cells can also transition to a mesenchymal phenotype via the process of endothelial to mesenchymal transition () [Citation13]. Other cells that may be involved in the pathogenesis of pulmonary fibrosis include innate and adaptive immune cells. Alveolar macrophages in the lung may undergo aberrant signaling in the context of IPF, which can lead to altered M1/M2 macrophage differentiation [Citation14]. Genetic studies of IPF have confirmed that mutations in genes expressed in type 2 alveolar cells are important for IPF development, including TERT, TERC, TINF2, DKC1, RTEL1, PARN, and NAF1 [Citation11]. Mutations in the MUC5B gene, which is important for mucociliary clearance, and TOLLIP, which codes for a component of Toll-like receptor signaling, are highly associated with increased IPF risk and poorer prognosis [Citation15]. Current ONTs under investigation for IPF treatment will be discussed later and are outlined in .
Figure 1. Pathophysiology of pulmonary fibrosis. Right healthy lung (light pink) in which alveoli is lined with epithelial cells which upon stimulation from inflammation or apoptosis recruits fibroblasts. These fibroblasts undergo activation and differentiation to myofibroblasts that results in the synthesis of extracellular matrix and airway remodeling to ultimately cause scarred tissue or thickened alveoli walls shown in left lung with fibrosis (dark pink). Created with Biorender.com.
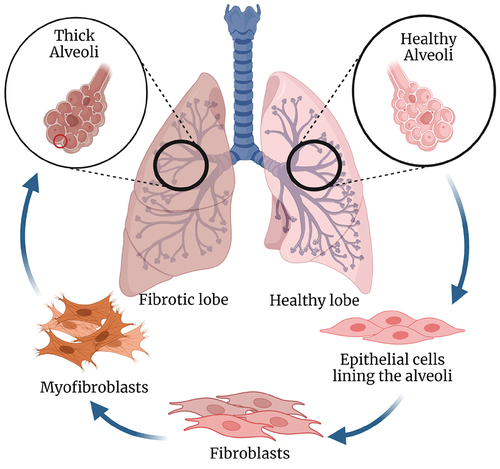
Chronic asthma, COPD, and cystic fibrosis can also result in pulmonary fibrosis. Asthma is a chronic disease of the airway that leads to airway hyperresponsiveness and lung remodeling. It is characterized by symptoms such as breathlessness, wheezing, and coughing. Structural changes in the airway due to chronic asthma lead to epithelial fibrosis and collagen deposition [Citation16]. Th2-high asthma is typically associated with increased numbers of IgE antibody-producing B cells, eosinophils, and IL-4, IL-5, and IL-13 cytokine production [Citation17]. A variety of transcription factors involved in Th2 differentiation have been discovered, including GATA-3, STAT-3, STAT-5, STAT-6, Gfi-1, c-Maf, and IRF4 [Citation18]. Epithelial cells and type-2 dendritic cells may also play a significant role in leading type-2 immunity. Mainstay treatments for Th2-high asthma include inhaled corticosteroids and β2-adrenoreceptor agonists. Biologics that target Th2-associated cytokines or their receptors, the innate immune cytokine thymic stromal lymphopoietin (TSLP), and IgE have recently been approved for the treatment of severe or eosinophilic-high Th2-dominant asthma [Citation19,Citation20]. There is still a significant population of individuals with Th2-dominant asthma that are unresponsive to mainstay treatments, indicating a need for alternate therapeutic strategies for this type of asthma. Little progress has been made in the treatment of neutrophilic (Th2-low) asthma, and patients in this endotype do not respond well to inhaled corticosteroids and other asthma treatments [Citation21]. Further understanding of the underlying pathogenesis of this endotype may lead to better treatment strategies.
Chronic obstructive pulmonary disease (COPD) is a complex, heterogeneous inflammatory disease of the lung characterized by chronic bronchitis and emphysema [Citation22]. It leads to irreversible limitation of airflow, airway remodeling/fibrosis, mucus overproduction, destruction of lung epithelial tissue, and loss of elastic recoil in the lung [Citation23]. Much remains to be elucidated about the pathophysiology and mechanisms underlying COPD. A large, non-specific inflammatory response occurs because of injury to airway epithelial cells, releasing damage-associated molecular patterns (DAMPs) which activate pattern recognition receptors such as TLR-4 and TLR-2 on AECs. This results in the production of inflammatory cytokines including TNFα, IL-1, and IL-8, causing inflammatory response, elastolysis, and fibrosis [Citation24]. The genetic component of COPD is still unclear, but there are likely a few polymorphisms that increase the likelihood of developing COPD in response to cigarette smoke [Citation25]. Current treatments for COPD include smoking cessation, inhaled long-acting β-2 agonists, and long-acting muscarinic antagonists [Citation22].
Cystic fibrosis (CF) is an autosomal recessive disease that is associated with CFTR gene mutations [Citation26]. The CFTR gene codes for cystic fibrosis transmembrane conductance regulator (CFTR). CFTR protein forms a calcium channel, and in CF, patients either have a mutation in CFTR that does not allow it to reach the membrane or a mutation that causes dysfunction of calcium transport by the CFTR protein, leading to less water outside of the cells [Citation26]. This leads to increased thick mucus production in the lungs, small intestine, reproductive tract, pancreas, and liver due to dysfunctional transport of chloride, bicarbonate, and other ions [Citation27]. In the lungs, there is marked inflammatory cell recruitment and lung damage/fibrosis, leading to productive, persistent coughing. Indeed, lung involvement in CF is predictive of mortality [Citation26].
Current approved treatments for CF include CFTR modulators (which correct the ion transport defect in CF), anti-inflammatory drugs, mucolytics, bronchodilators and corticosteroids, and physiotherapy [Citation28]. Despite advances in treatments in recent years, 10% of individuals with CF are not cured by current therapies [Citation29]. Thus, new therapeutic targets that will restore mucosal hydration are being sought out, one of which blocks the epithelial sodium channel (ENaC). In CF, there is less Cl− secretion and more absorption of Na+, which leads to airway dehydration [Citation30]. Currently, there are several ongoing clinical trials testing the efficacy of ENaC inhibitors, including ONT therapeutics targeting ENaC, which will be described in Section 4.
2. Biological barriers for delivering ONTs to the lung
The biological landscape of the respiratory tract can be divided into two sections: the air-conducting region (nasal cavity, pharynx, trachea, bronchi, and bronchioles) and the respiratory region responsible for the gaseous exchange (bronchioles and the alveoli). Thickening of the tissues between the alveolar walls is commonly observed in pulmonary fibrosis, therefore the therapeutic oligonucleotides ideally must reach the distal airway. To achieve this, the ONTs must first travel through the extensively branched bronchoalveolar tree (). Physical phenomena such as Brownian motion, gravitational sedimentation, and the ability to cross the lung surface area due to inertial impaction affect the delivery of ONTs to deep lung regions [Citation31]. The fluid lining of mucus and pulmonary surfactant poses another physical barrier for drug delivery. The gel-like nature of the mucus which acts as a sieve and filters particles (>500 nm) and the rapid mucociliary clearance impacts drug retention [Citation32]. Next, the compatibility between the lipophilicity of the drug and the high lipid content of pulmonary surfactant determines the permeability of the drug. Once the ONTs are introduced in the patient, they are also susceptible to nuclease degradation and certain chemistries (Section 3) have been proposed to improve stability.
Figure 2. Biological barriers for the oligonucleotide therapies (ONTs). To treat fibrosis, the ONTs have to travel the respiratory tract to reach deep lung cavity (the path shown in black arrows) and overcome the extracellular and intracellular barriers. created with Biorender.com.
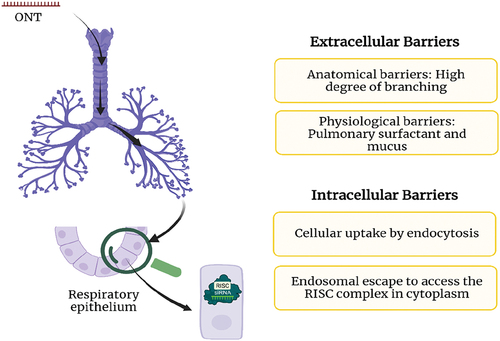
The hydrophilic, negatively charged ONTs must cross the hydrophobic cellular membranes after overcoming extracellular barriers. This is followed by cellular uptake of ONTs, which can occur by gymnosis, clathrin-coated endosomes, caveolin pits, and micropinocytosis. To exert their biological function, the ONTs must escape the endosomes to ultimately reach the RISC complex in the cytoplasm (). Currently, the endosomal escape remains a challenging problem and approaches such as acidification and manipulation of lipid profiles of endosomal organelles can offer promising solution. These topics have been described in detail by Bost et al. [Citation33].
3. Chemical basis of designing ONTs for the lung
To maximize the impact of ONTs for treating lung diseases, careful design considerations are needed for the following chemical aspects: backbone, nucleoside, and sequence design that are discussed in this section.
3.1. Backbone
The phosphate group (PO4−, ‘PO’) linking between the 5′ and 3′ position of RNA and DNA monomers is highly prone to hydrolytic cleavage by various enzymatic reactions and nucleases in vivo. Chemical modifications such as phosphorothioates (PS and PS2), amidation [Citation34], amination (PN) [Citation35], and stereochemically pure backbones [Citation36] () have been shown to reduce the nucleolytic degradation stability of said siRNA/ASO designs, intracellular uptake, and increase binding to plasma proteins in order to prevent renal excretion [Citation37]. Among these design choices for ASO technology, fully phosphorylated backbone (vide infra sequence design) seen across several different generational designs and the flanking phosphorothioates of the terminus of the guide strands in siRNAs, is the most commonly used [Citation38].
Figure 3. Chemical basis of ONT design. (a) Most common nucleotide modifications seen in siRNA and ASO design for lung-targeting oligos. GNA = Glycine nucleic acid, PMO = phosphorodiamidate morpholino oligomer, UNA = unlocked nucleic acid, PNA = peptide nucleic acid. (b) Examples of structures of Ionis’s first–third generation ASOs, a general design choice for 23/23 mer siRNA, and various derivatives of siRNA design [Citation40]. (c) sisiRNA = small integral segmented interfering RNA, containing a segmented sense strand to facilitate cleavage. shRNA = short-hairpin RNA, containing a short loop that acts as a DICER substrate before loading of RISC. aiRNA = asymmetric interfering RNA, a synthetically more attractive siRNA design that holds similar potency to canonical siRNA. Figures were created with Biorender.com. The molecule designs were created with ChemDraw and Biorender.com.
![Figure 3. Chemical basis of ONT design. (a) Most common nucleotide modifications seen in siRNA and ASO design for lung-targeting oligos. GNA = Glycine nucleic acid, PMO = phosphorodiamidate morpholino oligomer, UNA = unlocked nucleic acid, PNA = peptide nucleic acid. (b) Examples of structures of Ionis’s first–third generation ASOs, a general design choice for 23/23 mer siRNA, and various derivatives of siRNA design [Citation40]. (c) sisiRNA = small integral segmented interfering RNA, containing a segmented sense strand to facilitate cleavage. shRNA = short-hairpin RNA, containing a short loop that acts as a DICER substrate before loading of RISC. aiRNA = asymmetric interfering RNA, a synthetically more attractive siRNA design that holds similar potency to canonical siRNA. Figures were created with Biorender.com. The molecule designs were created with ChemDraw and Biorender.com.](/cms/asset/113ae437-97c8-4349-a5a7-d58105743363/iedc_a_2160439_f0003_oc.jpg)
3.2. Nucleoside modifications
Nucleoside modifications () are being extensively explored and continuously innovated to improve ADME, potency, and knockdown of both gene and protein expression and have been discussed in detail by Khvorova et al. [Citation38]. While designing lung targeting siRNA, the most popular choices are 2′-OMe and 2′-F modification to improve stability and potency. Further, these modifications favor the northern pucker conformation that helps in positioning the cleavage center in the RISC complex and enhances the activity. Among these two modifications, 2′-F is more ‘overwinding’ which results in pi-stacking as compared to the 2′-OMe, which can therefore provide additional stability [Citation38]. Other common nucleotides used in ASOs for lung delivery are the MOE (2′-O-methoxy ethyl), LNA (locked nucleic acid), and cEt (constrained ethyl) designs (). The methylene bridge between the 2′ and 4′ positions in the LNA and cEt introduces the desired N-type pucker that is ideal for attaining rigidity [Citation39].
3.3. Sequence design
The sequence design of the oligonucleotide is the most important design aspect. For the ONT to complementarily pair with target, mRNA sequence is crucial and has been described by Nur et al., at length [Citation40]. Here, we have summarized the design choices that have shown promising results in early-stage clinical trials for treating lung diseases with fibrotic indication.
3.3.1. ASO designs
Three generations of ASO sequence design have been proposed. The first generation includes all PS DNA ASOs, used in lengths of 14–25 nucleotides (nts) long. The second generation was the MOE-DNA ‘Gapmer,’ where nucleotides 1–5 were 2′-MOE, nucleotides 6–10 were comprised of DNA, and nucleotides 11–15 were 2′-MOE, such that a gap of DNA existed around the scissile bond to induce RNAse H cleavage. This improved potency, stability, and durability over the first generation. Further exploration has led to design choices to arrive at the 2.5/3rd generation cEt/LNA Gapmers () [Citation41]. This Gapmer design increased the potency by 20× time and has since become a staple in design choice by Ionis Pharmaceuticals. ION-827359 from Ionis is an inhalable Gapmer ASO formulation being developed for CF to assess the hyperactivity of ENaCs, though this trial has been paused due to safety concerns. The ASO sequence has the (S)-cEt modification that provides excellent target affinity and nuclease resistance. It binds to αENaC pre-mRNA via complementarity to the fourth intronic region to recruit RNAse-H1 for cleavage [Citation42]. The ASO was evaluated in a Phase 1/2a study involving healthy volunteers and CF patients. Single (3–100 mg) and multiple dosage (10–75 mg, 5 doses, weekly) of the drug were shown to be well tolerated with low systemic exposure [Citation43]. The inhaled formulation was absorbed rapidly and led to a dose-dependent reduction in ENaC mRNA levels in the airway epithelial cells.
Studies have reported that the optimization of synthetic efficiency/potency can be achieved with shorter ASO with 3–10-3 LNA-DNA-LNA design () [Citation39,Citation44]. In some instances, it was observed that 14mers were as potent as the respective 15, 16, and 20mers [Citation39]. The target specificity for ASO design for the lung can also be achieved by inserting systematic base pair mismatches in the middle of the sequence and further increasing the number of mismatches from there. Generally, a dramatic drop-off in knockdown is observed after the second mismatch is incorporated [Citation37,Citation45]. To mitigate cross-reactivity, A- and T-rich sequence design for 14–25 nucleotides ASOs can be adopted to avoid high G/C content [Citation46].
3.3.2. siRNA design
Previously published in-silico based studies have shown siRNA sequence designs guided by rules proposed by Ui-Tei, Amarzguioui, and Reynold [Citation40,Citation47]. Further, the sequence design choices account for circumventing immune response from Toll-like receptors (TLR) [Citation48]. Ui-Tei’s siRNA sequence designs are prominently used among chemists.
3.3.3. Splice switching ONTs (SSO)
Splice-switching ONTs (SSOs) are typically 15–30 nucleotides long that are directed toward splicing sites to correct an altered splicing event that results in the formation of an aberrant protein. SpliSense’s leading molecule, SPL84-23-1, is an SSO with 2′-OMe/PS chemical modification that targets the 3849+10 kb C-to-T mutation of CFTR gene to rescue CFTR expression in primary human bronchial and nasal epithelial cells at ascending doses of 0.1–100 nM of the SSO [Citation49]. On 4 January 2022, SPL84-23-1 received Orphan Drug Status for the treatment of CF in the EU and USA and currently planning Phase I/II studies beginning mid-2022. The drug will be delivered as an inhaled formulation with single ascending dose (SAD) and multiple ascending dose (MAD) of 5–40 mg. SpliSense is also developing SPL24-N and SPL23-2. These are two SSOs targeted toward other splice variants associated with CF, and they are expected to enter clinical trials by 2023.
3.3.4. DNAzyme
DNAzymes are single-stranded DNA molecules that bind to target mRNA by Watson–Crick base pairing and cause catalytic cleavage of target mRNA strands in the presence of metal ions (such as Na+, Mg2+, Ca2+, Pb2+ etc.). A typical DNAzyme contains an active size for catalytic cleavage surrounded by two binding arms responsible for mRNA recognition. SB-010 is an inhalable DNAzyme formulation that is being developed by Sterna Biologicals for the treatment of asthma and COPD. The ASO is administered in phosphate-buffered saline solution via an inhalation device at a dose of 5.0 mg/ml [Citation50]. GATA-3 is the master transcription factor in regulating the Th2 response. SB-010 prevents translation of GATA-3 by binding and cleaving the mRNA precursor of the protein. SB-010 was evaluated in Phase IIa clinical trials for asthma and COPD patients. Orally inhaled SB-010 improved lung function in both early and late-phase asthma patients. SB-010 utilized for the treatment of moderate-to-severe COPD patients showed that it was efficacious by reducing the relative sputum eosinophil count by 58.8% as compared to 11% in the placebo group and patients who received the standard therapy of inhaled corticosteroids. In both the studies, SB-010 was demonstrated to be safe, well tolerated with no serious adverse effects [Citation51].
4. Specialized vectors for ONT delivery to the lung
To treat the fibrotic diseases of the lung, chemically modified (naked) oligonucleotide can be aerosolized and delivered by inhalation and details have been covered in section 5.1. Both viral and non-viral vectors have also been utilized to deliver lung tropic ONTs.
4.1. Viral vectors
Viruses are naturally evolved transfecting vectors with high transfection capability to deliver functional copies of genes to target cells. 4D-Molecular Therapeutics, USA, has developed 4D-710 which is a viral vector-based gene therapy to deliver a transgene cassette encoding CFTR gene with a deletion in the regulatory domain (CFTRΔR) to lung airway cells for the treatment of cystic fibrosis patients irrespective of their mutation. The proprietary adeno-associated virus (AAV) vector, called 4D-A101, was designed through a directed evolution approach that employed non-human primates (NHP) to screen a library of 1 billion individual capsid protein sequences [Citation52]. In NHP model, A101 demonstrated enhanced tropism toward lung airway cells and robust transduction and subsequent transgene expression of CFTRΔR gene when delivered via clinically approved nebulizer. Based on positive NHP data, they have initiated an open-label Phase 1/2 in adults with CF who are ineligible or unable to tolerate CFTR modulator therapy [Citation53]. The primary outcome measure of the study is to monitor the incidence and severity of adverse events and dose limiting toxicities at two dose levels of 4D-710.
4.2. Non-viral vectors
4.2.1. Lipid Nanoparticles
Out of these platforms, lipid-based nanoparticles (LNPs) have recently received a lot of attention due to their success in COVID-19 mRNA vaccines. Both Spikevax® (Moderna) and Comirnaty® (Pfizer-BioNtech) have been approved by the FDA and have been key in battling the SARS-Cov-2 pandemic, and these are LNP formulations. This application has been extended to treat CF as well. Translate Bio (currently Sanofi Pasteur) is the first aerosolized LNP for direct pulmonary delivery. Their lead candidate MRT 5005 is an LNP-encapsulating codon-optimized CFTR mRNA capable of enhanced expression of functional CFTR protein. In the single ascending dose (8, 16, and 24 mg) arm of the study, four out of nine patients treated with MRT 5005 demonstrated marked improvement in the function of the lung. This suggests that the drug can cross the mucus barrier and aid in the production of functional CFTR protein. However, in the multiple ascending doses (five once-weekly doses of 8, 12, and 16 mg) arm involving 16 patients, no improvement in the lung function was observed and lack of efficacy halted the clinical trials [Citation54].
4.2.2. Exosomes
Exosomes are extracellular vesicles filled with biologically active moieties such as proteins, RNAs, and lipids that play a critical role in inter-cellular communication. Capricor Therapeutics (California, USA) is developing CAP-1002, a novel exosome platform, derived from proprietary Cardiosphere-derived cells (CDCs). In a Phase II study, 63 critically ill COVID-19 patients were injected 100 ml of 150 M CDCs with 5% Human Serum Albumin (HSA) via intravenous infusion. These exosomes were loaded with mRNAs encoding SARS-CoV-2 Spike and Nucleocapsid proteins. As a result, long lasting, dose-dependent development of anti-Spike and anti-Nucleocapsid antibody was observed validating the safety and tolerability of the intravenous formulation of CAP-1002 [Citation55].
4.2.3. Polymer-based delivery system
Besides lipid-based delivery systems, polymers are another main class of material that have shown promising delivery capabilities for oligonucleotides. Natural polymers such as chitosan have been widely investigated to complex with oligonucleotides because of their mucoadhesive and mucopermeable properties, allowing them to deliver RNA across mucus layer efficiently. Chitosan helped to enhance the mucus penetration, when coated to the surface of PLGA (poly-D, L-lactide-co-glycolide) nanoparticles [Citation56], while more water-soluble piperazine-substituted chitosan derivatives directly complexed with siRNA and facilitated pulmonary delivery of siRNA [Citation57]. Other than chitosan, PEI being the most investigated synthetic polymer, also played an important role in the delivery of oligonucleotide to the lung. The versatility of PEI allows incorporating with targeting ligands, leading to more specific delivery and reduced toxicity, as demonstrated by targeted delivery of siRNA to activated T cells in the lung through transferrin-PEI polyplexes [Citation58]. Although polymer-based delivery systems have not progressed to the clinic, their versatility continues to contribute to the evolution of delivery strategies.
5. Routes of administration
An important aspect of maximizing the therapeutic effect of the oligonucleotide with/without the appropriate delivery cargos is the identification of an efficient route of administration. We have presented here an overview of four popular routes of administration for lung targeted ONTs.
5.1. Inhalation
Inhalation is the preferred method to deliver therapies to the lungs in humans due to non-invasiveness and translatability. A caveat of inhalation for animal studies is that it has been difficult to get the entire administered dose into the lung of the animals due to a lack of effective nebulizing devices (around 99% of aerosol droplets are recycled back into the reservoir) [Citation59]. For human inhalation, well-developed inhaler devices with efficient drug delivery to deep lung tissue exist, including metered-dose inhalers (MDIs), dry powder inhalers (DPIs), and nebulizers, which are currently available devices that could be utilized for pulmonary delivery of ONTs. However, caveats of MDIs include that they need to use propellants to drive the drug to the respiratory tract that may create compatibility issues (i.e. chlorofluorocarbons or CFCs and hydrofluoroalkanes or HFAs), these propellants have environmental concerns, and there is poor lung deposition with MDIs. There has been some success at formulating stable chitosan-PEG 1000 nanoparticles less than 230 nm in size to HFA 227 which could possibly be used in MDIs to deliver siRNA in the future [Citation60].
DPIs allow for inhalation of aerosol clouds of dry particles, and these dry powders have better stability than liquid aerosols, and they do not have to be formulated with propellants. DPIs could be formulated to deliver ONTs to the lungs; however, the drug deposition could be dependent on patient inspiratory flow rate and the delivery device must account for this. There has been some success at forming dry powders of ONTs that could be used for this approach. Nebulizers generate liquid aerosols and can deliver large volumes of drug solutions to the lung via inhalation [Citation61]. There are several nebulization devices that can be used for pre-clinical models in mice including the nose-only chambers (inExpose, Scireq) or free-moving nebulizing chambers that can be used (these are less invasive, but less drug is deposited than in nose-only chambers) [Citation62]. Indeed, the inExpose device has been characterized for pulmonary delivery of liposomes and nucleic acids in mouse models of allergic inflammation [Citation63]. In addition, biomolecules tend to be less stable in liquid form than in dry powder form, and stress from nebulization could break down LNPs, so this should be considered in development. There are a variety of nebulizing devices already available for delivery of drugs to humans, including jet nebulizers, vibrating mesh nebulizers, smart nebulizers, ultrasonic nebulizers, and soft mist inhalers [Citation64]. Formulating ONTs and nanoparticles that can withstand nebulization may be a promising approach.
5.2. Intranasal
Intranasal drug delivery to the lung is popular because it is a minimally invasive route, easy to perform, and only requires light anesthetization. ONT formulations can be administered dropwise through the nares of the mouse via a catheter or a pipette. Zhang and colleagues were the first to show that naked siRNA intranasal delivery to the lung could knock down a gene in vivo. They did this through targeting of the heme-oxygenase-1 protein (HO-1) and naked siRNA for this gene significantly downregulated HO-1 expression in the whole lung [Citation65]. At the same time, Bitko and Barik showed that unencapsulated siRNA delivered intranasally could protect against RSV infection [Citation66]. Another study that used intranasal delivery of naked siRNA against PAI (plasminogen activator inhibitor)-1 to the lung showed decreased PAI-1 expression in the lung and attenuation of pulmonary fibrosis in a bleomycin mouse model of IPF [Citation67].
In humans, intranasal administration is not highly effective or transferable, due to absorption in the naso/oropharynx, mucociliary clearance, and other mucosal barriers, and because humans are not obligate nose-breathers. One study showed that only about 3% of 1–5 μM particles are deposited in the lung when humans inhale through the nose [Citation68]. In addition to these drawbacks, intranasal delivery may have an indirect impact on the central nervous system (CNS) as it has been shown that particles including nucleotides (in particular, nucleotides attached to nanoparticles) can bypass the blood–brain barrier through a paracellular path between tight junctions or through endocytic pathways in the nasal epithelium [Citation69]. One such study showed that siRNA against Beclin1 that was compounded to polyethyleneimine (PEI) as a carrier was able to effectively knockdown Beclin1 (65% after 24 hours and 43% after 48 hours with 16 μg treatment) in the brain when administered intranasally [Citation70]. Thus, it is important to keep in mind that although this route is effective for testing in animal models, off-target effects on the brain should be considered when testing ONTs delivered IN.
5.3. Intravenous
Intravenous (IV) delivery of ONTs to the lung is more systemic than the previously described routes above and challenging due to several factors. First, the accumulation of ONT in the liver and its subsequent rate of clearance. This not only determines the effective dose of the oligonucleotide to reach the lung but also the efficient dose for successful knockdown. Besides this, ONTs must overcome the innate immune response and maintain stability against nucleases in the vascular milieu. Additionally, the physical properties of ONTs such as size, hydrophilicity, and negative charge hamper their ability to cross cellular membranes, thereby making it difficult for localized delivery to the lung.
A unique delivery method of polymeric antisense oligonucleotide (ASO) against TGF-β mRNA to treat lung fibrosis along with cationic peptide of a dimeric human β-defensin peptide, found in epithelial cells, improved accumulation of ASO in the lung [Citation71]. A similar approach was adopted by another group to deliver siRNA complexed with K-β peptide with the capability to form intermolecular β sheet structure to form 60 nm complexes that resulted in passive accumulation and gene silencing in lung capillaries [Citation72]. In another study, siRNA encapsulated in PEGylated cationic LNPs (Staramine) when delivered intravenously showed preferential uptake and long retention times for siRNA in the mouse lung [Citation73]. Although several studies have accomplished IV delivery of oligonucleotides, two comparative studies have highlighted the drawbacks of this delivery method. Firstly, it was observed that aerosolized nanoparticles encapsulated with siRNA were more efficacious than IV delivery as they escaped accumulation in the liver [Citation74]. In another study, ASO and siRNA when administered intratracheally also showed longer retention and higher peak concentration in the lungs as compared to IV delivery [Citation75].
5.4. Intratracheal
Intratracheal (IT) administration is useful for drug delivery to the lung in animal models, though its translation to human use is lower than that of the inhalation route due to (1) the invasiveness of the procedure and (2) reduced drug loss due to oropharynx avoidance/physiological differences [Citation76]. For endotracheal administration, the rodent always must be anesthetized and a device such as an ET tube or needle must be inserted into the trachea for drug administration via suspension or aerosolized by a microsprayer device or nebulizing catheter [Citation76,Citation77]. In addition, this oro-tracheal route is difficult to learn as direct administration to the trachea and lungs rather than the esophagus is required [Citation78]. Despite these difficulties, IT administration is advantageous as there is high efficiency of drug delivery and because it can be used for proof-of-concept studies using ONTs.
Several groups have shown that unconjugated siRNA was able to reduce lung mRNA expression significantly without inducing local lung inflammation [Citation79]. In addition, direct IT lung delivery of siRNA through also improves therapeutic benefits and reduces systemic side effects [Citation80]. It has been shown that ONTs can be delivered to the lungs through nebulization catheters (Aeroprobe), nebulizers (Micro-Mist), and microsprayers (Penn-Century) to aerosolize dry powders using IT delivery [Citation81,Citation82]. It is also possible to aerosolize dry powders of nanoparticles for in vivo intratracheal delivery to the deep lung using devices like the Dry Powder Insufflator™ [Citation83].
6. Challenges of delivering ONTs to the lung
Designing therapeutic oligonucleotides for treating fibrotic lung diseases requires innovative design to overcome the biological barriers discussed above. In addition to naked delivery of ONTs, assisted delivery has also become popular among the scientific community. However, ONTs pose the following challenges discussed below.
6.1. Targeted delivery to the lung
The most important consideration while delivering the therapeutic drug to treat fibrotic lung diseases is to reach the distal lung. For nanoparticles to reach the distal lungs, their diameter sizes should lie within the range of 10–500 nm [Citation84]. Additionally, liver being the metabolic hub easily attracts the ONTs. A study conducted by Li et al. demonstrated that conjugating the antibody αPV1 to LNP surface enabled successful localization of mRNA for protein expression in mouse lung [Citation85]. Studies have shown that synthesizing lipids with properties similar to surfactant and combining with adjuvants [Citation86] and surfactant proteins from alveolar epithelial cells can help in avoiding clearance by the mucus lining and penetrating the surfactant lining of the respiratory tract [Citation87]. Oligonucleotides can also activate an inflammatory response in the lung; therefore, it is important to design sequences that avoid activating the STING pathway, interferons, and toll-like receptors.
6.2. Strategies for improved pharmacokinetic (PK) and pharmacodynamic (PD) properties
Various approaches have been adopted to achieve an ideal PK/PD profile for the delivery of ONTs. One such example is the Self-Assembled Micelle inhibitory RNA (SAMiRNA) nanoparticle technology developed by Bioneer, where the siRNA is conjugated with hydrophilic polymers and lipids that increased the stability and decreased the immunotoxicity since no noticeable induction of cytokines was observed in mouse PBMC (peripheral blood mononuclear cell) treated with SAMiRNA. The formulation demonstrated suitable pharmacokinetic profile since the SAMiRNA nanoparticle successfully reduced collagen accumulation in lung tissue and restored pulmonary function in the IPF animal model when administered via intratracheal and intravenous routes [Citation88]. Besides nanoparticles, Wang et al. reported emulsion polyplexes of perfluorocarbon with a fluorinated polymeric antagonist (F-PAMD) for chemokine receptor type 4 (CXCR4) as an siRNA carrier administered via pulmonary delivery in a bleomycin mouse model. These emulsion polyplexes were retained in the lung for up to 24 hours, with less availability to other organs and showed uniform distribution to all the lobes of the lung [Citation89].
Other suggested improvements in pharmacodynamic properties of siRNA delivery can be attained by chemical engineering of components conjugated to siRNA. In a study by Biscans et al., mono-, di-, and tri-meric fatty acids conjugated to siRNA were screened for their uptake by various tissues in mouse and they observed a higher retention of siRNA conjugated to fatty acid-Myristic acid dimeric (Myr-d) in the lung and spleen [Citation90]. Several research groups have utilized PEI for delivering siRNA systemically to the lung, and others have used deacetylated PEI or JetPEI containing siRNA or ssiRNA (RNA with sticky overhangs) to treat lung diseases and reported improved pharmacokinetics and biodistribution. Park et al. have provided a comprehensive summary of advancements in siRNA nanomedicines for lung diseases and their PK/PD profiles [Citation91].
6.3. Safety and toxicity considerations for pulmonary delivery
Traditional naked siRNA delivery approach is limited by factors that make them unviable for endocytosis or tissue delivery. Stability can be improved with nucleotide modifications that improve PK/PD profile for longer half-life and an increase in potential efficacy; however, the highly negative charge of these molecules makes them poor vehicles for cell internalization and substrate binding. As such, delivery strategies to target and/or encapsulate siRNA within LNPs have been developed to mitigate systemic delivery limitations, as well as using direct tissue delivery to the lung or trachea using nebulized or aerosolized formulations.
A recent murine study looked at cationic liposome (CDO14) toxicity noted no change in hepatic and immune toxicities at higher doses after complexation with IGF-1 R-siRNA, at higher doses and none at lower doses, indicating an effect by the vehicle [Citation92]. Compared to their neutral counterparts, cationic LNPs result in higher toxicity due to their ROS generation [Citation93], contribute to an enhanced interferon response [Citation94], and can attenuate or intensify the intended siRNA therapeutic effect [Citation95,Citation96]. Due to the stimulation of immune inflammatory responses by LNP, this may be problematic for chronic lung diseases.
Naturally, derived NP formulations have also been developed to reduce the toxic effects of LNPs. Bovine milk extracellular vesicles are naturally occurring vehicles thought to be ideal for siRNA delivery. They have a long circulating half-life, are biocompatible, and do not appear to stimulate an immune response. A recent study was able to effectively deliver siRNA against signaling substrates KRAS, VEGF, AKT, EGFR, and MAPK in lung cell lines using a milk derived exosome, and successfully reduced tumor volume with the siKRAS formulation in a murine xenograft model [Citation96].
Cationic LNP and lipoplexes generally have better transfection efficiencies but do not have reliable stability or reproducibility compared to other lipid-based siRNA delivery platforms due to issues with limited endosomal escape. One technology utilizing direct naked siRNA delivery represents a potentially viable alternative. A novel siRNA platform, termed PnkRNA, incorporates two proline derivatives into a linear sequence that self-anneal into a double-loop structure for stability was used for naked siRNA delivery to pulmonary tissue through aerosolization without the need for a delivery platform. This approach was able to inhibit ribophorin II, while avoiding significant toxicity associated with LNP formulations [Citation97].
While encapsulated formulations improve siRNA delivery, toxicological challenges present an opportunity for exploration and improvement. Delivery via the trachea, nasal passages, or lungs has been explored to circumvent first-pass metabolism associated with traditional administration routes [Citation98]. As such, aerosolized formulations have been developed to deliver siRNA directly to the lung tissue for relevant indications like COPD, fibrosis, or asthma, which also benefit from reduced risk for systemic toxicities. An ASO targeting IL-4α, ISIS 369645, was administered by inhalation in murine and NHP studies. Systemic toxicities outside the local tissue of administration were not observed between 15 and 50 mg/kg/exp, which exceed doses proposed for clinical trials by a factor of 20 to 100 [Citation99]. Thus, the use of delivery devices that directly administer ONTs to the lung would be beneficial as they would reduce off-target and systemic effects.
7. Expert opinion
RNA therapeutics have been of interest since the 1990s, with the discovery of RNA silencing in mammals [Citation100]. siRNAs and ONTs are particularly of interest due to their ability to therapeutically inhibit otherwise difficult to target genes. siRNAs have been in development over the past 20 years and in 2018 the FDA approved the first siRNA therapy, patisiran (Alnylam), to treat hereditary transthyretin amyloidosis. Following that, in 2019, givosiran (Alnylam) was approved to treat acute hepatic porphyria; in 2020, lumasiran (Alnylam) was approved to treat primary hyperoxaluria type 1; and in 2021, inclisiran (Novartis) was approved to treat high LDL cholesterol levels. These therapeutics have used GalNAc-conjugation or lipid nanoparticle systems to directly deliver siRNA to the liver.
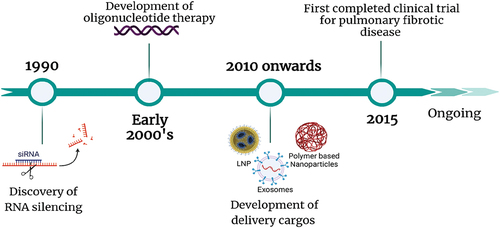
Despite these important advances in RNA therapeutics over the last 20 years, it has proven difficult to get them to the lung. shows the timeline of the development in the field of RNA therapeutics for pulmonary fibrotic diseases. There have been improvements in therapies for fibrotic lung diseases including chronic asthma, CF, and ILDs, which include the use of biologics (omalizumab, dupilumab, tezepelumab, and others), anti-CFTR modulators (ivacaftor, iumacaftor, tezacaftor, and exlexacaftor), and anti-fibrotic small molecules (nintedanib and pirfenidone). However, many of these treatments are not focused on the underlying causes, are systemic treatments, or only work in a specific subset of those with the disease, indicating a need for alternate therapeutic modalities for the treatment of these debilitating diseases. Due to the nature of biologics and small molecules, there are often off-target systemic effects or concerns with adverse side-effects (i.e. immunosuppression in the case of many biologics and tolerability concerns in the case of nintedanib and pirfenidone). Many groups have shown that direct delivery to lung can offset these systemic toxicity concerns. RNA therapeutics present a promising approach for the treatment of fibrotic lung disease. With the aid of informatics, robust sequence designs of oligonucleotides can be utilized to target undruggable proteins, and with direct delivery to the lung it may be possible to reduce systemic adverse effects. Progress has been made on this front, with recent clinical trials focusing on lung delivery of ONTs for cystic fibrosis.
Figure 4. Timeline of discovery and development of oligonucleotide therapy for pulmonary fibrotic diseases. created with Biorender.com.
However, several factors make it difficult for the effective delivery of ONTs to the lung in the clinic. The first and foremost challenge faced early in the discovery stage is to design chemically stable ONTs that can successfully navigate the endosomal pathway in the cell to achieve knockdown. Another important consideration for lung delivery would be to circumvent the activation of immune responses to ONTs, due to the presence of immune cells in the lung. The next major challenge is the successful delivery of ONTs to the lung via the routes of administration commonly used in pre-clinical studies. In the last three decades, we have seen an exponential rise in developing various delivery methods, and one of the key considerations for delivery is that they must be translatable to humans. IT administration is not used in humans, IN administration alone may not be effective in humans due to physiological differences, and it has been difficult to create inhaled ONTs encapsulated in LNPs, as the LNPs may not withstand the sheer force of nebulization. With recent advances in formulating dry powders that can be aerosolized and demonstrate successful gene knockdown using naked siRNAs, DPIs and spray-drying techniques may prove useful in successful administration of naked or conjugated ONTs to the lung via inhalation.
Peptide conjugated ONTs specifically provide a more effective approach to treat fibrotic diseases because this technology allows us to target the specific cell types that are involved in the pathogenesis of fibrotic diseases. This technique offers the advantage of potentially reducing the systemic effects and tolerability/immunogenicity concerns over delivery cargos. To our knowledge, as summarized in , ASOs are prevalent ONTs for treating fibrotic diseases. It will be interesting to see more prominent use of mRNA, various designs of siRNAs described in and additional exploration in using CRISPR/Cas 9 systems for treating fibrotic diseases.
Overall, the ongoing exploration of ONTs for lung fibrotic diseases may prove to be an effective strategy, by incorporating these considerations during drug development. Recent developments in formulating aerosolized ONTs that can reach the distal lung will improve the applicability of ONT therapy for the lung. Besides this, ONTs may prove especially promising in treating fibrotic lung diseases particularly due to their ability to target challenging drug pathways and reduce off-target systemic effects when delivered directly to the lung.
Article highlights
Fibrotic lung diseases represent a large disease subset with an unmet clinical need that could be targeted using oligonucleotide therapy.
Oligonucleotide therapies (ONT) hold promise for the treatment of pulmonary disease as they can inhibit pathways that are otherwise difficult to target.
ONTs directly delivered to the lung show little systemic side effects compared to current therapeutic strategies.
Dry powder aerosolized inhalers could be a potential means to deliver ONTs to human subjects.
Delivery methods that are able to assist in direct targeting may also improve the use of ONTs in lung fibrotic diseases.
This box summarizes key points contained in the article.
Abbreviations
| ADME | = | absorption, distribution, metabolism, and excretion |
| AEC1 | = | type 1 alveolar cells |
| AEC2 | = | type 2 alveolar cells |
| aiRNA | = | asymmetric, interfering RNA |
| ASO | = | antisense oligonucleotide |
| BNA | = | bridged nucleic acid |
| CCL2 | = | CC motif chemokine 2, also known as MCP-1 |
| CF | = | cystic fibrosis |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| CFC | = | chlorofluorocarbon |
| CNS | = | central nervous system |
| COPD | = | chronic obstructive pulmonary disease |
| CTGF | = | connective tissue growth factor |
| CTD-ILD | = | connective tissue-related ILD |
| CXCL12 | = | CXC motif chemokine, or stromal cell-derived factor 1 |
| DAMPs | = | damage-associated molecular patterns |
| DPI | = | dry powder inhalers |
| ECM | = | extracellular matrix |
| ENaC | = | epithelial sodium channel |
| ET | = | endotracheal |
| GNA | = | glycine nucleic acid |
| HFA | = | hydrofluoroalkanes |
| HO-1 | = | heme oxygenease-1 |
| IgE | = | immunoglobulin E |
| ILD | = | interstitial lung disease |
| IPF | = | idiopathic pulmonary fibrosis |
| M1 | = | classically activated macrophages |
| M2 | = | alternatively activated macrophages |
| MAD | = | multiple ascending dose |
| MCP-1 | = | monocyte chemoattractant protein, also known as CCL2 |
| MDI | = | metered dose inhalers |
| MMP | = | matrix metalloproteinases |
| NSCLC | = | non-small cell lung cancer |
| NTs | = | nucleotides |
| ONT | = | oligonucleotide therapy |
| PAI | = | plasminogen activator inhibitor |
| PBMC | = | peripheral blood mononuclear cell |
| PDGF | = | platelet-derived growth factor |
| PEI | = | polyethyleneimine |
| PIWI | = | P-element Induced Wimpy testis in Drosophila |
| PLGA | = | poly-D, L-lactide-co-glycolide |
| PMO | = | phosphorodiamidate morpholino oligomer |
| PNA | = | peptide nucleic acid |
| PTH | = | parathyroid hormone |
| RISC | = | RNA-induced silencing complex |
| RNAi | = | RNA interference |
| RSV | = | respiratory syncytial virus |
| SAD | = | single ascending dose |
| SAMiRNA | = | self-assembeled micelle inhibitory RNA |
| shRNA | = | short, hairpin RNA |
| siRNA | = | small, interfering RNA |
| sisiRNA | = | -small, integral segmented interfering RNA |
| TGF | = | transforming growth factor |
| Th1 | = | type 1 T helper cell |
| Th2 | = | type 2 T helper cell |
| Th17 | = | type 17 T helper cell |
| TLR | = | toll-like receptor |
| TNF | = | tumor necrosis factor |
| TSLP | = | thymic stromal lymphopoietin |
Author contributions
D.P. led the effort of compiling the review article by coordinating among the authors, designing, editing and wrote parts of the manuscript. M.M. provided insights into pulmonary fibrosis and wrote parts of the manuscript. K.T. and V.R.K conceptualized and helped in interpreting relevant literature. All the authors substantially contributed towards the revision, writing, and designing figures in the manuscript.
Declaration of interest
VR Krishnamurthy is an employee of Korro Bio. All other authors are employees of Eli Lilly and Company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors would like to thank Dr Malgorzata D Gosia for their critical review of the manuscript. Additionally, we would like to thank the support from Dr Jibo Wang, Dr MariJean Eggen, and Dr Pooja Gangras from Lilly Genetic Medicine, and Dr Henry Bryant from the Department of Biotechnology and Immunology.
Additional information
Funding
References
- Gbdcrd C, Kendrick PJ, Paulson KR. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020 Jun;8(6):585–596.
- Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020 Jun;55(6):2000085.
- Interstitial Lung Disease American Lung Association 2022. Available from: https://www.lung.org/lung-health-diseases/lung-disease-lookup/interstitial-lung-disease
- Bazdyrev E, Rusina P, Panova M, et al. Lung Fibrosis after COVID-19: treatment Prospects. Pharmaceuticals (Basel). 2021 Aug 17;14(8):807.
- Laporta Hernandez R, Aguilar Perez M, Lázaro Carrasco MT, et al. Lung transplantation in idiopathic pulmonary fibrosis. Medical sciences. (Basel Switzerland): 2018 Aug 23;6(3):68.
- Strens D, Bondue B, Dahlqvist C, et al. Long-term tolerability of real-life use of antifibrotic agents (AFA) in Idiopathic Pulmonary Fibrosis (IPF). Eur Respir J. 2021;58(suppl 65):A465.
- Ruigrok MJR, Frijlink HW, Hinrichs WLJ. Pulmonary administration of small interfering RNA: the route to go? J Control Release. 2016 Aug 10;235:14–23.
- Maher JM, Zhang R, Palanisamy G, et al. Lung-restricted ALK5 inhibition avoids systemic toxicities associated with TGFβ pathway inhibition. Toxicol Appl Pharmacol. 2022 Mar 1;438:115905.
- Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017 Oct 20;3(1):17074.
- Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014 Mar 13;507(7491):190–194.
- Glasser SW, Hardie WD, Hagood JS. Pathogenesis of Interstitial Lung Disease in Children and Adults. Pediatr Allergy Immunol Pulmonol. 2010 Mar;23(1):9–14.
- Chen H, Qu J, Huang X, et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat Commun. 2016 Aug 18;7(1):12564.
- Uhal BD, Joshi I, Hughes WF, et al. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol. 1998 Dec;275(6):L1192–9.
- Zhang L, Wang Y, Wu G, et al. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res. 2018 Sep 6;19(1):170.
- Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013 Jun;1(4):309–317.
- NAEPP: National Asthma Education and Prevention Program Tepotdamoa. Expert panel Report 3: guidelines for the diagnosis and management of asthma. Bethestda MD: National Heart, Lung, and Blood Institute (US).; 2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7232/.
- Wenzel S. Severe asthma: from characteristics to phenotypes to endotypes. Clin Exp Allergy. 2012 May;42(5):650–658.
- Kumar S, Jeong Y, Ashraf MU, et al. Dendritic cell-mediated Th2 Immunity and immune disorders. Int J Mol Sci. 2019 May 1;20(9):2159.
- Leon B, Ballesteros-Tato A. Modulating Th2 Cell Immunity for the treatment of asthma. Front Immunol. 2021;12:637948.
- Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled Asthma. N Engl J Med. 2021 May 13;384(19):1800–1809.
- Chen Y, Dales R, Krewski D, et al. Increased effects of smoking and obesity on asthma among female Canadians: the national population health survey, 1994-1995. Am J Epidemiol. 1999 Aug 1;150(3):255–262.
- Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017 May 13;389(10082):1931–1940.
- O’Donnell DE. Hyperinflation, dyspnea, and exercise intolerance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006 Apr;3(2):180–184.
- Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012 Apr 7;379(9823):1341–1351.
- Moll M, Sakornsakolpat P, Shrine N, et al. Chronic obstructive pulmonary disease and related phenotypes: polygenic risk scores in population-based and case-control cohorts. Lancet Respir Med. 2020 Jul;8(7):696–708.
- Chen Q, Shen Y, Zheng J. A review of cystic fibrosis: basic and clinical aspects. Animal Model Exp Med. 2021 Sep;4(3): 220–232.
- Elborn JS. Cystic fibrosis. Lancet. 2016 Nov 19;388(10059):2519–2531.
- Giron Moreno RM, Garcia-Clemente M, Diab-Caceres L, et al. Treatment of Pulmonary Disease of Cystic Fibrosis: a Comprehensive Review. Antibiotics (Basel). 2021 Apr 23;10(5):486.
- Bose SJ, Krainer G, Drs N, et al. Towards next generation therapies for cystic fibrosis: folding, function and pharmacology of CFTR. J Cyst Fibros. 2020 Mar;19 Suppl 1:S25–S32.
- Moore PJ, Tarran R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin Ther Targets. 2018 Aug;22(8):687–701.
- Darquenne C. Deposition Mechanisms. J Aerosol Med Pulm Drug Deliv. 2020;33(4):181–185.
- Qiu Y, Lam JK, Leung SW, et al. Delivery of RNAi Therapeutics to the Airways-From Bench to Bedside. Molecules. 2016 Sep 20;21(9):1249.
- Bost JP, Barriga H, Holme MN, et al. Delivery of oligonucleotide therapeutics: chemical modifications, lipid nanoparticles, and extracellular vesicles. ACS Nano. 2021 Sep 28;15(9):13993–14021.
- Egli M, Manoharan M. Re-Engineering RNA molecules into therapeutic agents. Acc Chem Res. 2019 Apr 16;52(4):1036–1047.
- Kandasamy P, Liu Y, Aduda V, et al. Impact of guanidine-containing backbone linkages on stereopure antisense oligonucleotides in the CNS. Nucleic Acids Res. 2022 Feb 2;50(10):5401–5423.
- Iwamoto N, Butler DCD, Svrzikapa N, et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol. 2017 Sep;35(9):845–851.
- Karras JG, Crosby JR, Guha M, et al. Anti-inflammatory activity of inhaled IL-4 receptor-alpha antisense oligonucleotide in mice. Am J Respir Cell Mol Biol. 2007 Mar;36(3):276–285.
- Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 2017 Mar;35(3):238–248.
- Seth PP, Siwkowski A, Allerson CR, et al. Short antisense oligonucleotides with novel 2′−4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem. 2009 JAN 08;52(1):10–13.
- Nur SM, Al Amin M, Alam R, et al. An in silico approach to design potential siRNA molecules for ICP22 (US1) gene silencing of different strains of human herpes simplex 1. J Young Pharm. 2013 Jun;5(2):46–49.
- Hong D, Kurzrock R, Kim Y, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015 Nov 18;7(314):314ra185.
- Ionis Pharmaceuticals I A Phase 1/2a Study to Assess the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of IONIS-ENaCRx in Healthy Volunteers and Patients With Cystic Fibrosis. 2018. https://ClinicalTrials.gov/show/NCT03647228
- Sutharsan S, Fischer R, Gleiber W, et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of ION-827359, an Antisense Oligonucleotide Inhibitor of ENaC, in Healthy Volunteers and Patients with Cystic Fibrosis: a Double-Blind, Placebo-Controlled, Dose-Escalation, Phase 1/2a Study. Healthy Volunteers and Patients with Cystic Fibrosis: A Double-Blind, Placebo-Controlled, Dose-Escalation, Phase.1.
- Siwkowski AM, Madge LA, Koo S, et al. Effects of antisense oligonucleotide-mediated depletion of tumor necrosis factor (TNF) receptor 1-associated death domain protein on TNF-induced gene expression. Mol Pharmacol. 2004 Sep;66(3):572–579.
- Crosby JR, Guha M, Tung D, et al. Inhaled CD86 antisense oligonucleotide suppresses pulmonary inflammation and airway hyper-responsiveness in allergic mice. J Pharmacol Exp Ther. 2007 Jun;321(3):938–946.
- Daniel C, Takabatake Y, Mizui M, et al. Antisense oligonucleotides against thrombospondin-1 inhibit activation of tgf-beta in fibrotic renal disease in the rat in vivo. Am J Pathol. 2003 Sep;163(3):1185–1192.
- Kobayashi Y, Fukuhara D, Akase D, et al. siRNA Seed Region Is Divided into Two Functionally Different Domains in RNA Interference in Response to 2’-OMe Modifications. ACS Omega. 2022 Jan 18;7(2):2398–2410.
- Oh SY, Ju Y, Park H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol Cells. 2009 Oct 31;28(4):341–345.
- Oren YS, Irony-Tur Sinai M, Golec A, et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J Cyst Fibros. 2021 [2021 Oct 01];20(5):865–875.
- Bhindi R, Fahmy RG, Lowe HC, et al. Brothers in arms: DNA enzymes, short interfering RNA, and the emerging wave of small-molecule nucleic acid-based gene-silencing strategies. Am J Pathol. 2007;171(4):1079–1088.
- Greulich T, Hohlfeld JM, Neuser P, et al. A GATA3-specific DNAzyme attenuates sputum eosinophilia in eosinophilic COPD patients: a feasibility randomized clinical trial. Respir Res. 2018;19(1):55–55.
- Calton M, Kotterman M, Schmitt C, et al. A10. A010 NOVEL DISCOVERIES IN LUNG BIOLOGY. In: Identification and Characterization of a Novel AAV Capsid and Product for the Treatment of Cystic Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2021;A1043–A1043.
- 4D-710 in Adult Patients With Cystic Fibrosis. https://ClinicalTrials.gov/show/NCT05248230.
- Study to Evaluate the Safety & Tolerability of MRT5005 Administered by Nebulization in Adults With Cystic Fibrosis. https://ClinicalTrials.gov/show/NCT03375047.
- CAP-1002 in Severe COVID-19 Disease. https://ClinicalTrials.gov/show/NCT04338347.
- Haque AKMA, Dewerth A, Antony JS, et al. Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci Rep. 2018 Nov 13;8(1):16776.
- Capel V, Vllasaliu D, Watts P, et al. Water-soluble substituted chitosan derivatives as technology platform for inhalation delivery of siRNA. Drug Deliv. 2018 Jan 01;25(1):644–653.
- Xie Y, Kim NH, Nadithe V, et al. Targeted delivery of siRNA to activated T cells via transferrin-polyethylenimine (Tf-PEI) as a potential therapy of asthma. J Control Release. 2016 May 10;229:120–129.
- Agu RU, Ugwoke MI, Armand M, et al. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res. 2001;2(4):198–209.
- Sharma K, Somavarapu S, Colombani A, et al. Crosslinked chitosan nanoparticle formulations for delivery from pressurized metered dose inhalers. Eur J Pharm Biopharm. 2012 May;81(1):74–81.
- Sharma K, Somavarapu S, Colombani A, et al. Nebulised siRNA encapsulated crosslinked chitosan nanoparticles for pulmonary delivery. Int J Pharm. 2013 Oct 15;455(1–2):241–247.
- Mainelis G, Seshadri S, Garbuzenko OB, et al. Characterization and application of a nose-only exposure chamber for inhalation delivery of liposomal drugs and nucleic acids to mice. J Aerosol Med Pulm Drug Deliv. 2013 Dec;26(6):345–354.
- Aich J, Mabalirajan U, Ahmad T, et al. Loss-of-function of inositol polyphosphate-4-phosphatase reversibly increases the severity of allergic airway inflammation. Nat Commun. 2012 Jun 6;3(1):877.
- Youngren-Ortiz SR, Gandhi NS, Espana-Serrano L, et al. Aerosol Delivery of siRNA to the Lungs. Part 1: rationale for Gene Delivery Systems. Kona. 2016 Feb 28;33:63–85.
- Zhang X, Shan P, Jiang D, et al. Small interfering RNA targeting heme oxygenase-1 enhances ischemia-reperfusion-induced lung apoptosis. J Biol Chem. 2004 Mar 12;279(11):10677–10684.
- Bitko V, Musiyenko A, Shulyayeva O, et al. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med. 2005 Jan;11(1):50–55.
- Senoo T, Hattori N, Tanimoto T, et al. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax. 2010 Apr;65(4):334–340.
- Heyder J, Gebhart J, Rudolf G, et al. Deposition of particles in the human respiratory tract in the size range 0.005–15 μm. J Aerosol Sci. 1986 Jan 01;17(5):811–825.
- Illum L. Nasal drug delivery–possibilities, problems and solutions. J Control Release. 2003 Feb 21;87(1–3):187–198.
- DeVincenzo J, Cehelsky JE, Alvarez R, et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res. 2008 Mar;77(3):225–231.
- Kim J, Jeon S, Kang SJ, et al. Lung-targeted delivery of TGF-β antisense oligonucleotides to treat pulmonary fibrosis. J Control Release. 2020 Jun 10;322:108–121.
- Zhou J, Li D, Wen H, et al. Inter-molecular β-sheet structure facilitates lung-targeting siRNA delivery. Sci Rep. 2016 Mar 9;6(1):22731.
- Polach KJ, Matar M, Rice J, et al. Delivery of siRNA to the mouse lung via a functionalized lipopolyamine. Mol Ther. 2012 Jan;20(1):91–100.
- Yan Y, Zhou K, Xiong H, et al. Aerosol delivery of stabilized polyester-siRNA nanoparticles to silence gene expression in orthotopic lung tumors. Biomaterials. 2017 Feb;118:84–93.
- Garbuzenko OB, Saad M, Betigeri S, et al. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharm Res. 2009 Feb;26(2):382–394.
- Lam JK, Liang W, Chan HK. Pulmonary delivery of therapeutic siRNA. Adv Drug Deliv Rev. 2012 Jan;64(1):1–15.
- Bivas-Benita M, Zwier R, Junginger HE, et al. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur J Pharm Biopharm. 2005 Oct;61(3):214–218.
- Sakagami M. In vivo, in vitro and ex vivo models to assess pulmonary absorption and disposition of inhaled therapeutics for systemic delivery. Adv Drug Deliv Rev. 2006 Oct 31;58(9–10):1030–1060.
- Perl M, Chung CS, Lomas-Neira J, et al. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol. 2005 Dec;167(6):1545–1559.
- Xie Y, Merkel OM. Pulmonary Delivery of siRNA via Polymeric Vectors as Therapies of Asthma. Arch Pharm (Weinheim). 2015 Oct;348(10):681–688.
- Koping-Hoggard M, Issa MM, Kohler T, et al. A miniaturized nebulization catheter for improved gene delivery to the mouse lung. J Gene Med. 2005 Sep;7(9):1215–1222.
- Nielsen EJ, Nielsen JM, Becker D, et al. Pulmonary gene silencing in transgenic EGFP mice using aerosolised chitosan/siRNA nanoparticles. Pharm Res. 2010 Dec;27(12):2520–2527.
- Sinsuebpol C, Chatchawalsaisin J, Kulvanich P. Preparation and in vivo absorption evaluation of spray dried powders containing salmon calcitonin loaded chitosan nanoparticles for pulmonary delivery. Drug Des Devel Ther. 2013;7:861–873.
- Zhang Y, Almazi JG, Ong HX, et al. Nanoparticle Delivery Platforms for RNAi Therapeutics Targeting COVID-19 Disease in the Respiratory Tract. Int J Mol Sci. 2022 Feb 22;23(5):2408.
- Li Q, Chan C, Peterson N, et al. Engineering caveolae-targeted lipid nanoparticles to deliver mRNA to the lungs. ACS Chem Biol. 2020 Apr 17;15(4):830–836.
- Sigurdsson HH, Kirch J, Lehr CM. Mucus as a barrier to lipophilic drugs. Int J Pharm. 2013 Aug 30;453(1):56–64.
- Moschos SA, Usher L, Lindsay MA. Clinical potential of oligonucleotide-based therapeutics in the respiratory system. Pharmacol Ther. 2017 Jan;169:83–103.
- Lee K, Jang B, Lee YR, et al. The cutting-edge technologies of siRNA delivery and their application in clinical trials. Arch Pharm Res. 2018 Sep;41(9):867–874.
- Wang Y, Ding L, Li Z, et al. Treatment of acute lung injury and early- and late-stage pulmonary fibrosis with combination emulsion siRNA polyplexes. J Control Release. 2019 Nov 28;314:12–24.
- Biscans A, Coles A, Echeverria D, et al. The valency of fatty acid conjugates impacts siRNA pharmacokinetics, distribution, and efficacy in vivo. J Control Release. 2019 May 28;302:116–125.
- Park J, Park J, Pei Y, et al. Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv Drug Deliv Rev. 2016 Sep 1;104:93–109.
- Zhu Y, Meng Y, Zhao Y, et al. Toxicological exploration of peptide-based cationic liposomes in siRNA delivery. Colloids Surf B Biointerfaces. 2019 Jul 1;179:66–76.
- Dokka S, Toledo D, Shi X, et al. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000 May;17(5):521–525.
- Ma Z, Li J, He F, et al. Cationic lipids enhance siRNA-mediated interferon response in mice. Biochem Biophys Res Commun. 2005 May 13;330(3):755–759.
- Omidi Y, Barar J, Akhtar S. Toxicogenomics of cationic lipid-based vectors for gene therapy: impact of microarray technology. Curr Drug Deliv. 2005 Oct;2(4):429–441.
- Aqil F, Munagala R, Jeyabalan J, et al. Milk exosomes - Natural nanoparticles for siRNA delivery. Cancer Lett. 2019 May 1;449:186–195.
- Fujita Y, Takeshita F, Mizutani T, et al. A novel platform to enable inhaled naked RNAi medicine for lung cancer. Sci Rep. 2013 Nov 25;3(1):3325.
- Laube BL, Janssens HM, de Jongh FH, et al. What the pulmonary specialist should know about the new inhalation therapies. Eur Respir J. 2011 Jun;37(6):1308–1331.
- Fey RA, Templin MV, McDonald JD, et al. Local and systemic tolerability of a 2ʹO-methoxyethyl antisense oligonucleotide targeting interleukin-4 receptor-alpha delivery by inhalation in mouse and monkey. Inhal Toxicol. 2014 Jul;26(8):452–463.
- Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001 May 01;411(6836):494–498.