
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Introduction
Prediction of pharmacokinetic (PK) properties is crucial for drug discovery and development. Machine-learning (ML) models, which use statistical pattern recognition to learn correlations between input features (such as chemical structures) and target variables (such as PK parameters), are being increasingly used for this purpose. To embed ML models for PK prediction into workflows and to guide future development, a solid understanding of their applicability, advantages, limitations, and synergies with other approaches is necessary.
Areas covered
This narrative review discusses the design and application of ML models to predict PK parameters of small molecules, especially in light of established approaches including in vitro-in vivo extrapolation (IVIVE) and physiologically based pharmacokinetic (PBPK) models. The authors illustrate scenarios in which the three approaches are used and emphasize how they enhance and complement each other. In particular, they highlight achievements, the state of the art and potentials of applying machine learning for PK prediction through a comphrehensive literature review.
Expert opinion
ML models, when carefully crafted, regularly updated, and appropriately used, empower users to prioritize molecules with favorable PK properties. Informed practitioners can leverage these models to improve the efficiency of drug discovery and development process.
1. Introduction
Drug discovery is a long, expensive, and risky endeavor: It takes billions of dollars and years to bring a new molecular entity from early discovery to patients, while approximately 90% of drug candidates fail to reach the market [Citation1,Citation2]. Increasing complexity of therapeutic molecules contributes to an exponential rise in the development costs [Citation3]. Poor pharmacokinetics (PK) or pharmacodynamics (PD) profiles of the molecule are the cause of at least half of the failures [Citation4]. A better estimation of PK parameters and rational prioritization of drugs with favorable PK profiles, therefore, is key to improve the efficiency of pharma research and development [Citation5].
Poor PK properties and failure to mitigate PK-related risks have severe consequences. For instance, both low bioavailability and high clearance result in insufficient exposure at the site of action. Excessive variability in drug metabolism and drug–drug interactions may lead to inadequate PK profiles or complicate the design of dosing regimen. Examples from our recent experience include a TAAR1 agonist whose clinical development had to be paused due to large PK variability associated with a polymorphism [Citation6], as well as other small molecules that displayed unanticipated PK behavior [Citation7].
With the growth in both amount of data and computational power in the last decades [Citation8], in silico approaches to drug discovery are playing an increasingly important role [Citation9–13]. Besides activities that focus on ligand–target interaction and potency [Citation14–17], prediction of absorption, distribution, metabolism, and excretion (ADME) and PK properties remains a challenging but attractive venue of research.
Many methods and tools have been proposed and developed to predict ADME and PK properties [Citation18]. In this review, we introduce three archetypal approaches: machine learning (ML) models, in vitro-in vivo extrapolation (IVIVE), and physiologically based pharmacokinetic (PBPK) modeling (). In particular, we highlight the development and application of ML models. We argue for a hybrid approach to PK prediction that integrates all three archetypes in order to address different questions raised along the value chain of drug discovery. The goal of the hybrid approach is to provide project teams with fit-for-purpose predictive models that address the question at hand in a timely and cost-effective manner, by integrating knowledge, existing data, and newly generated sparse data.
Figure 1. Three archetypes of methods for PK parameter prediction, and visual presentations of selected PK parameters. Panel A: graphical representation of the three archetypal methods for PK property prediction: machine learning (ML), in vitro-in vivo extrapolation (IVIVE), and physiologically based PK (PBPK) modeling. Panels B-E: graphical representations of selected PK parameters that are of common interest. See for definitions and explanations. Panel a was created with BioRender.com. Panels B-E were made by JD Zhang and released under GNU public license. The source code and the figure can be found at https://github.com/Accio/2024-02-PK-parameters-visualized. JD Zhang explicitly authorizes the use of his work for this review.
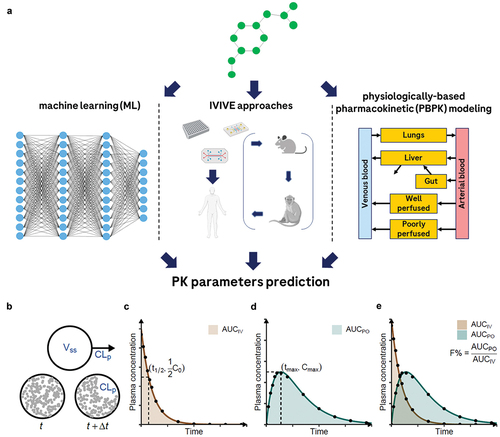
Table 1. Selected PK parameters obtainable through a single-dose PK experiment.
2. PK parameter prediction in preclinical drug discovery
2.1. Aims of PK parameter estimation
PK parameter prediction is desired at three stages of preclinical discovery and early development: during lead optimization, prior to in vivo studies, and prior to entry into humans.
During the ligand optimization phase, medicinal chemists may wish to prioritize molecules with good PK parameters for synthesis.
Once a molecule shows promising overall properties in vitro, PK prediction is an essential task prior to any in vivo pharmacokinetics, pharmacology, or toxicology studies. Comparing data derived from in vivo studies with predictions, we may increase our confidence in the PK prediction, or identify issues in either the predictive model or the in vivo study.
Prior to entry-into-human studies, human PK parameter prediction based on in vitro or in vivo findings is indispensable for the design and optimization of dosage regimens. Well-predicted PK properties are key for successful safety and efficacy assessment as well as biomarker discovery [Citation19,Citation20]. PK readout from clinical studies can be compared with preclinical predictions, bringing valuable learnings for ongoing and future projects.
One may wonder whether it is possible to predict in vivo or even human PK parameters directly with machine-learning approaches using chemical structures, and optionally data derived from in vitro and/or in vivo studies, as input. Such models may save time and resources, prioritize molecules with the most promising properties, and refine, reduce, or replace animal studies. While the first attempts toward this ambitious goal have been made [Citation21–23], the real-world performance of such models and their impact on discovery projects remain to be assessed.
2.2. Main PK parameters of interest
Most in vivo and first-in-human pharmacokinetic studies are single-dose studies, in which a single dose of the drug candidate is administered. summarizes the PK parameters of common interest that are either directly measured or derived from such studies. In case of systemic administration via intravenous injection (IV), two parameters are of primary interest: the plasma clearance (CLp), i.e. the volume of plasma from which a substance is completely removed per unit time [Citation24], and the volume of distribution at steady state (Vss), which describes the apparent total volume in which a drug is distributed in the body [Citation25] (, panel B). Many other PK parameters can be calculated and reported. For instance, the area under the curve (AUC), which is calculated from the concentration-versus-time plot (, panel C and D), can be used to calculate CLp and Vss together with the dose (and vice versa) [Citation26].
For non-IV administration routes such as oral administration (PO), bioavailability (F, or F%) is another important PK parameter [Citation27] (, panel E). In these cases, the amount of drug entering systemic circulation is generally lower than that for the IV case. Bioavailability can be defined as the ratio of the AUC of the tested administration route to that of IV, and its value is usually smaller than 100%.
Multiple-dose PK studies offer additional information about PK parameters of the drug when it is administered multiple times, for instance the steady-state concentration, the time to reach the steady state, and the trough concentration, i.e. the concentration in the blood immediately before the next dose is administered. These parameters can be predicted from PK parameters derived from single-dose PK studies. Discrepancies between predictions and observations may reveal unexpected PK behavior.
Having introduced the key PK parameters of interest, we wish to briefly illustrate the principles of absorption, distribution, metabolism, and excretion (ADME) properties. ADME properties determine PK parameters of the molecule, and most predictive models, either explicitly or implicitly, consider ADME properties in the prediction process.
2.3. ADME properties and processes
ADME summarizes major processes governing the pharmacokinetics of compounds throughout the body. Besides physiology of the living organism and the environment it lives in, physicochemical properties of compounds influence their ADME properties. For example, the process of intestinal absorption is governed by chemical stability, solubility, and permeability of the compound, as well as rates of drug metabolism and transport in the intestinal tissues.
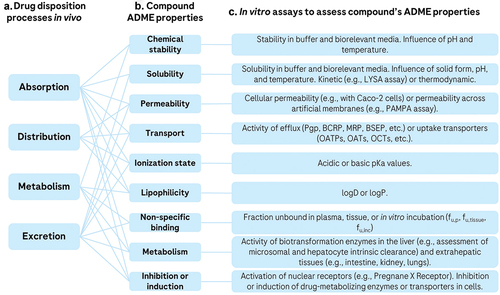
It is common to measure ADME-related properties in vitro in order to predict and explain ADME outcomes in vivo. depicts relationships between drug disposition processes in vivo, ADME properties, and various in vitro assays available to assess them.
Figure 2. Relationship between ADME processes in vivo (a), compound’s ADME properties (b), and selected in vitro assays available to assess ADME properties (c). The in vivo outcomes are typically governed by multiple ADME compound properties. Many in vitro experiments are available to measure these properties, including physicochemical, biochemical, and cellular assays. in vitro assays can be used to deconvolute observed ADME processes or to predict ADME properties prospectively.
summarizes advantages and limitations of using assays to predict ADME properties. A key strength of the assays is that they offer mechanistic insight and allow an understanding of individual properties deconvoluted from other concomitant and confounding phenomena. Furthermore, ADME assays can be standardized and automated, allowing for high-throughput (HT) and systematic testing of compounds. We believe that the value of HT assays should be rigorously scrutinized, since experience suggests that not all assays stay robust during up-scaling, and data quality may substantially deteriorate.
Table 2. Advantages and potential limitations of ADME assays as predictors of drug disposition in vivo and tools for rational drug design, as well as possible mitigation measures.
We face two types of complexity when using in vitro assays to predict in vivo outcomes. First, as long as we are dealing with models instead of the reality of human physiology, model-intrinsic biases and variances are unavoidable. For instance, in-vitro rate of liver metabolism, often expressed as intrinsic clearance (CLint) [Citation28], is only partially predictive of in vivo liver (hepatic) metabolic clearance (CLh) [Citation29].
Second, summing readouts from individual ADME assays does not necessarily equate the outcome in vivo. While the strength of an individual ADME assay partly stems from the deconvolution of mechanisms, in the physiological context, mechanisms interact with each other and show nonlinear behaviors typical of complex systems.
Despite the complexities, ADME properties measured by in vitro assays, when interpreted appropriately, inform how a compound may behave physiologically. Therefore, ADME measurements and predictions have been widely used to generate IVIVE hypotheses.
3. Three archetypes of PK prediction methods
3.1. In vitro-in vivo extrapolation (IVIVE)
In vitro and ex vivo systems that aim to model human biology are indispensable for drug discovery. Within the context of PK property prediction, the hope is that they mimic relevant aspects of the interaction between drug candidates and the human organism. Simpler systems include tissue extracts such as liver microsomes and cellular systems such as hepatocytes in suspension. More complex in vitro systems are often referred to as microphysiological systems (MPS) and sometimes summarized under the general-term New Approach Methodologies (NAM). For instance, one type of MPS, organ-on-a-chip (OoC), has been implemented in many studies for the extrapolation of human PK parameters [Citation30]. Organoids, in which adult stem cells are differentiated toward desired lineages to form a simplified biological system, provide another vivid example of such systems which can be leveraged for PK prediction [Citation31].
The creation of a proper IVIVE model requires simulating physiological and biochemical conditions with constraints, such as the size and specific blood flow of the organ, how the drug partitions, and how the compounds are eliminated from the organism [Citation32]. Calibration with compounds with known PK profiles reveals the bias and the variance of the system. Once calibrated and validated, the prediction of PK parameters is derived from statistical modeling and the implementation of species-specific scaling factors.
Classically, extrapolation of PK parameters to human individuals was performed with allometric scaling. The method is based on the observation that certain physiological parameters of different mammalian species can be described empirically by an exponential equation. Extrapolation from observations in animal studies is used to predict human PK parameters [Citation32,Citation33]. This method has been gradually replaced by more sophisticated PBPK models as described below.
Despite much progress, simulating a multifaceted organic system with in vitro systems in a reproducible and unbiased manner remains a challenge, even if the goal is limited to mimicking a subset of biological processes that are relevant for PK. To address the challenge, we consider both company internal research and community-based approaches, for instance the TEX-VAL Tissue Chip Testing Consortium, important to identify robust and predictive models [Citation34]. Rigorous experimental design, extensive calibration with compounds with well-described PK profiles, and sharing of experimental and analytical details are key prerequisites to allow adoption and further development of such models for drug discovery and development.
3.2. PBPK models
Physiologically based pharmacokinetic (PBPK) models are a well-established approach to human PK prediction. Torsten Teorell made seminal contributions as early as 1937 [Citation35]. In the early 1990s, arguments for more rational approaches leveraging pharmacokinetic principles emerged [Citation36]. The rise of in vitro technologies further accelerated mechanistic approaches for in vitro to in vivo scaling [Citation37]. The trend continued as more efficient and high-throughput in vitro approaches were developed [Citation38].
Over the past two decades, PBPK modeling has become a powerful and versatile tool for pharmaceutical research and drug development [Citation39]. PBPK provides a framework to integrate knowledge on physiology with drug properties and leverages IVIVE methods to scale in vitro ADME processes to in vivo. PBPK has gained widespread acceptance and is endorsed by regulatory agencies including the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).
PBPK has represented one of the most exploited methods for PK prediction in the latest decades [Citation40–42]. Besides simulating time-based drug concentration changes in the body of healthy volunteers and patients, PBPK models are particularly powerful for simulating PK profiles for specific populations such as pregnancy [Citation43], elderly [Citation44], and children [Citation45]. Indeed, one of the great advantages of PBPK is its adaptability to allow simulation in specific populations which are challenging to study clinically such as people affected by rare diseases, different ethnic groups or neonatal babies. Furthermore, efforts coupling PBPK with IVIVE techniques may have the potential to improve PK prediction [Citation46,Citation47].
Development of PBPK models also benefits from progress in better biological models of human physiology. Besides the NAM approaches discussed above, improved animal models offer another evidence of the synergy. While data used to be almost exclusively collected from wild-type animals as input for PBPK models, gene-edited animal models offer a new venue. Gene-editing techniques such as CRISPR-Cas9 [Citation48] are regularly applied to build relevant in vitro and in vivo models for DMPK evaluation and prediction [Citation49]. Such models are used to stimulate the behavior of drug candidates in a physiological condition mimicking that of humans. Examples include knock-out animals [Citation50], humanized animals [Citation51]. We refer interested readers to further case studies [Citation52–54].
3.3. Machine-learning methods
Over the years, the wide and consistent application of assays for the determination of physicochemical and ADME properties of small molecules has generated a large volume of data [Citation55–57]. At the same time, PK data derived from in vivo and clinical studies accumulate, too. As in other fields where abundant data are available, machine learning approaches have demonstrated promising results [Citation58].
In order to predict ADME and PK properties, machine-learning models are trained from a large collection of small molecules and matching values of the property. A well-trained and validated model makes good predictions for the property of interest for a new, unseen molecule. For readers not familiar with the basic concepts and practices of machine learning, we recommend our open-access introduction to the topic [Citation59]. We also present in Supplementary Table S1 major families of algorithms that we refer to in this review. For readers interested in more details, we recommend relevant literature [Citation60–63].
We consider machine-learning models as generalizations of quantitative structure–activity relationship (QSAR) models and their variants (QSPR models for any property, and QSPKR for PK parameters) [Citation64,Citation65]. All these methods attempt to quantitatively correlate a chemical structure with a property or potential of the compound.
Machine-learning approaches have been applied to many tasks of drug discovery, for instance to predict on-target binding potency [Citation66], off-target profiles [Citation67], physicochemical properties [Citation68], and, relevant for this work, ADME and PK parameters [Citation69,Citation70]. In some cases, the predictive performance has reached such a high level that they have largely replaced in vitro measurements, for instance permeability (by replacing the PAMPA assay) and hydrophilicity (by predicting logP). At the same time, we struggle to construct good prediction models for some parameters. Challenges include different definitions of the property, a myriad of experimental details that lead to large variability of measurements, factors other than the chemical structure that contribute to the property (for instance physiology), as well as missing data [Citation71].
Despite the challenges, the research community has witnessed encouraging achievements. For instance, Di Lascio et al. constructed ML models for 10 different ADME endpoints, using 112 drug discovery projects covering more than 800’000 compounds as input. Comparing local models, which were trained with compounds designed for the same project or chemical series, with global models, which were trained with a larger set of diverse compounds in the larger, union space, the authors observed that global models tend to perform better than the local ones [Citation72]. This study supports the wide-held view that machine-learning models trained with diverse compounds may work well for PK property prediction, especially when the molecule of interest falls within its application domain, i.e. is covered by the chemical space on which the model was trained.
Corroborating these findings, Kumar et al. reported that graph convolutional network (GNN), which learns patterns from small-molecule structures represented as graphs (i.e. sets of atoms as nodes and bonds as edges), could predict 18 ADME properties proficiently when trained on several thousand compounds. It is noteworthy that the model is used by more than 200 scientists in Janssen [Citation73].
We observe an exponential growth of publications reporting applications of machine learning to PK prediction (Supplementary Figure S1). Even though this review is primarily narrative, to help practitioners gain an overview of the ongoing developments and trends, we performed a comprehensive survey of the relevant literature up to August 2023 (see Supplementary Material for search criteria and details). We screened 323 articles, read 137 key publications, and synthesized our high-level findings below. For readers interested in more details, we include a summary table in the Supplementary Material.
We highlight state-of-the-art development and trends of machine-learning models for ADME and PK predictions below, stratified by the type of models and the properties of interest.
3.3.1. Classification models
Output of ML models can be either a label (e.g. soluble or not) or a number (e.g. the intrinsic solubility in mg/mL). The former task is known as classification, and many models for ADME and PK prediction that we reviewed are classification models. Usually, classification models are preferred when a rough classification is good enough, or when a satisfactory regression model is unavailable for reasons such as discontinuity in the property of interest. Even though results of classification are more qualitative than quantitative, they can be useful for the purpose of selecting or excluding molecules.
Below are a few examples of classification models stratified by the target property of interest:
Permeability: Czub et al. developed a classification model for the discrimination of high-permeable compounds for human intestinal absorption, featuring a SHAP (SHapely Additive exPlanation) analysis that estimates the impact of each input variable to the prediction [Citation74]. Zhang et al. built classification ML models for blood–brain barrier (BBB) permeation for more than 10.000 molecules from traditional Chinese medicine [Citation75].
Active transport: Prachayasittikul et al. compared the performance of different classifiers in the discrimination of p-Glycoprotein (Pgp) inhibitors from non-inhibitors, as well as Pgp substrates from non-substrates [Citation76]. Belekar et al. benchmarked various ML methods for the classification of inhibitors for Breast Cancer Resistant Protein (BCRP), another very relevant transporter in the ADME field [Citation77].
Metabolism: More classification models predicting drug metabolism have been reported than regression models, with neural networks (NN) and random forest (RF) as dominating algorithms [Citation78]. Holmer et al. built a set of models to distinguish substrates from non-substrates of nine human CYP enzymes [Citation79]. Multiple open-source tools have been created, for instance the MetaClass tool for metabolism reaction estimation [Citation80], GutBug, a web-based tool that predicts which bacterial enzymes can metabolize small molecules [Citation81], and PredPS, which predicts plasma stability based on the attention mechanism of graph learning [Citation82]. Software that require licenses for commercial use include XenoSite [Citation83] and Stardrop (Optibrium Ltd., Cambridge, UK) [Citation84].
Bioavailability: Ng et al. built a classifier based on GNN for human oral bioavailability prediction, obtaining an average accuracy of 0.797 [Citation85].
3.3.2. Regression models
Numeric predictions obtained through a regression algorithm can be used to compare and rank molecules. Therefore, such models can be better suited for molecule design and optimization than classification models. Note that a regression model can be turned into a classification model by introducing one or more threshold values.
Common goodness-of-fit metrics to measure the performance of regression models include root-mean-square error (RMSE), average fold error (AFE), and absolute average of fold error (AAFE). A particular variant of the coefficient of determination (R2), which quantifies the discrepancy of predictions and observations against a null model where the average value of observations is used as prediction, is also often used and reported.
The main advantage of regression models over classification models, in our opinion, is not only the quantitative output. Both the magnitude of the predicted value and the direction of change when the molecular structure changes matter, too.
Examples of regression models include:
Permeability: Ràcz et al. developed a model based on neural networks (NN) to estimate PAMPA permeability from molecular descriptors [Citation86]. Bhatia et al. proposed a quantum machine learning (QML) pipeline to predict different ADME-Tox endpoints starting from molecular SMILES representations [Citation87].
Bioavailability: Schneckener et al. exploited NN architectures to predict oral bioavailability in rats [Citation88].
CLp and Vss: Iwata et al. leveraged in vitro data and ML models to predict human CLp and Vss [Citation89]. They observed that missing value imputation for in vitro properties was beneficial.
3.3.3. Multi-task learning
Multi-task models offer the possibility to use the same set of input variables in order to make multiple predictions simultaneously in one model. Research shows that individual tasks may be better learned by taking advantage of the learnings gained from performing other tasks [Citation90]. The output of such models can be numbers, classifications, or a combination of both.
Conceptually, such methods are particularly interesting for ADME/PK predictions, among others due to the physiological interconnections between ADME and PK endpoints shown in . Practically, multi-task learning is a common practice in industrial settings, where high-quality data of many properties are systematically collected for many compounds.
One recent example of multi-task learning is Chemi-Net, a model that predicts ADME properties with a deep graph convolutional neural network [Citation91]. Another example was the study reported by Stoyanova et al., which compared several architectures and methods, including multi-task modeling, for preclinical PK prediction [Citation22].
3.3.4. Other learning techniques: unsupervised, semi-supervised, and federated learning
Most machine-learning models used for ADME and PK property prediction exploit supervised learning approaches, which means that the model is trained with pairs of structures and properties, known as the input and target variables, respectively. If we train a model that only has access to the structures but not the matching properties, the models are known as unsupervised models. In this context, being ‘supervised’ meaning that the model tries to predict a target variable using input features, while being ‘unsupervised’ meaning that the model does not try to predict a target variable, instead it tries to learn patterns from the molecule structures.
Unsupervised learning approaches are therefore not meant to be used to predict ADME or PK parameters directly. Nevertheless, they can be used to cluster inputs, to detect outliers, or to transform the representation of input features in order to make them better suitable for supervised methods. They may also help us better understand the performance and limitations of supervised models. For instance, Lou and Hageman exploited principal component analysis (PCA) to inform the prediction of monoclonal antibody subcutaneous availability [Citation92].
Our literature review did not recover applications of semi-supervised machine learning, which uses a small portion of labeled data and unlabeled data to train a predictive model. It is probable that such methods have been applied and our search has failed in retrieving relevant publications.
Besides individual studies, the federated-learning strategy has been used to build machine-learning models. The basic idea of federated learning is that ML models can be trained across companies that hold raw data (chemical structures and ADME/PK parameters) individually, without having to exchange the data. It has the appeal of ensuring data privacy and increasing the size and heterogeneity of data simultaneously. The MELLODDY project, which applies such a strategy, reported optimized neural networks for the prediction of ADME and other molecular properties [Citation93].
3.3.5. ML models for time-based profiles and other clinical parameters
We can use ML models to predict PK/PD time-based profiles. Some model architectures are particularly suited for this purpose, for instance Recurrent Neural Networks (RNN), and specifically the Long-Short Term Memory (LSTM) architecture, as well as transformer models.
In this context, Tang used four different RNN setups to model irregularly sampled PK/PD data, which successfully extrapolated PD profiles to twice-daily dosing. The author noted, though, that the model performed worse when extrapolating to dose levels outside the training set [Citation104]. With a similar architecture, Khusial et al. developed a model for the prediction of olanzapine drug concentrations reported in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study. This setup performed better than a population pharmacokinetics (PopPK) model [Citation105].
ML methods have also been often used in tandem with other mathematical and computational modeling approaches, such as PBPK and PK-PD modeling. Several studies have reported the value of using output of ML models as input for PBPK models [Citation106,Citation107]. Empirical study showed that PBPK models with predicted values as input performed well for highly lipophilic compounds [Citation108]. A hybrid ML-PBPK model was proposed to predict oral bioavailability from chemical structures, and the performance was evaluated with 184 molecules [Citation109]. Similar hybrid approaches have also shown promising performance in predicting both single parameters and entire pharmacokinetic time-based profiles [Citation110,Citation111].
Much ongoing research aims to address the grand challenge of predicting clinical pharmacological and safety parameters with compound structures and optionally in vitro and in vivo data as input. For instance, Hughes et al. developed a hybrid approach of PK/PD modeling and machine learning to predict the occurrence of neutropenia from a large real-world dataset of 9121 patients [Citation112]. Du et al. created a model to predict the concentration of the drug tacrolimus after liver transplantation, using the genetic and biochemical indexes of 31 liver recipients as input [Citation113]. Damnjanović et al. combined PopPK with machine learning to investigate the PK of valproic acid, lamotrigine, and levetiracetam. The combination allowed them to build a predictive model for epileptic seizures for the pediatric population [Citation114]. Jaber et al. reported a successful case study of using neural-network models as a prescreening tool to assign individualized absorption modes in pharmacokinetic analysis [Citation115]. Last but not least, Lu et al. proposed a neural ordinary differential equation (neural-ODE) method for the prediction of dosing treatment regimens in humans [Citation116].
3.3.6. Model benchmarks
In benchmark studies, different models are applied to the same set of datasets and their performances are compared with each other. Consistent with observations from other fields of computer science, benchmark studies are fundamental for understanding strengths and limitations of existing methods, distilling best practices, and developing new methods.
Selected benchmark studies that we reviewed include:
ADME: McCoubrey et al. compared the performance of 11 algorithms in the classification of gut microbiome drug depletion [Citation75]. Sakiyama et al. benchmarked different algorithms for the discrimination of 1952 molecules based on their metabolic stability [Citation76]. Fang et al. compared the performance of different ML models to predict six different ADME endpoints, among others human and rat liver microsomal stability, of molecules from over 120 internal datasets. They found that models based on neural networks and the gradient boosting decision tree algorithm performed best [Citation94]. In a comparable setting, different models were benchmarked to predict multiple ADME properties including aqueous solubility, lipophilicity, and membrane permeability by Wang et al. The results favored the approach that they developed known as multi embedding-based synthetic network (MESN) [Citation95]. Another benchmark study for ADME-Tox properties prediction was presented by Orosz et al. [Citation96]
PK: Obrezanova et al. compared the performance of several algorithms in predicting in vivo rat clearance and oral bioavailability starting from chemical information and in vitro data collected in the AstraZeneca company [Citation97]. Consistent with results of an independent study [85], they observed good performance of GNN-based models, which the authors built with the open-source Chemprop package [Citation98,Citation99] and the proprietary Alchemite software [Citation100]. Miljković et al. proposed an efficient data curation protocol for the prediction of 12 in vivo PK parameters from the chemical structures of 1001 unique compounds [Citation23]. Human Vss were predicted by a decision tree-based regression model exploiting estimated tissue:plasma partition coefficients, based on a dataset of more than 600 molecules [Citation101]. Direct prediction of Vss from chemical structure with ML methods was also highlighted as the most promising method among the ones benchmarked by Murad et al. [Citation102]. Cauvin et al. benchmarked nine different ML algorithms for the prediction of cisplatin PK parameters in a clinical setting [Citation103].
In most published cases, the authors demonstrate the non-inferiority of ML methods compared to alternative approaches. While we are convinced of the potential of ML methods in general, we would like to remind readers of potential influences by confirmation bias, report bias, data leakage (i.e. possible overlap between training and test data), implementation details, and other factors that influence the reproducibility of scientific publications.
3.3.7. Evolution of ML-based ADME and PK prediction
We wondered how key features of ML models, especially the size of dataset used and the performance, evolve with time. Focusing on three essential PK parameters – CLp, Vss and F% - we visualize the temporal patterns of these features with data extracted from regression-based models in .
Figure 3. Evolution of machine-learning models for PK parameter prediction. The panels illustrate key parameters of ML models, including the year of publication (x-axis) and performance metrics (y-axis), species of predictions (colors), number of compounds used (size of the bubbles), and root-mean-square errors (RMSE) indicated as numbers near the bubbles. In cases RMSE values are not available, NA (not available) is indicated. Panel a, c, and e visualize models reviewed that predict Vss, CLp, and bioavailability, respectively, using AAFE (absolute average fold error), which is also known as Geometric mean Fold error (GMFE), as the metric of performance. Panel b, d, and f show models that predict the same properties as panel a, c, and e, however using coefficients of determination (R2) as the metric. Some studies only report either AAFE or R2 but not the other, therefore the studies included in pairs of panels for the same property (for instance panel a and c for Vss prediction) overlap only partially. Red dashed lines indicate boundary conditions at which an ideal model that makes perfect predictions would perform: an AAFE of 1.0 or an R2 of 1.0.
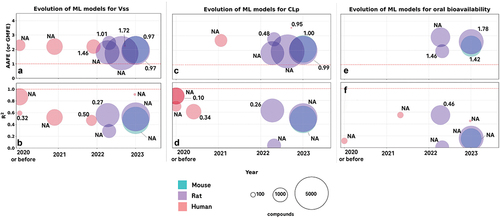
We consider two metrics, Absolute Average Fold Error (AAFE), also known as Geometric Mean Fold Error (GMFE) (formula reported in the Abbreviations section), and the coefficient of determination (R2) as measures of models’ performances. Both large AAFE/GMFE values and small R2 values suggest poor performance of models.
When stratifying models by the target variable of prediction, we observe that models predicting CLp show comparable predictability with those predicting Vss, and both tend to fare better compared to those predicting the oral bioavailability. The conclusion holds both when using AAFE/GMFE (panels A, C, and E of ) and R2 (panels B, D, and F of ) as the metric.
shows that the number of studies reporting ML models for PK prediction has significantly increased in the last 5 years, and recent studies tend to use larger datasets. Intriguingly, we did not observe a steady and substantial improvement in model predictivity. Given potentially huge differences between studies with regard to chemical spaces, methodology, and technical details, and the relatively short-time window that we considered, we believe that more observations and critical analysis are needed to address the question whether newer models offer other advantages (for instance larger applicability domain or better interpretability). High-quality benchmark datasets, a minimal standard to report results, as well as crowd-sourcing approaches such as DREAM challenges [Citation117] may be helpful for future studies to study the evolution of model performance.
Given that models predicting CLp and Vss worked in general better than models predicting oral bioavailability, we think it is likely that the performance of machine-learning models is primarily determined by the target property, while the choice of methodology of ML-models plays a secondary role. Comparing different algorithms trying to predict the same target, we note that support vector regression, random forest, and GNNs tend to show better performance. The observation is consistent with our experience, though we note that empirical studies are at least sometimes required to choose the best methodology for each property. An alternative approach is to use a multi-task model that delivers overall good predictivity.
We report results of further analyses with models predicting other ADME/PK properties in the Supplementary Material. We observe that for AUC prediction, most models report a moderate performance with an R2 value between 0.4 and 0.6, and/or an AAFE value that spans a wider range, reaching values even above 4.0 (Supplementary Figure S2). At the same time, AFE tends to be close to 1, indicating no specific trend of over- or under-prediction. For plasma half life, we found only a few studies, which reported R2 values that spanned a large range (Supplementary Figure S3).
In conclusion, our analysis reveals patterns in recent evolution of ML models for PK prediction with regard to their data size and performance. We observe a trend of using larger datasets over the years, and in comparison with CLp and Vss prediction, room for improvement for ML-based bioavailability prediction.
3.4. Perspectives of development of machine-learning models
ML-based PK prediction may benefit from multiple lines of research. Most apparently, having more paired data of molecules and PK parameters may lead to better models. Based on our experience, access to PK parameters, especially those measured in vivo, may require additional efforts, since they may be incompletely or inconsistently captured in database systems. Programmatic access to such data may allow expansion of the datasets and potential improvement of the ML models.
Besides data availability, the choice of algorithm affects the performance of ML models. More complicated methods do not necessarily lead to better outcomes: For instance, both previous studies and our literature review suggest that kernel-based and tree-based methods often perform better than or as well as neural-network based methods, especially when they are applied to tabular data [Citation118], which is often the case for PK parameter prediction. In this sense, benchmark datasets are valuable to assess new methodologies and algorithms.
Furthermore, we believe that future models may benefit from considering applicability domain, uncertainty quantification, and model interpretability [Citation119]. The applicability domain of a model refers to the range of inputs in which the model is able to produce reliable predictions [Citation120,Citation121]. To build useful predictive models, it is obligatory to assess its applicability domain and to inform users about predictions made outside of the domain so that users can adjust their confidence in the prediction. A rigorous analysis of a model that predicts human clearance, for instance, showed a much poorer performance for unseen, dissimilar compounds compared with the training set than for similar compounds [Citation122]. The authors reckoned that their model yielded a more realistic assessment of how well human clearance can be predicted prospectively.
Applicability domain and uncertainty quantification are inherently connected. The further the distance of the inputs from the applicability domain, the higher the uncertainty associated with predictions. But the applicability domain is not the only source of uncertainty. Many factors such as model design, dissimilarity between molecules in the training and test set (judged by scaffolds or Tanimoto similarity [Citation123], for example), as well as variability in experimental data contribute to uncertainty. Uncertainty estimates for both a predictive model and for individual predictions help users to estimate the confidence in the prediction and to gauge its value for decisions [Citation124].
Model interpretability refers to the provision of justification for predictions [Citation125]. For PK predictions, especially those that inform in vivo or clinical studies, we believe it is helpful to develop interpretable models that help the expert to interpret the prediction. The principle of parsimony further suggests that given similar performance, simpler, interpretable models should be prioritized over more complex, ‘black-box’ models.
Various metrics are available to assess whether a model is confidently giving outputs close to the already labeled data. Some approaches are based on ensemble models, which combine many simple methods to perform a consensus prediction. Examples include random forests and ensemble neural networks, which have been used for uncertainty determination for property prediction of small molecules [Citation126]. Uncertainty quantification methods based on deep Bayesian neural networks and sparse Gaussian Processes are emerging, too [Citation127].
In contrast to complementary methods for PK prediction, classical machine-learning methods are mostly structure-based and ‘physiology-agnostic.’ Consequently, they share three major bottlenecks: (1) limited amount of high-quality data for diverse chemical structures, (2) lack of interpretability, and (3) missing power to explain PK profile changes due to specific physiological conditions. To address the further issues, transfer learning, fine tuning, and federated learning have been tried, although with mixed results [Citation22,Citation128]. To improve the interpretability, we share the view that next-generation models need to favor parsimony, integrate prior knowledge (for instance with Bayesian methods and representation-learning techniques like GNNs [Citation129]), and consider causality [Citation125,Citation130]. One way to achieve this is to combine ML models with the other two archetypes, i.e. IVIVE and PBPK models.
3.5. Archetypal models for PK prediction in a nutshell
The requirements, limitations, and advantages of ML-based and complementary methods for PK parameter predictions are outlined in .
Table 3. Overview of the three approaches for PK predictions.
4. Expert opinion
Predicting the PK parameters of small molecules is a challenging yet rewarding problem. Both theoretical considerations and empirical evidence underpin the value of getting preclinical and clinical PK prediction right. Given the importance of predictive validity in decision-making [Citation131], we foresee that improving PK prediction with interpretable machine-learning models, fit-for-purpose in vitro models, and calibrated PBPK models has the potential to improve the productivity of pharma R&D.
Multiple pharmaceutical companies have reported increased productivity by implementing measures that improve the prediction of preclinical and clinical PK parameters. Besides the examples from Novartis and Janssen cited above, Pfizer enhanced data-driven decision-making principles, which included better PK prediction, and observed a ten-fold increase in their end-to-end clinical success rate in the last decade [Citation132]. AstraZeneca included PK-PD modeling as a key factor in their ‘5 R framework,’ and observed a 15% increase in success rate in the period between 2010 and 2016 [Citation133]. Looking forward, we believe that further developments in machine-learning, IVIVE, and PBPK approaches to PK prediction will unleash further potentials.
Though we discussed the archetypal methods for PK prediction separately, they complement and enhance each other. In vitro data generation can produce larger datasets which provide the basis for training ML models, and dedicated ML models can guide the improvement of in vitro models thanks to automation and robotics [Citation134]. PBPK can consume ML-predicted data as well as in vitro data, leading to high-throughput PBPK (HT-PBPK), which is able to predict PK profiles for many compounds at one time [Citation106]. The three approaches inform and empower each other. A hybrid approach combining different strategies may better leverage knowledge and data than isolated individual approaches.
In practice, data of a new molecule or a new chemical series are often scarce in the early phases of discovery, which limits the power of PBPK models. It is not a coincidence that machine-learning models are becoming more important in the early phases. The presence of historical data points for training, together with the possibility of fast validation cycles, makes ML the optimal choice for the prediction of physicochemical, ADME and PK parameters, perhaps even before a molecule is synthesized.
As a project approaches the entry-into-human phase, PBPK models gain more relevance, among others because more data are accumulated for a chemical series or a molecule. The development of better in vitro models and ML methods offer the opportunity to impute the missing data with prior knowledge and limited observations in order to use PBPK models.
How can we further develop ML models to help with human dose and PK prediction? Currently, ML models can be used to corroborate or challenge other models in order to help practitioners to make informed decisions with impact on clinical studies, for instance dose prediction and adjustment. Despite the great progress in the last years, it still seems imprudent to rely purely on machine learning to make such decisions due to several reasons. First, it is challenging to gather even a moderate amount of high-quality human PK data. Second, both the models and the compounds tested in humans are biased. The new compounds may lie outside of the applicability domain of the models. Even if they do fall in the domain, since compounds tested in humans are mostly filtered to have good overall PK properties, models trained with such molecules may just reinforce the filters that we already know. Last but not least, the regulatory path toward using machine learning alone to guide dose selection and clinical trial design still seems obscure. While future research will undoubtedly address these challenges, we believe it is now practical and sensible to keep further developing machine learning models that enhance and complement IVIVE and PBPK models for human PK and dose prediction.
We consider for now PBPK modeling as a converging point of the three archetypal methods for human PK prediction. It makes explicit, interpretable statements about mechanisms determining the observed PK profiles of drugs and is able to model population variance by adapting the causal parameters of the model to reflect variability. Just like complex in vitro systems have the potential to inform human-relevant parameters used by PBPK models, we believe further development of machine-learning methods that enhance PBPK models, for instance Neural-ODE [Citation135], reinforcement learning [Citation136], and generative models that propose PBPK model structures, may further enhance our ability to model populational and individual PK profiles.
Though the focus of this review is PK estimation in preclinical research and development, we wish to emphasize the importance of reverse translation, namely to use clinical and real-world observations to validate and guide improvement of IVIVE, PBPK, and ML models [Citation137–139]. Prediction errors are valuable, because they suggest potential for learning. Understanding causes of differences between predictions and clinical or real-world observations makes it possible to adapt the models to reality.
In summary, we are convinced that further development of ML, IVIVE, and PBPK approaches, as well as an intelligent integration of methods depending on the question and the project need, have the potential to improve preclinical and clinical PK prediction of small molecules.
Article highlights
Predicting pharmacokinetic (PK) properties of drug candidates in animals and in humans is an essential task for drug discovery and development.
Machine learning is increasingly applied to predict absorption, distribution, metabolism, and excretion (ADME) and PK properties of small molecules.
Machine-learning (ML) models complement established methods including in vitro-in vivo extrapolation (IVIVE) and physiologically based pharmacokinetic (PBPK) modeling, enhancing the ability to design and prioritize molecules with favorable PK properties.
Successful predictions of PK parameters with ML models require high-quality and continuously updated data, a reliable infrastructure, mechanisms to assess model’s performance regularly and to retrain the model when necessary, feedback and retrospective analysis comparing predictions and observations, as well as research and education on how to integrate them into drug discovery workflows.
ML-based PK prediction warrants further research, in particular enriching data, improving models’ interpretability, reducing bias, and exploring synergies with other models, especially in clinical settings.
The integration of ML models with IVIVE and PBPK approaches can provide a more comprehensive understanding of drug’s behavior, potentially improving the efficiency of drug discovery and development.
List of abbreviations
| AAFE | = | Absolute average fold error, a value indicating the closeness of the model prediction to the real value. The value is a positive number equal or greater than 1. The higher the value, the further the predictions to the real values (therefore the poorer the performance). The definition is given below (ABS=absolute). |
| AFE | = | Average fold error, a measure of the average over/under estimation of a predicted property by a model. Its value is positive, values above 1 indicate a tendency to overprediction, while below 1, a tendency to underprediction. The definition is given below. |
| ADME | = | Absorption, distribution, metabolism, and excretion. |
| AUC | = | Area under the (time/concentration) curve. It is defined as the definite integral of the concentration of a drug in plasma as a function of time. |
| CLp | = | Plasma clearance, the amount of plasma which is cleared from the drug in a defined time frame. |
| Cmax | = | Maximum plasma concentration, also written as Cmax. |
| DL | = | Deep learning. A term used to describe machine-learning methodologies that use neural network architectures with multiple layers. |
| DNN | = | Deep neural network. |
| F% | = | Bioavailability. It is expressed as the percentage of the administered compound which reaches the blood systemic circulation. It is calculated as the ratio between the AUC of the administration route of interest and the AUC of the intravenous route, which has F% = 100% by definition. |
| GMFE | = | Geometric mean fold error, synonymous with AAFE. |
| GNN | = | Graph neural network, a neural network architecture for graph-based learning. For instance, 2D or 3D structures of small molecules can be represented as a graph, i.e. a collection of nodes (atoms) and edges (bonds). |
| HT | = | High-throughput. |
| HT-PBPK | = | High-throughput PBPK modeling. |
| IVIVE | = | In vitro-in vivo extrapolation. |
| MESN | = | Multi embedding-based synthetic network. |
| MLP | = | Multilayer perceptron, a classical architecture of an artificial neural network, in which every neuron is fully connected to all the neurons in the previous and next layer. |
| MPS | = | Microphysiological systems. |
| NAM | = | New approach methodologies. |
| NN | = | Neural network, synonymous with artificial neural network in this context. |
| PBPK | = | Physiologically based pharmacokinetic modeling. |
| PCA | = | Principal component analysis. |
| PD | = | Pharmacodynamics. |
| PK | = | Pharmacokinetics. |
| ML | = | Machine learning. |
| ODE | = | Ordinary differential equation. |
| PopPK | = | Population pharmacokinetics. |
| QML | = | Quantum machine learning. |
| RMSE | = | Root mean square error, a measure of goodness of the model fit. It is defined as the root mean square of the residuals, using the following equation (pred = prediction; obs = observation, n = number of predictions/observation evaluated). |
| R2 | = | Coefficient of determination. There are multiple definitions of R2. In our context, it is defined with the equation below (the bar over observations means the average observation), which is a relative measure of discrepancy between predictions and observations, compared with a null model which uses just the average value of the observations as prediction. |
| RF | = | Random forest, a class of machine learning models built with an ensemble of individual decision trees. Each decision tree makes its prediction, and the random forest model makes predictions by pooling individual predictions. |
| SVR | = | Support vector regression, a variant of a machine-learning algorithm known as the support vector machine (SVM). SVM is a supervised learning algorithm for classification tasks. It works by mapping input data into higher dimensions and finding a decision boundary (known as hyperplanes) there to separate classes of input data. The term ‘support vector’ refers to the data point(s) that lie closest to the decision boundary. SVR is derived from SVM and addresses regression tasks. |
| t1/2 | = | Half-life. Time necessary for a substance to reach a plasma concentration equal to half of its initial value. |
| Tmax | = | Time point in which the Cmax is measured in the concentration/time curve of a substance in the plasma, also written as Tmax or tmax. |
| Vss | = | Volume of distribution at steady state. It represents a theoretical volume into which a drug is distributed at steady-state conditions. |
Declaration of Interest
D Bassani was kindly supported by the Roche Postdoc Fellowship (RPF). All authors are employees of Hoffmann-La Roche. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
SupplementaryTable2_Literature_ML_for_ADME_and_PK.xlsx
Download MS Excel (93.9 KB)Davide_PK_review_20240422 - Supplementary Document.docx
Download MS Word (521.8 KB)Acknowledgments
The authors would like to thank Stephen Fowler, Andrea Andrews-Morger, Julia Pletz, and Leonid Komissarov for their valuable comments and feedback. The authors are indebted to the input of many colleagues in the department of Pharmaceutical Science, and the support of Fabian Birzele, Sherri Dudal and Marianne Manchester. The authors also thank Matthew Wright from Genentech, who shared with us valuable experience and helpful suggestions.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17460441.2024.2348157.
Additional information
Funding
References
- Martin L, Hutchens M, Hawkins C. Clinical trial cycle times continue to increase despite industry efforts. Nat Rev Drug Discov. 2017;16(3):157–157. doi: 10.1038/nrd.2017.21
- Simoens S, Huys I. R&D Costs of new medicines: a landscape analysis. Front Med. 2021;8. doi: 10.3389/fmed.2021.760762
- Rennane S, Baker L, Mulcahy A. Estimating the cost of industry investment in drug research and development: a review of methods and results. Inq J Heal Care Organ Provis Financing. 2021;58:00469580211059731. doi: 10.1177/00469580211059731
- Sun D, Gao W, Hu H, et al. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022;12(7):3049–3062. doi: 10.1016/j.apsb.2022.02.002
- Schuhmacher A, Hinder M, von A, et al. Analysis of pharma R&D productivity – a new perspective needed. Drug Discov Today. 2023;28(10):103726. doi: 10.1016/j.drudis.2023.103726
- Fowler S, Kletzl H, Finel M, et al. A UGT2B10 splicing polymorphism common in African populations may greatly increase drug exposure. J Pharmacol Exp Ther. 2015;352(2):358–367. doi: 10.1124/jpet.114.220194
- Smith DA, van Waterschoot RAB, Parrott NJ, et al. Importance of target-mediated drug disposition for small molecules. Drug Discov Today. 2018;23(12):2023–2030. doi: 10.1016/j.drudis.2018.06.010
- Thompson NC, Ge GF, Manso S. The importance of (exponentially more) computing power. Aca Manag Proceed. cited 2023 Jul. 25;2023(1). Available from: https://arxiv.org/abs/2206.14007
- Pavan M, Menin S, Bassani D, et al. Implementing a scoring function based on interaction fingerprint for Autogrow4: protein kinase CK1δ as a case study. Front Mol Biosci. 2022;9. doi: 10.3389/fmolb.2022.909499
- Carbone D, De Franco M, Pecoraro C, et al. Discovery of the 3-amino-1,2,4-triazine-based library as selective PDK1 inhibitors with therapeutic potential in highly aggressive pancreatic ductal adenocarcinoma. Int J Mol Sci. 2023;24(4):3679. doi: 10.3390/ijms24043679
- Bassani D, Moro S. Past, present, and future perspectives on computer-aided drug design methodologies. Molecules. 2023;28(9):3906. doi: 10.3390/molecules28093906
- Sabe VT, Ntombela T, Jhamba LA, et al. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: a review. Eur J Med Chem. 2021;224:113705. doi: 10.1016/j.ejmech.2021.113705
- Leelananda SP, Lindert S. Computational methods in drug discovery. Beilstein J Org Chem. 2016;12:2694–2718. doi: 10.3762/bjoc.12.267
- Sliwoski G, Kothiwale S, Meiler J, et al. Computational methods in drug discovery. Pharmacol Rev. 2014;66(1):334–395. doi: 10.1124/pr.112.007336
- Choudhuri S, Yendluri M, Poddar S, et al. Recent advancements in computational drug design algorithms through machine learning and optimization. Kinases Phosphatases. 2023;1(2):117–140. doi: 10.3390/kinasesphosphatases1020008
- Bassani M, Scarso A, Drago M, et al. Endo-1-phenylborneol as a novel, alternative chiral auxiliary for the aza-diels-alder reaction. Tetrahedron Lett. 2020;61(31):152165. doi: 10.1016/j.tetlet.2020.152165
- Pavan M, Bassani D, Bolcato G, et al. Computational strategies to identify new drug candidates against neuroinflammation. CMC. 2022;29(27):4756–4775. doi: 10.2174/0929867329666220208095122
- Obrezanova O. Artificial intelligence for compound pharmacokinetics prediction. Curr Opin Struc Biol. 2023;79:102546. doi: 10.1016/j.sbi.2023.102546
- Polson AG, Fuji RN. The successes and limitations of preclinical studies in predicting the pharmacodynamics and safety of cell‐surface‐targeted biological agents in patients. Br J Pharmacol. 2012;166(5):1600–1602. doi: 10.1111/j.1476-5381.2012.01916.x
- “Review of the use of two species in regulatory toxicology studies | NC3Rs.” [ Online]. [cited 2023 Aug 21]. Available from: https://www.nc3rs.org.uk/our-portfolio/review-use-two-species-regulatory-toxicology-studies
- Zou P, Yu Y, Zheng N, et al. Applications of human pharmacokinetic prediction in first-in-human dose estimation. Aaps J. 2012;14(2):262–281. doi: 10.1208/s12248-012-9332-y
- Stoyanova R, Katzberger PM, Komissarov L, et al. Computational predictions of nonclinical pharmacokinetics at the drug design stage. J Chem Inf Model. 2023;63(2):442–458. doi: 10.1021/acs.jcim.2c01134
- Miljković F, Martinsson A, Obrezanova O, et al. MachinE learning models for human in vivo pharmacokinetic parameters with In-house validation. Mol Pharmaceut. 2021;18(12):4520–4530. doi: 10.1021/acs.molpharmaceut.1c00718
- Toutain PL, Bousquet‐Mélou A. Plasma clearance. J Vet Pharmacol Ther. 2004;27(6):415–425. doi: 10.1111/j.1365-2885.2004.00605.x
- Yates JWT, Arundel PA. On the volume of distribution at steady state and its relationship with two‐compartmental models. J Pharm Sci. 2008;97(1):111–122. doi: 10.1002/jps.21089
- Lappin G, Rowland M, Garner RC. The use of isotopes in the determination of absolute bioavailability of drugs in humans. Expert Opin Drug Metab Toxicol. 2006;2(3):419–427. doi: 10.1517/17425255.2.3.419
- Toutain PL, Bousquet‐Mélou A. Bioavailability and its assessment. J Vet Pharmacol Ther. 2004;27(6):455–466. doi: 10.1111/j.1365-2885.2004.00604.x
- Watanabe R, Esaki T, Kawashima H, et al. Predicting fraction unbound in human plasma from chemical structure: improved accuracy in the low value ranges. Mol Pharm. 2018;15(11):5302–5311. doi: 10.1021/acs.molpharmaceut.8b00785
- Boxenbaum H. Interspecies variation in liver weight, hepatic blood flow, and antipyrine intrinsic clearance: extrapolation of data to benzodiazepines and phenytoin. J Pharmacokinet Biopharm. 1980;8(2):165–176. doi: 10.1007/bf01065191
- Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet. 2022;23(8):467–491. doi: 10.1038/s41576-022-00466-9
- Zhao Z, Chen X, Dowbaj AM, et al. Organoids. Nat Rev Methods Prim. 2022;2(1):94. doi: 10.1038/s43586-022-00174-y
- Choi G-W, Lee Y-B, Cho H-Y. Interpretation of non-clinical data for prediction of human pharmacokinetic parameters: In vitro-in vivo extrapolation and allometric scaling. Pharmaceutics. 2019;11(4):168. doi: 10.3390/pharmaceutics11040168
- Dedrick RL. Animal scale-up. J Pharmacokinet Biopharm. 1973;1(5):435–461. doi: 10.1007/bf01059667
- Rusyn I, Sakolish C, Kato Y, et al. Microphysiological systems evaluation: experience of TEX-VAL tissue chip testing consortium. Toxicol Sci. 2022;188(2):143–152. doi: 10.1093/toxsci/kfac061
- Paalzow LK. Torsten Teorell, the father of pharmacokinetics. Upsala J Méd Sci. 2010;100(1):41–46. doi: 10.3109/03009739509178895
- Peck CC, Barr WH, Benet LZ, et al. Opportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development. Clin Pharmacol Ther. 1992;51(4):465–473. doi: 10.1038/clpt.1992.47
- Li AP. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 2001;6(7):357–366. doi: 10.1016/s1359-6446(01)01712-3
- Kerns E. Editorial [hot topic: high throughput in vitro ADME/Tox profiling for drug discovery (Guest Editor: Edward H. Kerns)]. Curr Drug Metab. 2008;9(9):845–846. doi: 10.2174/138920008786485074
- Lin W, Chen Y, Unadkat JD, et al. Applications, challenges, and outlook for PBPK modeling and simulation: a regulatory, industrial and academic perspective. Pharm Res. 2022;39(8):1701–1731. doi: 10.1007/s11095-022-03274-2
- Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6(5):430–440. doi: 10.1016/j.apsb.2016.04.004
- Belubbi T, Bassani D, Stillhart C, et al. Physiologically based biopharmaceutics modeling of food effect for Basmisanil: a retrospective case study of the utility for formulation bridging. Pharmaceutics. 2023;15(1):191. doi: 10.3390/pharmaceutics15010191
- Sager JE, Yu J, Ragueneau-Majlessi I, et al. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and Model verification. Drug Metab Dispos. 2015;43(11):1823–1837. doi: 10.1124/dmd.115.065920
- Gaohua L, Abduljalil K, Jamei M, et al. PBPK for pregnancy with time‐varying physiological parameters. Br J Clin Pharmacol. 2012;74(5):873–885. doi: 10.1111/j.1365-2125.2012.04363.x
- Chetty M, Johnson TN, Polak S, et al. Physiologically based pharmacokinetic modelling to guide drug delivery in older people. Adv Drug Deliv Rev. 2018;135:85–96. doi: 10.1016/j.addr.2018.08.013
- Calvier EAM, Krekels EHJ, Johnson TN, et al. Scaling drug clearance from adults to the young children for drugs undergoing hepatic metabolism: a simulation study to search for the simplest scaling method. Aaps J. 2019;21(3):38. doi: 10.1208/s12248-019-0295-0
- Rostami‐Hodjegan A. Reverse translation in PBPK and QSP: going backwards in order to go forward with confidence. Clin Pharmacol Ther. 2018;103(2):224–232. doi: 10.1002/cpt.904
- Rostami‐Hodjegan A. Physiologically based pharmacokinetics joined with in vitro–in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012;92(1):50–61. doi: 10.1038/clpt.2012.65
- Rodríguez-Rodríguez DR, Ramírez-Solís R, Garza-Elizondo MA, et al. Genome editing: a perspective on the application of CRISPR/Cas9 to study human diseases (review). Int J Mol Med. 2019;43(4):1559–1574. doi: 10.3892/ijmm.2019.4112
- Lu J, Liu J, Guo Y, et al. CRISPR-Cas9: a method for establishing rat models of drug metabolism and pharmacokinetics. Acta Pharm Sin B. 2021;11(10):2973–2982. doi: 10.1016/j.apsb.2021.01.007
- Hall B, Limaye A, Kulkarni AB. Overview: generation of gene knockout mice. Curr Protoc Cell Biol. 2009;44(1):.19.12.1–.19.12.17. doi: 10.1002/0471143030.cb1912s44
- Yong KSM, Her Z, Chen Q. Humanized mice as unique tools for human-specific studies. Arch Immunol Ther Exp. 2018;66(4):245–266. doi: 10.1007/s00005-018-0506-x
- Liu J, Lu J, Yao B, et al. Construction of humanized CYP1A2 rats using CRISPR/Cas9 to promote drug metabolism and pharmacokinetic research. Drug Metab Dispos. 2023;52(1):DMD-AR-2023–001500. doi: 10.1124/dmd.123.001500
- Liu J, Shang X, Huang S, et al. Construction and characterization of CRISPR/Cas9 knockout rat model of carboxylesterase 2a gene. Mol Pharmacol. 2021;100(5):MOLPHARM-AR-2021–000357. doi: 10.1124/molpharm.121.000357
- Lu J, Chen A, Ma X, et al. Generation and characterization of cytochrome P450 2J3/10 CRISPR/Cas9 knockout rat Model. Drug Metab Dispos. 2020;48(11):DMD-AR-2020–000114. doi: 10.1124/dmd.120.000114
- Petitet F, Barberan O, Dubus E, et al. Development of an ADME and drug–drug interactions knowledge database for the acceleration of drug discovery and development. Expert Opin Drug Discov. 2006;1(7):737–751. doi: 10.1517/17460441.1.7.737
- Przybylak KR, Madden JC, Covey-Crump E, et al. Characterisation of data resources for in silico modelling: benchmark datasets for ADME properties. Expert Opin Drug Metab Toxicol. 2018;14(2):169–181. doi: 10.1080/17425255.2017.1316449
- Bhhatarai B, Walters WP, Hop CECA, et al. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat Mater. 2019;18(5):418–422. doi: 10.1038/s41563-019-0332-5
- Hansen JU, Quinon P. The importance of expert knowledge in big data and machine learning. Synthese. 2023;201(2):35. doi: 10.1007/s11229-023-04041-5
- Badillo S, Banfai B, Birzele F, et al. An introduction to machine learning. Clin Pharmacol Ther. 2020;107(4):871–885. doi: 10.1002/cpt.1796
- Friedman JH. Recent advances in predictive (machine) learning. J Classif. 2006;23(2):175–197. doi: 10.1007/s00357-006-0012-4
- Atz K, Grisoni F, Schneider G. Geometric deep learning on molecular representations. Nature Mach Intell. 2021;3(12):1023–1032. doi: 10.1038/s42256-021-00418-8
- Zhou J, Cui G, Hu S, et al. Graph neural networks: a review of methods and applications. AI Open. 2020;1:57–81. doi: 10.1016/j.aiopen.2021.01.001
- Morger A, Garcia de Lomana M, Norinder U, et al. Studying and mitigating the effects of data drifts on ML model performance at the example of chemical toxicity data. Sci Rep. 2022;12(1):7244. doi: 10.1038/s41598-022-09309-3
- Yap CW, Li ZR, Chen YZ. Quantitative structure–pharmacokinetic relationships for drug clearance by using statistical learning methods. J Mol Graph Model. 2006;24(5):383–395. doi: 10.1016/j.jmgm.2005.10.004
- Xu C, Mager DE. Quantitative structure–pharmacokinetic relationships. Expert Opin Drug Metab Toxicol. 2011;7(1):63–77. doi: 10.1517/17425255.2011.537257
- Janela T, Takeuchi K, Bajorath J. Predicting potent compounds using a conditional variational autoencoder based upon a new structure–potency fingerprint. Biomolecules. 2023;13(2):393. doi: 10.3390/biom13020393
- Naga D, Muster W, Musvasva E, et al. Off-targetP ML: an open source machine learning framework for off-target panel safety assessment of small molecules. J Cheminformatics. 2022;14(1):27. doi: 10.1186/s13321-022-00603-w
- Prezhdo OV. Advancing physical chemistry with machine learning. J Phys Chem Lett. 2020;11(22):9656–9658. doi: 10.1021/acs.jpclett.0c03130
- Lim MA, Yang S, Mai H, et al. Exploring deep learning of quantum chemical properties for absorption, distribution, metabolism, and excretion predictions. J Chem Inf Model. 2022;62(24):6336–6341. doi: 10.1021/acs.jcim.2c00245
- Bräm DS, Parrott N, Hutchinson L, et al. Introduction of an artificial neural network–based method for concentration‐time predictions. CPT Pharmacom Syst Pharma. 2022;11(6):745–754. doi: 10.1002/psp4.12786
- Pietersma D, Lacroix R, Lefebvre D, et al. Performance analysis for machine-learning experiments using small data sets. Comput Electron Agric. 2003;38(1):1–17. doi: 10.1016/s0168-1699(02)00104-7
- Lascio ED, Gerebtzoff G, Rodríguez-Pérez R. Systematic evaluation of local and global machine learning models for the prediction of ADME properties. Mol Pharm. 2023;20(3):1758–1767. doi: 10.1021/acs.molpharmaceut.2c00962
- Kumar K, Chupakhin V, Vos A, et al. Development and implementation of an enterprise-wide predictive model for early absorption, distribution, metabolism and excretion properties. Futur Med Chem. 2021;13(19):1639–1654. doi: 10.4155/fmc-2021-0138
- Czub N, Szlȩk J, Pacławski A, et al. Artificial intelligence-based quantitative structure–property relationship model for predicting human intestinal absorption of compounds with serotonergic activity. Mol Pharm. 2023;20(5):2545–2555. doi: 10.1021/acs.molpharmaceut.2c01117
- Zhang X, Liu T, Fan X, et al. In silico modeling on ADME properties of natural products: classification models for blood-brain barrier permeability, its application to traditional Chinese medicine and in vitro experimental validation. J Mol Graph Model. 2017;75:347–354. doi: 10.1016/j.jmgm.2017.05.021
- Prachayasittikul V, Worachartcheewan A, Shoombuatong W, et al. Classification of P-glycoprotein-interacting compounds using machine learning methods. Excli J. 2015;14:958–970. doi: 10.17179/excli2015-374
- Belekar V, Lingineni K, Garg P. Classification of breast cancer resistant protein (BCRP) inhibitors and non-inhibitors using machine learning approaches. Comb Chem High Throughput Screen. 2015;18(5):476–485. doi: 10.2174/1386207318666150525094503
- Banerjee P, Dunkel M, Kemmler E, et al. SuperCYPsPred—a web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020;48(W1):W580–W585. doi: 10.1093/nar/gkaa166
- Holmer M, de C, Kops B, et al. Cypstrate: a set of machine learning models for the accurate classification of cytochrome P450 enzyme substrates and non-substrates. Molecules. 2021;26(15):4678. doi: 10.3390/molecules26154678
- Mazzolari A, Scaccabarozzi A, Vistoli G, et al. MetaClass, a comprehensive classification system for predicting the occurrence of metabolic reactions based on the MetaQSAR database. Molecules. 2021;26(19):5857. doi: 10.3390/molecules26195857
- Malwe AS, Srivastava GN, Sharma VK. GutBug: a tool for prediction of human gut bacteria mediated biotransformation of biotic and xenobiotic molecules using machine learning. J Mol Biol. 2023;435(14):168056. doi: 10.1016/j.jmb.2023.168056
- Jang WD, Jang J, Song JS, et al. PredPS: attention-based graph neural network for predicting stability of compounds in human plasma. Comput Struct Biotechnol J. 2023;21:3532–3539. doi: 10.1016/j.csbj.2023.07.008
- Zaretzki J, Matlock M, Swamidass SJ. XenoSite: accurately predicting CYP-Mediated sites of metabolism with neural networks. J Chem Inf Model. 2013;53(12):3373–3383. doi: 10.1021/ci400518g
- StarDrop: small molecule drug discovery & data visualisation software. 2024 Mar 19. Available from: https://optibrium.com/stardrop/
- Ng SSS, Lu Y. Evaluating the use of graph neural networks and transfer learning for oral bioavailability prediction. J Chem Inf Model. 2023;63(16):5035–5044. doi: 10.1021/acs.jcim.3c00554
- Rácz A, Vincze A, Volk B, et al. Extending the limitations in the prediction of PAMPA permeability with machine learning algorithms. Eur J Pharm Sci. 2023;188:106514. doi: 10.1016/j.ejps.2023.106514
- Bhatia AS, Saggi MK, Kais S. Quantum Machine learning predicting ADME-Tox properties in drug discovery. J Chem Inf Model. 2023. doi: 10.1021/acs.jcim.3c01079 63 21 6476–6486
- Schneckener S, Grimbs S, Hey J, et al. Prediction of oral bioavailability in rats: transferring insights from in vitro correlations to (deep) machine learning models using in silico Model outputs and chemical structure parameters. J Chem Inf Model. 2019;59(11):4893–4905. doi: 10.1021/acs.jcim.9b00460
- Iwata H, Matsuo T, Mamada H, et al. Predicting total drug clearance and volumes of distribution using the machine learning-mediated multimodal method through the imputation of various nonclinical data. J Chem Inf Model. 2022;62(17):4057–4065. doi: 10.1021/acs.jcim.2c00318
- Caruana R. Multitask learning. Mach Learn. 1997;28(1):41–75. doi: 10.1023/a:1007379606734
- Liu K, Sun X, Jia L, et al. Chemi-Net: a molecular graph convolutional network for accurate drug property prediction. Int J Mol Sci. 2019;20(14):3389. doi: 10.3390/ijms20143389
- Lou H, Hageman MJ. Machine learning attempts for predicting human subcutaneous bioavailability of monoclonal antibodies. Pharm Res. 2021;38(3):451–460. doi: 10.1007/s11095-021-03022-y
- Heyndrickx W, Mervin L, Morawietz T, et al. MELLODDY: cross-pharma Federated Learning at unprecedented scale unlocks benefits in QSAR without compromising proprietary information. J Chem Inf Model. 2023. doi: 10.1021/acs.jcim.3c00799
- Fang C, Wang Y, Grater R, et al. Prospective Validation of Machine Learning Algorithms for absorption, distribution, metabolism, and excretion prediction: an industrial perspective. J Chem Inf Model. 2023;63(11):3263–3274. doi: 10.1021/acs.jcim.3c00160
- Wang X, Liu M, Zhang L, et al. Optimizing pharmacokinetic property prediction based on integrated datasets and a deep learning approach. J Chem Inf Model. 2020;60(10):4603–4613. doi: 10.1021/acs.jcim.0c00568
- Orosz Á, Héberger K, Rácz A. Comparison of descriptor- and fingerprint sets in machine learning models for ADME-Tox targets. Front Chem. 2022;10:852893. doi: 10.3389/fchem.2022.852893
- Obrezanova O, Martinsson A, Whitehead T, et al. Prediction of in vivo pharmacokinetic parameters and Time–exposure curves in rats using machine learning from the chemical structure. Mol Pharm. 2022;19(5):1488–1504. doi: 10.1021/acs.molpharmaceut.2c00027
- Yang K, Swanson K, Jin W, et al. Analyzing learned molecular representations for property prediction. J Chem Inf Model. 2019;59(8):3370–3388. doi: 10.1021/acs.jcim.9b00237
- Heid E, Greenman KP, Chung Y, et al. Chemprop: a machine learning package for chemical property prediction. J Chem Inf Model. 2024;64(1):9–17. doi: 10.1021/acs.jcim.3c01250
- Irwin BWJ, Levell JR, Whitehead TM, et al. Practical applications of deep learning to impute heterogeneous drug discovery data. J Chem Inf Model. 2020;60(6):2848–2857. doi: 10.1021/acs.jcim.0c00443
- Freitas AA, Limbu K, Ghafourian T. Predicting volume of distribution with decision tree-based regression methods using predicted tissue: plasma partition coefficients. J Cheminformatics. 2015;7(1):6. doi: 10.1186/s13321-015-0054-x
- Murad N, Pasikanti KK, Madej BD, et al. Predicting volume of distribution in humans: performance of in silico methods for a large set of structurally diverse clinical compounds. Drug Metab Dispos. 2020;49(2):DMD-AR-2020–000202. doi: 10.1124/dmd.120.000202
- Cauvin C, Bourguignon L, Carriat L, et al. Machine-learning exploration of exposure-effect relationships of cisplatin in head and neck cancer patients. Pharmaceutics. 2022;14(11):2509. doi: 10.3390/pharmaceutics14112509
- Tang A. Machine learning for pharmacokinetic/pharmacodynamic modeling. J Pharm Sci. 2023;112(5):1460–1475. doi: 10.1016/j.xphs.2023.01.010
- Khusial R, Bies RR, Akil A. Deep learning methods applied to drug concentration prediction of olanzapine. Pharmaceutics. 2023;15(4):1139. doi:10.3390/pharmaceutics15041139
- Naga D, Parrott N, Ecker GF, et al. Evaluation of the success of high-throughput physiologically based pharmacokinetic (HT-PBPK) modeling predictions to inform early drug discovery. Mol Pharm. 2022;19(7):2203–2216. doi: 10.1021/acs.molpharmaceut.2c00040
- Adachi K, Shimizu M, Yamazaki H. Updated in silico prediction methods for fractions absorbed and key input parameters of 355 disparate chemicals for physiologically based pharmacokinetic models for time-dependent plasma concentrations after virtual oral doses in humans. Biol Pharm Bull. 2022;45(12):1812–1817. doi: 10.1248/bpb.b22-00502
- Parrott N, Manevski N, Olivares-Morales A. Can we predict clinical pharmacokinetics of highly lipophilic compounds by Integration of Machine Learning or in vitro data into physiologically based models? A feasibility study based on 12 development compounds. Mol Pharmaceut. 2022;19(11):3858–3868. doi: 10.1021/acs.molpharmaceut.2c00350
- Fagerholm U, Hellberg S, Spjuth O. Advances in predictions of oral bioavailability of Candidate drugs in man with new machine learning methodology. Molecules. 2021;26(9):2572. doi: 10.3390/molecules26092572
- Destere A, Marquet P, Labriffe M, et al. A hybrid algorithm combining population pharmacokinetic and machine learning for isavuconazole exposure prediction. Pharm Res. 2023;40(4):951–959. doi: 10.1007/s11095-023-03507-y
- Destere A, Marquet P, Gandonnière CS, et al. A hybrid Model associating population pharmacokinetics with machine learning: a case study with iohexol clearance estimation. Clin Pharmacokinet. 2022;61(8):1157–1165. doi: 10.1007/s40262-022-01138-x
- Hughes JH, Tong DMH, Burns V, et al. Clinical decision support for chemotherapy‐induced neutropenia using a hybrid pharmacodynamic/machine learning model. CPT: Pharmacomet Syst Pharmacol. 2023;12(11):1764–1776. doi: 10.1002/psp4.13019
- Du Y, Zhang Y, Yang Z, et al. Artificial neural network analysis of determinants of tacrolimus pharmacokinetics in liver transplant recipients. Ann Pharmacother. 2023;58(5):469–479.
- Damnjanović I, Tsyplakova N, Stefanović N, et al. Joint use of population pharmacokinetics and machine learning for optimizing antiepileptic treatment in pediatric population. Ther Adv Drug Saf. 2023;14:20420986231181336. doi: 10.1177/20420986231181337
- Jaber MM, Yaman B, Sarafoglou K, et al. Application of deep neural networks as a prescreening tool to assign individualized absorption models in pharmacokinetic analysis. Pharmaceutics. 2021;13(6):797. doi: 10.3390/pharmaceutics13060797
- Lu J, Deng K, Zhang X, et al. Neural-ODE for pharmacokinetics modeling and its advantage to alternative machine learning models in predicting new dosing regimens. iScience. 2021;24(7):102804. doi: 10.1016/j.isci.2021.102804
- Choobdar S, Ahsen ME, Crawford J, et al. Assessment of network module identification across complex diseases. Nat Methods. 2019;16(9):843–852. doi: 10.1038/s41592-019-0509-5
- Grinsztajn L, Oyallon E, Varoquaux G. Why do tree-based models still outperform deep learning on typical tabular data? [cited Jan 2024 19]. Available from: https://arxiv.org/pdf/2207.08815.pdf
- Rudin C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat Mach Intell. 2019;1(5):206–215. doi: 10.1038/s42256-019-0048-x
- Sheridan RP. The relative importance of domain applicability metrics for estimating prediction errors in QSAR varies with training set diversity. J Chem Inf Model. 2015;55(6):1098–1107. doi: 10.1021/acs.jcim.5b00110
- Morger A, Mathea M, Achenbach JH, et al. KnowTox: pipeline and case study for confident prediction of potential toxic effects of compounds in early phases of development. J Cheminformatics. 2020;12(1):24. doi: 10.1186/s13321-020-00422-x
- Lombardo F, Bentzien J, Berellini G, et al. Prediction of human clearance using in silico models with reduced bias. Mol Pharmaceutics. 2024;21(3):1192–1203. doi: 10.1021/acs.molpharmaceut.3c00812
- Tanimoto K. An elementary mathematical theory of classification and prediction. New York (USA): McGraw-Hill; 1958.
- Hirschfeld L, Swanson K, Yang K, et al. Uncertainty quantification using neural networks for molecular property prediction. J Chem Inf Model. 2020;60(8):3770–3780. doi: 10.1021/acs.jcim.0c00502
- Vasudevan RK, Ziatdinov M, Vlcek L, et al. Off-the-shelf deep learning is not enough, and requires parsimony, Bayesianity, and causality. Npj Comput Mater. 2021;7(1):16. doi: 10.1038/s41524-020-00487-0
- Busk J, Jørgensen PB, Bhowmik A, et al. Calibrated uncertainty for molecular property prediction using ensembles of message passing neural networks. Mach Learn Sci Technol. 2022;3(1):015012. doi: 10.1088/2632-2153/ac3eb3
- Li Y, Rao S, Hassaine A, et al. Deep Bayesian Gaussian processes for uncertainty estimation in electronic health records. Sci Rep. 2021;11(1):20685. doi: 10.1038/s41598-021-00144-6
- Bassani D, Brigo A, Andrews-Morger A. Federated learning in computational toxicology: an industrial perspective on the Effiris Hackathon. Chem Res Toxicol. 2023;36(9):1503–1517. doi: 10.1021/acs.chemrestox.3c00137
- Crouzet SJ, Lieberherr AM, Atz K, et al. G-PLIP: knowledge graph neural network for structure-free protein-ligand bioactivity prediction. bioRxiv. 2023 Sep 1;555977. doi: 10.1101/2023.09.01.555977
- Michoel T, Zhang JD. Causal inference in drug discovery and development. Drug Discov Today. 2023;28(10):103737. doi: 10.1016/j.drudis.2023.103737
- Scannell JW, Bosley J, Hickman JA, et al. Predictive validity in drug discovery: what it is, why it matters and how to improve it. Nat Rev Drug Discov. 2022;21(12):915–931. doi: 10.1038/s41573-022-00552-x
- Fernando K, Menon S, Jansen K, et al. Achieving end-to-end success in the clinic: Pfizer’s learnings on R&D productivity. Drug Discov Today. 2022;27(3):697–704. doi: 10.1016/j.drudis.2021.12.010
- Morgan P, Brown DG, Lennard S, et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov. 2018;17(3):167–181. doi: 10.1038/nrd.2017.244
- Schneider G. Automating drug discovery. Nat Rev Drug Discov. 2018;17(2):97–113. doi: 10.1038/nrd.2017.232
- Lin Z, Chou W-C. Machine learning and artificial intelligence in toxicological sciences. Toxicol Sci. 2022;189(1):7–19. doi: 10.1093/toxsci/kfac075
- Ribba B, Bräm DS, Baverel PG, et al. Model enhanced reinforcement learning to enable precision dosing: a theoretical case study with dosing of propofol. CPT: Pharmacomet Syst Pharmacol. 2022;11(11):1497–1510. doi: 10.1002/psp4.12858
- Edginton AN. Using physiologically based pharmacokinetic modeling for mechanistic insight: cases of reverse translation. Clin Transl Sci. 2018;11(2):109–111. doi: 10.1111/cts.12517
- Shakhnovich V. It’s time to reverse our thinking: the reverse translation research paradigm. Clin Transl Sci. 2018;11(2):98–99. doi: 10.1111/cts.12538
- Kasichayanula S, Venkatakrishnan K. Reverse translation: the art of cyclical learning. Clin Pharmacol Ther. 2018;103(2):152–159. doi: 10.1002/cpt.952