
ABSTRACT
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons. Several animal models have been generated to understand ALS pathogenesis. They have provided valuable insight into disease mechanisms and the development of therapeutic strategies.
Areas covered
In this review, the authors provide a concise overview of simple genetic model organisms, including C. elegans, Drosophila, zebrafish, and mouse genetic models that have been generated to study ALS. They emphasize the benefits of each model and their application in translational research for discovering new chemicals, gene therapy approaches, and antibody-based strategies for treating ALS.
Expert opinion
Significant progress is being made in identifying new therapeutic targets for ALS. This progress is being enabled by promising animal models of the disease using increasingly effective genetic and pharmacological strategies. There are still challenges to be overcome in order to achieve improved success rates for translating drugs from animal models to clinics for treating ALS. Several promising future directions include the establishment of novel preclinical protocol standards, as well as the combination of animal models with human induced pluripotent stem cells (iPSCs).
1. Introduction
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disorder affecting approximately 4–6 persons per 100 000 worldwide, with only 20% of patients living for five years or more after diagnosis [Citation1,Citation2]. ALS is characterized by a progressive degeneration of motor neurons in the spinal cord and the brain [Citation3]. These clinical manifestations lead to progressive muscle denervation, muscle weakness, muscle atrophy, paralysis and ultimately death. Considerable diversity exists within the disease, with heterogeneity of initial presentation, spreading of disease, progression rates, and survival [Citation4]. Indeed, in addition to motor symptoms, ALS patients can also experience non-motor symptoms including cognitive and/or behavioral impairment, including degeneration in the brain extending to interneurons in frontotemporal lobar degeneration (FTLD) [Citation5,Citation6]. The initial manifestation of ALS varies; some patients present symptoms originating in the spinal region, such as muscular weakness of the limbs (limb onset), while others present symptoms originating in the bulbar region (bulbar onset), characterized by difficulties in speaking and swallowing, known as dysarthria and dysphagia.
The majority of ALS cases (approximately 90%) are sporadic (sALS), and the remaining 10% of cases are familial (fALS). Mutations in more than 40 of them have been associated with ALS and the most common mutated genes are superoxide dismutase-1 (SOD1), chromosome 9 open reading frame 72 (C9ORF72), fused in sarcoma (FUS), and TAR DNA-binding protein (TARDBP) (see reviews [Citation7,Citation8]). Several animals have been used to model the major genetic cases of ALS, and they have been instrumental in our mechanistic understanding of ALS and for drug discovery. There are several pathologic mechanisms such as excitotoxicity, oxidative stress, protein misfolding and aggregation, axonal transport dysfunction and degeneration, neuroinflammation, mitochondrial dysfunction, altered RNA processing, endoplasmic reticulum stress and dysregulated nucleocytoplasmic transport that have been associated with ALS onset and progression. Interestingly, at the cellular level, the cytoplasmic mislocalization and aggregation of TAR DNA-binding protein 43 (TDP-43) has been reported in 98% of all ALS cases. Abnormal TDP-43 aggregation results in proteasomal dysfunction, stress granule formation, cryptic splicing, mitochondrial dysfunction and altered RNA metabolism including cryptic splicing-polyadenylation.
Out of over 80 human clinical trials, only riluzole [Citation9], edaravone [Citation10] and more recently Sodium phenylbutyrate and taurursodiol combination [Citation11] modestly slowed disease progression. Moreover, oxidative stress and excitotoxicity are the only two nonspecific pathways targeted by these FDA-approved drugs. Since 2023, the FDA-approved Tofersen (also known as BIIB067, QALSODY), an antisense oligonucleotide (ASO) drug, is indicated for the treatment of ALS in adults who have the mutation in SOD1 and proved to reduce the amount of SOD1 protein [Citation12,Citation13]. The success of Tofersen suggests that this therapeutic strategy holds great promise, particularly for correcting other ALS-related mutations ([Citation14] for review). Given the rarity of each genetic variant and the unknown causes of most ALS cases, there is still an urgent need to identify disease-causing pathways to develop better treatment targets for this complex motor neuron (MN) disorder.
Translation of findings from animal models to therapy have largely been focused on the discovery of small molecules targeting pathogenic pathways in ALS, such as excitotoxicity, oxidative stress, mitochondrial function and neuroinflammation. However, over the past decade, novel techniques, such as ASOs, have been developed to target disease-causing mutations and/or restore gene expression (e.g. STMN2). In this review, we will focus on the animal models created over the years by the scientific community to study ALS and for drug discovery. We will highlight how the so-called simple models, and more complex models, have been a springboard toward the identification of several drugs with therapeutic potential for this complex disease.
2. Genetics of ALS
Most ALS cases (90%) are of sporadic origin (sALS), appearing spontaneously with no family history, while the remaining 10% are familial (fALS) involving causative genetic mutations. The study of these familial forms has revealed numerous mutations, mainly transmitted in an autosomal dominant mode, and affecting more than 50 genes, involved in the triggering of ALS [Citation2]. In this review, we will focus on the four causative gene responsible for the majority of fALS cases: SOD1, TARDBP, FUS and C9ORF72. Apart from those genes that have been clearly implicated in ALS, there are other potentially causative and susceptibility genes that we will not discuss further (see [Citation15] for review).
The GGGGCC hexanucleotide repeat expansion (HRE) mutation in the 5’ non-coding region of C9ORF72 (encoding chromosome 9 open reading frame 72) is responsible for most ALS cases, as it has been identified in ~ 40% and ~ 5% of familial and sporadic cases respectively [Citation16,Citation17]. This HRE comprises 2–20 tandem copies in healthy conditions but is expanded to hundreds or thousands copies in C9ORF72 patients. Repeat expansion was first associated with reduced C9ORF72 gene expression [Citation18], suggesting a haploinsufficiency mechanism. Two other disease mechanisms of toxic gain-of-function were then proposed [Citation19,Citation20], associated with 1) toxicity from aggregation-prone dipeptide-repeat proteins (DPRs) translated from the GGGGCC repeat region in by a start codon-independent RAN translation manner and 2) neurotoxic RNA foci nucleated by the GGGGCC repeat. The function of C9ORF72 protein is still unknown, but a role in autophagy and vesicular trafficking has been proposed based on sequence homology with the DENN family of regulators of endocytosis [Citation21,Citation22].
A toxic gain-of-function mutation in the Superoxide dismutase 1 (SOD1) gene was found in ~ 20% of fALS and ~ 1–7% of sALS [Citation23]. Most of SOD1 mutations are missense mutations, but deletions and insertions have also been reported. The main physiological function of SOD1 is to protect cells against oxidative damage [Citation24,Citation25]. To date, it is still unclear how mutations in SOD1 lead to ALS. It has been proposed that SOD1 mutations can result in either a detrimental loss of its enzymatic activity or gain of toxic function due to protein misfolding and aggregation. Mechanistically, excitotoxicity, mitochondrial dysfunction, axonopathy, apoptosis, microglial activation and endosomal trafficking have been reported in ALS SOD1 models [Citation26–29].
Mutations in TAR-DNA Binding Protein (TARDBP) are responsible for ~ 4–6% of fALS and ~ 1.5% of sALS cases [Citation30]. This gene encodes for TDP-43, a highly conserved and ubiquitously expressed DNA/RNA-binding protein [Citation31]. TDP-43 is involved in many steps of RNA processing, including transcription, splicing, transport, translation and stress granules formation [Citation32]. Over 50 different dominant heterozygous mutations have been found, mainly affecting the C-terminal domain containing the nuclear localization signal sequence. Mutations in TARDBP result in ubiquitin-positive inclusions of TDP-43 protein, predominantly observed in the cytoplasm of neurons and glial cells, and correlate with nuclear depletion of TDP-43 [Citation33,Citation34]. To date, it remains unclear whether loss or gain of function, or a combination of both, is responsible for the pathophysiology of TDP-43.
Finally, mutations in the Fused in Sarcoma (FUS) gene account for around 5% of fALS and < 1% of sALS [Citation35,Citation36]. Like TDP-43, FUS is an DNA/RNA binding protein. Mutations in FUS, mostly located in the exon 15 which encodes for the nuclear localization signal at the C-terminal region of the protein, are known to cause its redistribution in the cytoplasm and accumulation of protein inclusions [Citation37–40]. ALS-associated FUS mutations have been reported to cause defective DNA damage repair and RNA splicing [Citation41,Citation42].
The identification of these mutations has led to the development of animal models of ALS, with the aim of understanding the physiological and molecular mechanisms underlying the disease.
3. Simple model organisms of ALS
Simple animal models such as worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), and zebrafish (Danio rerio) offer several advantages. They have relatively simple genomes compared to mammals, making them highly amenable to genetic manipulation. Their short generation times, small size and cost-effectiveness facilitates their maintenance. Simple model organisms are particularly suited for high-throughput screening of chemical libraries, as embryos and larvae can be accommodated in 96-well microplates, and drugs can be easily administered at appropriate developmental stages. Overall, simple animal models offer valuable tools for studying basic biological processes, modeling human diseases, and identifying potential therapeutic.
3.1. C. elegans ALS models
Caenorhabditis elegans (C. elegans) is a non-parasitic nematode that is approximately one millimeter long in the adult, with an average lifespan of 2–3 weeks. The nervous system of the worm consists of 302 neurons in the hermaphrodite, and neuronal connectivity has been well mapped and described [Citation43–45]. Indeed, neurons have been found to communicate via approximately 6400 chemical synapses, 900 gap junctions, and 1500 neuromuscular junctions [Citation46] and use most of the known neurotransmitters in the mammalian nervous system. Full genomic sequence comparisons have shown that approximately 35% of the C. elegans genome has an orthologous equivalent in humans [Citation47]. C. elegans is a valuable genetic model because it is amenable to a wide range of genetic manipulations including forward genetic screens, reverse genetic RNAi screens, rapid mutation mapping, and transgenesis. C. elegans assays are fast, low cost, suitable for high-throughput analysis, and genetically tractable with a fully sequenced genome, making the worm an excellent model to study ALS and other neurological diseases [Citation48] ().
Figure 1. Overview of simple genetic models (zebrafish, C. elegans and Drosophila) and rodent models that recapitulate ALS pathology. Abbreviations: LOF, loss of function; GOF, gain of function.
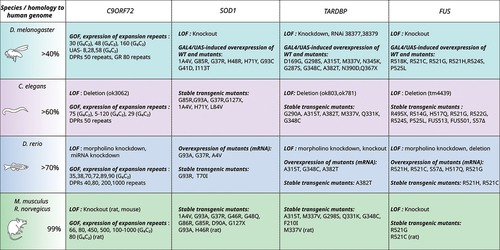
Human SOD1 and C. elegans sod-1 are highly homologous, sharing 71% protein similarity [Citation49]. The toxic gain-of-function have first been observed in transgenic C. elegans by introducing human wild-type and ALS-related mutant forms. Overexpression of disease-associated SOD1 mutations (A4V, G73R and G93A) under the control of a muscle promoter (unc-54) in C. elegans resulted in low toxicity, including mild cellular dysfunction and protein aggregates with distinct morphological characteristics [Citation50]. Another overexpression C. elegans model with pan-neuronal expression of SOD1G85R (from the snb-1 promoter) display insoluble SOD1 aggregates in neurons which are associated with locomotor defects, reduced axonal length and impaired neuronal transmission [Citation51,Citation52]. More recently, overexpression of human SOD1G93A (from the unc-25 promoter) in the worm’s motor neurons also leads to SOD1 aggregates, axon guidance failure, and motor defects [Citation53,Citation54]. Later, another SOD1G93A model was described as associated with the same motor deficits, in addition to a reduced lifespan [Citation55].
The only model of sod-1 deletion in C. elegans reports no effect on survival, only on fertility and resistance to oxidative stress [Citation56]. Recently, single-copy ALS SOD1 knock-in models in C. elegans have been used as a different genetic approach. Besides modeling the main characteristics of ALS, these models enabled discrimination between the effect of gain and loss of SOD1 function on cholinergic and glutamatergic degeneration [Citation49].
Human TDP-43 and C. elegans tdp-1 share 57% protein similarity. C. elegans with pan-neuronal expression of TARDP mutants (G290A, A315T, Q331K, M337V) exhibit neurotoxic features including uncoordinated locomotor phenotypes and abnormal motor neuron synapses including defasciculation of GABAergic motor neurons [Citation57,Citation58]. These phenotypes were particularly correlated with the identification of calcineurin-dependent phosphorylated protein accumulation [Citation59]. Interestingly, loss-of-function mutations in the C. elegans TDP-43 homolog tdp-1 (tdp-1(ok803) or tdp-1(ok781)) do not induce motor and neurodegenerative phenotypes, but rather increase sensitivity to DNA damage and oxidative stress [Citation60–63]. The expression of the mutant TDP-43A315T in GABAergic motor neurons results in age-dependent motility defects, cytoplasmic insoluble aggregates, neurodegeneration and increased endoplasmic reticulum (ER) stress. These findings recapitulate important features of ALS pathogenesis [Citation64–66].
C. elegans models have also been used to study ALS-associated mutations. fust-1 shares 38% identity and 56% similarity with human FUS. Pan-neuronal overexpression of human FUS mutations (S57Δ, R514G, R521G, R522G, R524S and P525L) in C. elegans results in cytosolic FUS aggregates, locomotor impairment, synaptic dysfunction, reduced lifespan [Citation65,Citation67]. FUS C-terminal pathological form (FUS501) overexpression in worm also leads to impaired synaptic ultrastructure of GABAergic motor neurons and neuromuscular junction (NMJ) vesicles, reduced postsynaptic currents [Citation68]. This elegant study done in C. elegans implicates a role of FUS in the organization of synaptic vesicles and synaptic transmission at the NMJ. The first single-copy transgenic human FUS nematode (S57∆) results in age-dependent paralysis and hypersensitivity to acetylcholinesterase inhibitor, treatment suggesting dysfunction in the GABAergic system [Citation69]. A more recent study reported the creation of a novel knock-in model in worm (R524S and P525L mutations). These mutations are associated with stress-induced locomotor defects, impaired neuronal and muscle autophagy and neuromuscular dysfunction [Citation70]. A deletion model fust-1 (tm4439) has also been described. It is characterized by motor deficits and GABAergic motor neuron degeneration [Citation71].
Finally, alfa-1, the conserved homolog of C9ORF72 in C. elegans, allowed the research community to model all three pathways caused by the repeat expansion in the gene. Deletion mutations in alfa-1 result in motility defects leading to an age-dependent paralysis, impaired nuclear transport, increased stress sensitivity, degeneration of motor neurons [Citation72] and dysregulation in endolysosomal homeostasis [Citation73]. From a translational perspective, this result is important because it confirms the functional similarity of alfa-1 to its human counterpart C9ORF72 [Citation74] and thus the model’s relevance. In addition to modeling C9ORF72 loss-of-function, the introduction of C9ORF72 hexanucleotide repeats or DPRs also cause neurotoxicity and motor impairments. For instance, the transgenic C. elegans expressing 75 G4C2 repeats developed a shortened lifespan, locomotor defects and distinct dipeptide repeat protein aggregates [Citation75]. Muscle expression of G4C2 repeats (from 5 to 120) under the myo-3 promoter [Citation76] in worms leads to the formation of RNA foci. Similarly, pan-muscular expression of certain DPRs accelerates paralysis [Citation77].
Overall, the C. elegans model offers a wide variety of genetic tools that can be adapted to the genetic diversity of ALS pathology. However, it has several limitations, especially in phenotype analysis related to ALS pathophysiology. The short life cycle of C. elegans allows for large-scale, high-throughput in vivo drug screening studies – as discussed in the next section. However, it does not permit to reproduce the progression of neurodegenerative symptoms that characterize ALS disease, in particular the degeneration of motor neurons leading to progressive motor phenotypes, and the slow onset of abnormal protein conformations and inclusions. Neurotransmission is simplified, and the worms lack a conserved neuroinflammatory system. The model cannot be used to study neuroinflammation, which is a major contributor to ALS disease.
3.2. Drosophila ALS models
The fruit fly Drosophila was introduced as an animal model in the early 20th century and has since become one of the most widely used models in biology ever since [Citation78]. Its small size, ease of breeding, short life cycle (10 days from fertilized embryo to adult fly), and the power of available genetic tools [Citation79] make it an ideal model for studies in biology. Complete genome sequence comparisons have shown that around 75% of the Drosophila genome has an orthologous equivalent in humans [Citation80–82]. Its nervous system contains approximately 100,000 neurons [Citation83], whose synaptic transmission uses mechanisms similar to those found in humans [Citation84]. Among the genetic tools most commonly used in Drosophila, the most important are: chemical and insertional mutagenesis, the inducible UAS/GAL4 system [Citation85], the temporal and regional gene expression targeting (TARGET) method, allowing precise control of Gal4 system [Citation86]. A genomic-comprehensive transgenic RNA interference (RNAi) library has been successfully used to identify genetic modifiers associated with human disease [Citation87]. More recently, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and Clustered Regularly Interspaced Short Palindromic Repeats with associated Cas 9 protein (CRISPR/Cas9) have been successfully employed in Drosophila. Several fly models have provided new insights into the cellular and molecular pathways underlying ALS progression (see [Citation88,Citation89] for review) ().
The first Drosophila models of ALS were created shortly after the identification of SOD1 as the main causative gene. The first study reported shortened lifespans and infertility in Drosophila SOD1 (dSod1)-null animals [Citation90]. This phenotype is notably rescued by overexpression of human SOD1 in adult motor neurons [Citation91]. Later, flies expressing the human SOD1 mutation (A4V or G85R) [Citation92] exhibit motor impairment, synaptic dysfunction and accumulation of human SOD1 protein in motor neurons, as well as increased heat shock proteins (HSP) in glial cells. Interestingly, expression of the human SOD1G85R in specific cell types, either motor neurons or glia, shortened Drosophila longevity when exposed to ALS-linked environmental insults [Citation93,Citation94]. Interestingly, the expression of SOD1G93A in Drosophila thoracic muscles causes ALS phenotype comprising motor behavior defects, decreased lifespan, and causes mitochondrial pathology [Citation95].
The knock-in Drosophila model for SOD1 (G37R, H48R, H71Y, and G85R) results in a strong lethality [Citation96]. Escaper adult flies show severe locomotion defect, muscle atrophy and denervation [Citation96]. At the NMJ level, dSod1G85R adult mutants are characterized by BMP ligand-dependent reduction in bouton number, mEPSP frequency, and muscle capacitance and locomotion defect [Citation97].
Deletion mutations in the fly TARDBP ortholog TBPH results in larval lethality and reduced lifespan in adults escapers, as well as locomotion impairment [Citation98–102]. At the level of the NMJs, some groups observed a reduction in the number of axonal branching and synaptic boutons [Citation98,Citation103], or an increase in synaptic boutons [Citation100], and impaired synaptic transmission [Citation101]. Overexpression of both TBPH and TDP-43 in Drosophila results in toxic phenotypes with a reduced lifespan and age-progressive locomotor defects and NMJ degeneration [Citation99,Citation101,Citation104–110].
Similarly to TDP-43, expression of wild-type and mutant human FUS (R524S or P525L) in eyes results in progressive retinal degeneration, associated with axonal loss [Citation111–113]. Gain-of-function mutants have been shown to be either lethal when FUS is ubiquitously expressed [Citation114,Citation115], or associated with reduced lifespan when pan-neuronal [Citation112,Citation113]. At the NMJ synaptic level, few phenotypes have been described in flies : a reduction in the number of synaptic boutons, associated with functional deficits, increased apoptosis in motor neurons, synaptic transmission impairment and disturbed locomotion [Citation111,Citation114,Citation116,Citation117]. Several studies have hypothesized a role for FUS in degeneration, notably through reduced mitochondrial transport [Citation118,Citation119]. Characterization of cabeza (caz) mutant, the fly homolog of FUS, reveals a key function for the gene in neuronal development, which is not responsible for maintaining adult neuronal function [Citation120–122].
The study of the C9orf72 gene in Drosophila model has focused on gain-of-function models, due to the absence of an ortholog. The toxicity of RNA foci nucleated by the GGGGCC repeat has first been modeled in Drosophila. These studies showed that the expression of expanded repeat RNA is sufficient to cause neurodegeneration in the eye and motor neurons [Citation123]. For instance, the expression of a (G4C2)48 RNA repeat in Drosophila recapitulates ALS-associated phenotypes such as locomotion defect and branching defects [Citation124]. Interestingly, a Drosophila model expressing 160 G4C2 repeats flanked by human intronic and exonic sequences formed nuclear RNA foci in glia and neurons [Citation125].
Since then, the Drosophila model has been used extensively to assess the pathogenicity of C9ORF72-associated ALS (see [Citation126] for review). C9ORF72 DPRs toxicity has also been studied in Drosophila. Flies expressing arginine-rich dipeptide-repeats [Citation127], specifically in glutamatergic neurons [Citation128], shows toxicity-related phenotypes and severe neurodegeneration, whose underlying mechanisms have been investigated in several studies [Citation129–134]. Pan-neuronal expression of DPRs leads to neurodegeneration and cell death in the Drosophila central brain, and causes age-related motor impairment and neurodegeneration [Citation135]. At the synaptic level, the expression of arginine-rich dipeptide-repeats leads to a decrease in the number and function of neuromuscular synapses, associated with reduced muscle size, and locomotion defects [Citation136,Citation137]. Even more recently, the specific expression of polyGR in Drosophila muscle [Citation138] resulted in morphological and functional mitochondrial defect.
Drosophila has played a key role in the modeling of ALS. However, as with C. elegans, the progressive onset of different motor phenotypes or pathophysiological markers of the disease cannot be accurately described due to the relatively short life cycle. In the absence of a C9ORF72 ortholog, current fly models are limited to the analysis of HRE-induced toxicity gain. The limited selection of motor and behavioral tests is another limitation. As a result, eye degeneration has been and is still commonly utilized as a phenotypic marker in the model. We want to stress the importance of thoroughly confirming the specificity of this phenotype in genetic analysis to avoid misleadingly suggestive induced toxicity.
3.3. Zebrafish ALS models
Zebrafish have become increasingly popular as a simple vertebrate model organism for studying vertebrate biology, physiology and human disease due to several advantages it offers [Citation139–142]. Zebrafish shares approximately 70% of human genes, with a high degree of identity conservation [Citation143,Citation144]. Zebrafish embryos develop ex utero, so rapidly that at 3 days post fertilization (dpf), embryogenesis is complete, reaching adulthood at 3 months post fertilization (mpf). The transparency of zebrafish embryos facilitates imaging and makes observation of organ development, cell migration, and ALS-relevant pathology possible. Zebrafish stereotypes movement patterns enable behavioral tests to examine changes in motor activity. In particular, it offers many advantages for the study of neuromuscular diseases [Citation141], with the increasing development of new methods for pharmacological screening and quantitative analysis of the neuromuscular junction [Citation145,Citation146].
Finally, one of the main advantages of the zebrafish model is the ease of genetic modification, fostered by the sequencing of the entire zebrafish genome in 2013. The genetic loss-of-function includes the expression of dominant negative constructs by (i) antisense morpholino oligonucleotides (AMO) transient knockdown of gene expression [Citation147] or (ii) knockout using several genome editing methods such as ZFNs, TALENs and CRISPR-Cas9 (see [Citation140,Citation148] for review). The genetic gain-of-function includes (i) injecting blastocysts with mRNA for global expression or (ii) by driving expression in small sets of neurons from DNA constructs using neural-specific or inducible genomic regulatory elements (e.g. the UAS/Gal4 system) [Citation149]or (iii) by CRISPR/Cas9 knock-in of point mutations [Citation150–152] and more recently by using CRISPR-based cytosine base editors [Citation153]. Several ALS-causing genes have been studied in zebrafish using loss- or gain-of-function approaches ().
Transgenic sod1 zebrafish larvae expressing mutants (G93R, G93A) exhibit abnormal NMJ and mitochondrial dysfunction, while adults show a progressive locomotor decline, altered muscle respond and motor neuron loss [Citation154,Citation155]. Co-injection of three SOD1 mutants (G93A,G37R,A4V) induces a motor axonopathy specific for motor neurons in zebrafish larvae [Citation156]. Another group reported comparable results, including increased sensitivity to oxidative stress, in a transgenic zebrafish line expressing the missense T70I sod1 mutation [Citation157]. At the functional level, a study conducted on the transgenic SOD1G93R zebrafish model has allowed elucidating the role of interneuron dysfunction in the early stages of ALS [Citation158]. In addition to an NMJ loss, the group described a reduced glycinergic transmission (spontaneous miniature glycinergic excitatory postsynaptic currents (mEPSCs)) onto motor neurons of sod1 zebrafish larvae.In zebrafish embryos, overexpression of either wild-type or mutant (A315T, G348C, A382T) human TARDBP with mRNA injection led to motor axonopathy and locomotor defects [Citation159,Citation160]. At NMJ level, expression of TARDBPG348C mRNA resulted in impaired neuromuscular synaptic transmission, reduced frequency of mEPCs, reduced quantal transmission and orphaned presynaptic and postsynaptic structures. Interestingly, similar phenotypes were observed upon zebrafish tardbp knockdown with an antisense morpholino oligonucleotide, suggesting that both loss- and toxic gain-of-function might be involved in the molecular mechanism of pathogenesis [Citation160]. As for C. elegans model, motor impairments in zebrafish expressing mutant TDP-43 (G348C) were partially ameliorated by compounds that mitigate endoplasmic reticulum stress, such as methylene blue and salubrinal [Citation66]. Zebrafish knockout models were then created for the two orthologous TARDBP genes (tardbp and tardbpl) [Citation161,Citation162]. Interestingly, reduced survival, motor and axonal defects described in double knockouts can be compensated with a novel tardbpl splice form (Tardbpl-FL). To date, only one CRISPR/Cas9-based knock-in of FUS- and TARDBP-related missense mutation has been created in zebrafish [Citation151].
The overexpression of human FUS mutants (R495X or G515X) leads to disrupted transportin-mediated nuclear import and accumulation in cytosolic stress granules [Citation37,Citation163]. Cytosolic mislocalization and accumulation in stress granules has been also described in cultured zebrafish cells from larvae expressing mutant FUS (R521C) [Citation164].
Transient knockdown of zebrafish fus with an AMO were shown to have impairments in locomotor activity with abnormal motor axon projections [Citation165]. Functionally, this model shows increased motoneuron excitability, reduced fidelity of synaptic transmission and quantal content and abnormal NMJ structure [Citation166]. However, the first CRISPR-Cas9-based fus knockout zebrafish lack motoneuronal and locomotor defects [Citation167]. Another mutagenesis-generated fus knock-out model, summarizes ALS-associated phenotypes such as behavioral deficits, NMJ defects including reduced motor neurons axonal length, and altered tau transcription [Citation168]. Overall, despite these conflicting results, mechanisms involving loss of function or haploinsufficiency are likely relevant to FUS- associated pathology in zebrafish model.
Different zebrafish models that have been developed to study the pathological mechanisms of ALS related to the C9orf72 gene (see [Citation169] for review). Expression of G4C2 repeats in c9orf72 with DNA constructs injections results in toxic RNA foci formation and apoptotic cell death [Citation170]. Sense and antisense RNA repeats induce motor axonopathy in zebrafish larvae [Citation171]. This C9orf72 zebrafish model permitted to decipher a new potential pathogenic mechanism by demonstrating that RNA toxicity and RNA cytoplasmic formation can contribute to the pathogenesis of C9orf72-associated ALS/FTD.
The role of DPR protein toxicity in C9ORF72 ALS has also been demonstrated in the zebrafish using transgenic UAS responder lines forcing the translation of poly-GA or poly-GR protein [Citation172,Citation173]. The sustained expression of G4C2-linked DPR proteins increases mortality, impairs locomotion, leads to motor axonopathy, and promotes DPR protein aggregation in zebrafish larvae. Similarly, a stable transgenic zebrafish line expressing C9ORF72-related hexanucleotide repeats recapitulate the motor behavioral deficits, muscle atrophy, motor neuron loss and early mortality [Citation174].
Models of C9ORF72 loss of function or haploinsufficiency have also been established in zebrafish. The transient knockdown with AMO injection [Citation175] or overexpression of a nonfunctional mutant [Citation176] results in motor axonopathy, increased apoptotic death and abnormalities of spontaneous and evoked swimming. Our group recently developed a new loss-of-function zebrafish line using the stable expression of a specific miRNA targeting the 3’-UTR region of C9orf72 gene [Citation177]. C9-miR fish replicates the hallmarks of ALS as it causes locomotor deficits, motor neuron axonopathy and degeneration, muscle atrophy and TDP-43 mislocalization. At NMJ level, this model showed reduced mEPCs frequency and impaired release of quantal synaptic vesicles. Lastly, a new loss of function zebrafish line was generated by targeting exon 2 using CRISPR/Cas9 [Citation178]. The model lacks a neurogenerative phenotype in the spinal cord but is characterized by neuronal loss in the retina.
Among the three simple animal models selected for this review, the zebrafish stands out as the sole vertebrate and therefore the most complex organism. However, it is important to acknowledge that there are some limitations associated with this model. First, the zebrafish lineage experienced a complete duplication of the genome, making it challenging to model gene silencing due to the presence of multiple orthologs for some human genes. It is necessary to consider the genetic interactions between duplicated genes, as has been shown for the tardbp genes [Citation161,Citation162].
4. Rodent models of ALS
Prior to the development of simple models, mouse and rat models have been important tools to better understand the early and late physiological mechanisms of ALS-associated and to evaluate potential therapeutic strategies ([Citation179] for review) ().
The first ALS mouse model was created shortly after the discovery of the first SOD1 causative gene [Citation23]. The dominant gain-of-function mutation SOD1-G93A causes motor neuron disease in transgenic mice [Citation24], which has since been widely used, notably to identify ALS-related cellular changes [Citation180–184]. Symptoms include loss of spinal motor neuron, paralysis, and early death. The different models created express a varying number of copies of the transgene, and the time of onset of symptoms is variable [Citation185–187]. Other mouse models were then developed with human SOD1 mutation overexpression: G85R [Citation188], G37R [Citation189], G86R [Citation190], D90A [Citation191], H46R [Citation192] and D83G [Citation193]. These mice develop progressive degeneration of lower and upper motor neurons, distal axonopathy, intraneuronal and glial inclusions and muscle wasting and atrophy, sometimes leading to paralysis.
Early descriptions of the first SOD1 KO mouse indicated an absence of motor phenotype. Further studies showed this to be a model of chronic oxidative stress [Citation194]. SOD1 null mouse models are much rarer, but those described later also show progressive and oxidative stress-mediated distal motor axonopathy [Citation195], reduction in muscle mass and oxidative damage in skeletal muscle leading to reduced motor performance [Citation196]. Importantly, rat models overexpressing human SOD1 mutant (hSOD1G93A) also display pathological features such as motor neuron axonal loss, muscle wasting and paralysis of both hindlimbs and one forelimb [Citation197,Citation198].
Many murine models of TDP-43 mutations have been generated. The first models were created by ubiquitous overexpression, under the control of the Prnp promoter, of wild-type and mutant (A315T, M337V, Q331K) human cDNA [Citation199–202]. This method has proven to be very severe, with early onset of the motor phenotype, aggregates of ubiquitinylated proteins and premature death, but no or very few TDP-43 inclusions. Conversely, expression of TDP-43 mutants (A315T and G298S) under the control of an endogenous TDP-43 promoter [Citation203] results in ubiquitinylated TDP-43 inclusions, but no paralysis-inducing motor phenotype or reduced life span. The same BAC transgenic approach was used in rats, which rapidly developed paralysis and died after a month [Citation204]. To limit the extremely severe effect, probably due to peripheral toxic effects of the ubiquitous expression, other models of neuronal TDP-43 overexpression driven by the Thy1.2 promoter have been established [Citation205–207]. The results show a toxic effect of wild-type human protein in phenotypic mice, which is exacerbated by mutant overexpression. Different groups have used an inducible overexpression model at different developmental stages, using the CaMK2 promoter [Citation208–210]. The mice are characterized by moderate motor neuron loss, progressive motor and cognitive impairment, and slightly earlier death. Finally, conditional expression of the TDP-43 mutant in the brain and spinal cord, through the NEFH (neurofilament heavy chain) promoter, results in an ALS-specific neurodegenerative phenotype with loss of motor neurons, denervation in the NMJ and TDP-43 mislocalization in mice [Citation211] and motor neuron loss, loss of hindlimb grip strength and paralysis in rats [Citation212]. Although rarer, TDP-43 knockout models also exist in murine models. Because complete loss of function is lethal, two groups created a model knocking out TDP-43 specifically in the postmitotic motor neurons [Citation213,Citation214]. This model results in modest loss of motor neurons, astrogliosis and accumulation of ubiquitinated material in motor neurons. The knock-in method has also been used for the Tardbp gene, in particular for the M323K mutation, which causes motor neuron death [Citation215], the Q331K mutation which causes cognitive deficits [Citation216], the M337V and G298S mutations, which specifically affect neuromuscular junctions without overt neurodegeneration [Citation217], and the N390D mutation which covers several important phenotypes including TDP-43 aggregation and motor neuron degeneration [Citation218].
TDP-43 plays a crucial role in mRNA splicing of many transcripts and mislocalization of TDP-43 in ALS leads to aberrant splicing of transcripts, foremost Stathmin2 (STMN2). Cryptic exon inclusion in STMN2 mRNAs results in a decreased STMN2 protein level in many patients with ALS. Interestingly, Stmn2± mice display NMJ denervation and motor deficits [Citation219]. Recently, Baughn and colleagues [Citation220] engineered mice to carry a Stmn2 gene partially humanized by insertion of the human STMN2 cryptic splice and polyadenylation sequences but without TDP-43 binding. Using this mouse model, they showed the efficacy of ASOs to correct Stmn2 pre-mRNA misprocessing and restored its protein levels. However, the effects of these ASOs have yet to be tested on functional/behavioral outcomes in animal models. Nevertheless, restoration of STMN2 expression using an ASO (QRL-201) is currently a strategy being tested in a Phase 1 clinical trial (NCT05633459).
As in the case of TDP-43, many FUS mouse models have been designed [Citation221]. Interestingly, only a gain-of-function of the mutated protein (conditional knock-in mutation) mimics ALS phenotypes, with progressive motor neuron degeneration, neuromuscular junction defects, and FUS mislocalization without aggregation [Citation222–225]. Mice with ubiquitous overexpression of human wild-type FUS [Citation226], truncated variant FUS 1–359 under control of the prion Thy1 promotor [Citation227], and FUS R521C mutation [Citation41] develop such a severe motor and neuroinflammatory phenotypes that premature death generally occurs between 3 and 4 months of age. Overexpression of the FUS R521C mutation also leads to ALS-like phenotypes in rats with motor neuron degeneration, denervation atrophy of skeletal muscle and progressive paralysis [Citation228]. Loss of FUS function, conversely, leads to a pathological and behavioral phenotype that is not characteristic of ALS [Citation229].
Most C9ORF72 knockdown [Citation230] and knockout [Citation231–237] in mice mainly develop an inflammatory phenotype, sometimes associated with a reduced lifespan, but no locomotor deficits or motor neuron degeneration. A more recent study has shown that knockdown of C9ORF72 in mice, using a miR-RNAi sequence targeting a region located in exon 8 of the gene, causes mild strength loss and deficits in the neuromuscular junction and impaired social interaction, but still no motor neuron disease [Citation238]. Interestingly, several studies in mice [Citation239,Citation240] and rats [Citation241] support the hypothesis of a synergistic effect of c9orf72 loss- and gain-of-function mechanisms in promoting ALS pathology. In this sense [Citation239], have shown that loss of C9orf72 and haploinsufficiency exacerbate motor behavior deficits in a dose-dependent manner. Viral G4C2 repeat expansion mouse models recapitulate hallmark features including motor and cognitive deficits, TDP-43 inclusions, cortical neuron loss [Citation242,Citation243] and NMJ abnormalities [Citation244]. DPR formation has been described in an inducible gain-of-function model that exhibits locomotor phenotype and muscular dystrophy [Citation245].
Here, we provide a brief overview of the large number of mouse models that have been developed for ALS. This heterogeneity in the phenotypes described, and their onset complicates the selection of the most appropriate model for subsequent translational studies. For instance, the use of the SOD1 model predominates in mouse preclinical studies. It has led to significant progress in describing some of the cellular mechanisms leading to selective MN degeneration. However, these animals do not reproduce an essential marker of pathology, namely TDP-43 inclusions, and show a rapid progression of the disease. These characteristics question their relevance in a preclinical study. Moreover, the development of new knock-in models including humanized mouse, in which the mouse locus is replaced by the human orthologous sequence, is increasingly being used to study ALS. This strategy can generate clinically relevant animal models. However, a humanized mouse gene can have undesirable effect on splicing and phenotypic data must be analyzed with caution.
5. Translational value of ALS animal models for therapy
5.1. Small molecules
In addition to provide a better comprehension of pathogenic mechanisms, the simple ALS models have been used to screen therapeutic compounds [Citation246] (). An in vivo chemical screening platform for ALS genes, conducted in worms and fish, revealed that methylene blue efficiently attenuated the TDP-43 and FUS phenotypes, notably motor impairments, by promoting a protective ER stress response [Citation64,Citation66]. The efficacy of this compound has since been demonstrated in a mouse model of ALS [Citation247]. Interestingly, methylene blue has been shown to be an effective inhibitor of TDP-43 aggregation in a human cell model. In addition, Vaccaro and colleagues showed that compounds structurally related to methylene blue, such as salubrinal, guanabenz and phenazine, also exhibited potent suppression of mutant TDP-43 protein proteotoxicity [Citation66].
Figure 2. Translational value of ALS animal models for therapy. Summary of small compounds and other therapeutic strategies that have been identified in various animal models of ALS and are currently in clinical trials, with the corresponding phase of development.
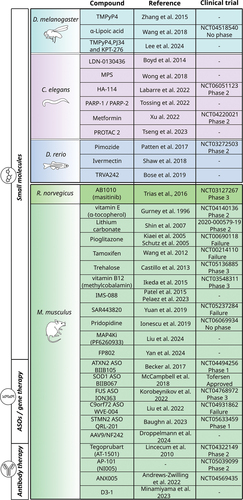
A drug screening in both C. elegans and zebrafish models (TARDBPA315T and TARDBPG348C, respectively) identified a potential therapeutic compound, pimozide, which, after validation in SOD1G37R mice, is currently in clinical trial (NCT03272503) [Citation248]. The pimozide, a derivative-TRVA242 T-type calcium channel antagonist, was shown to increase synaptic transmission at the NMJ, reduce motor defect and rescue pathological phenotypes at the NMJ [Citation248]. Later, another high-throughput screen reported a novel molecule, TRVA242 as effective in restoring motor phenotypes in worms and zebrafish [Citation249]. Additionally, the compound TRVA242 was found to ameliorate morphological and synaptic abnormalities of NMJ in zebrafish and mouse ALS models [Citation249].
The ease of pharmacological phenotype analyses in the TARDBPA315T worm model allowed the selection of the compound α-methyl-α-phenylsuccinimide (MPS) as an effective neuroprotector to ameliorate locomotor defects and reduce GABAergic motor neurons loss [Citation250]. The same worm model TARDBPA315T [Citation65], together with the model FUSS57Δ [Citation69] also allowed the repositioning of the probiotic Lacticaseibacillus rhamnosus HA-114 as a neuroprotector restoring motor defects by promoting mitochondrial β-oxidation [Citation251]. The same group proposes an interesting regenerative approach to rescue axonal abnormalities using the poly (ADP-ribose) (PAR) polymerases (PARP) PARP-1 and PARP-2 in the mFUSS57∆ worm model [Citation252]. A screening strategy using the SOD1G93A C. elegans model identified novel genetic modifiers that prevent SOD1 protein aggregation and suppress toxicity [Citation253]. The SOD1G93A worm model was pharmacologically studied with the diabetes drug metformin. It proved effective in reducing neurodegeneration and extending life expectancy by upregulating autophagy [Citation55]. Finally, the C. elegans model has also been used to validate hits from cell-based screens, such as the compounds LDN-0130436 [Citation254] and PROTAC2 [Citation255], which decreases GABAergic motor neuron degeneration and decrease TDP-43 aggregation, respectively.
A behavior-based drug screening system indiscriminately identified 12 compounds in another C. elegans TDP-43 toxicity model not described in the review, dnc-1/dynactin 1 knockdown [Citation256]. This study provides strong support for the use of simple organisms, as among the compounds that improved the motor defects and axonopathy were two FDA-approved drugs for ALS : riluzole and nifedipine [Citation257].
Finally, McGown and colleagues created a neuronal stress reporter line, lying on the increased expression of the fluorescent heat shock stress protein hsp70 following neuronal stress (hsp70-DsRed) [Citation158]. This model was used as an additional in vivo model to validate the efficacy of the FDA-approved ALS drug riluzole in stabilizing sodium channels and reducing excitotoxicity, hence providing new insight into a novel mechanism of this treatment [Citation258]. After reporting a new zebrafish model expressing C9orf72 HRE that recapitulates ALS phenotypes, the same group described for the first time and in several other C9orf72 expansion models an abnormal activation of the heat shock response (HSR) that correlates with disease progression. Importantly, ivermectin, like riluzole, reduced HSR activation in both C9orf72 and SOD1 zebrafish models, offering new therapeutic perspectives [Citation174].
The Drosophila ALS model was also subjected to pharmacological screening, leading to the identification of the PPARγ agonist pioglitazone as a neuroprotective compound improving locomotor function but not the lifespan of TDP-43-expressing flies [Citation259]. This new compound was then tested in a phase II clinical trial [Citation260]. The results showed that pioglitazone did not produce any significant improvements in terms of slowing the progression of the disease. More recently, α-Lipoic acid was shown to attenuate oxidative stress, neurotoxicity and improve motor activity in the hSOD1G85R fly model [Citation261]. In a recent study using LDS-(G4C2)44 and (G4C2)30 transgene constructs in flies, the authors evaluated the ability of three compounds (TMPyP4, PJ34 and KPT-276) to reduce G4C2 toxicity which is manifested by reduced lifespan, eye degeneration and motor defects [Citation262]. TMPyP4 and PJ34 are the most potent compounds and ameliorate the disease phenotype by extending lifespan and improving climbing ability. It has previously been reported that TMPyP4 attenuates G4C2 repeat-dependent neurotoxicity in Drosophila by suppressing hexanucleotide repeat-mediated nuclear import deficits by modifying the structure of the repetitive RNA. These results represent a major breakthrough in the therapeutic potential of this compound [Citation263].
Mouse model remains one of the most widely used ALS animal models, especially for the preclinical studies. There have been as many preclinical studies on mouse models as there are potential pathophysiological mechanisms for this complex pathology ([Citation264] for review). Here, we review the most significant translational studies resulting from work done on mouse models of ALS ().
Shortly after its creation, the mSOD1 model was the subject of a study aimed at reducing oxidative stress using vitamin E (α‐tocopherol) [Citation265], which failed to show clinical efficacy [Citation266]. The same conclusion has been reached for other compounds with antioxidant and anti-inflammatory properties [Citation267,Citation268]. More recently, vitamin B12 (methylcobalamin) has been reevaluated for its antiglutamatergic and neuroprotective properties in mice [Citation269]. Its clinical study is still underway and shows promising results in early stage ALS ([Citation270]. The motor performance of SOD1-G93A mouse model was evaluated to assess the effect of the main FDA-approved drugs : riluzole [Citation271] and edavarone [Citation272], together with the rat SOD1- H46R model [Citation273]. In the light of riluzole, other compounds aimed at modulating the glutamatergic pathway have been tested in the clinic without much success. A very recent study, partially conducted in the SOD1G93A mouse model, used a recent pharmacological principle that, in contrast to classical NMDA receptor (NMDAR) pharmacology, allows selective elimination of extra-synaptic NMDAR-mediated sensing without disrupting synaptic function [Citation274]. The compound, named FP802, which thus selectively inhibits the TwinF interface, is a very promising new therapeutic avenue as it improves motor performance, extends lifespan, and prevents motor neuron loss in SOD1G93A mouse model.
Some autophagic enhancers are currently being developed in the clinic, after being validated in mouse models of ALS. We can notably cite the example of Tamoxifen, which has been shown to reduce TDP43 protein aggregation in motor neurons of the FTLD-U mouse model [Citation275].
A very important part of current translational research is focused on the inflammatory component of the pathology [Citation276]. Much of this development involves mouse models of ALS, particularly to test the efficacy of anti-inflammatory treatments such as pioglitazone [Citation277–279]. For example, Pelaez and colleagues show that inhibiting the NF-κB pathway with IMS-088, derived from withaferin A (ImStar Therapeutics Inc.) improves cognitive and motor function, increases motor neuron dendritic branching and restores synapses in a newly generated mouse model hFUSR521G/Syn1 [Citation280]. Another anti-inflammatory compound, MN-166 (ibudilast), targets several phosphodiesterases and macrophage migration inhibitory factor and is currently in clinical trials (NCT02238626 and a biomarker study NCT02714036). An in vitro study has shown that an ibudilast treatment could clear TDP-43 and SOD1 aggregates and prevent TDP-43-induced neurotoxicity in cells [Citation281]. Its efficacy has been demonstrated, particularly in the SOD1G93A and FUSR521C mouse models, in prolonging survival, reducing spinal glial neuroinflammation and MN degeneration [Citation282].
In a recent study, a high-throughput pharmacological screen using human induced motor neurons (hiMNs) identified a compound (F6260933) that inhibits MAP4 kinases (MAP4ks), previously implicated in neurodegeneration in ALS [Citation283], and is able to improve the survival of ALS hiMNs and their ability to form NMJs [Citation284]. Importantly, a neuroprotective effect was observed in vivo in SOD1G93A mice after MAPK4 inhibition, with prolonged survival, an increase in the number of motor neurons compared to untreated control mice, and a significant decrease in nuclear mislocalization of TDP-43 protein [Citation284]. A clinical study is currently underway for a similar compound, a MAP4k inhibitor named Prosetin and Phase 1 has been completed (NCT05279755). There are many other examples of therapeutics identified in the ALS rodent models that are currently in clinical trials. Among these, the tyrosine kinase inhibitor masitinib (AB1010), now in clinical study (NCT03127267), has emerged as a promising compound in the SOD1G93A rat model, preventing microglia-associated inflammation, particularly the number of aberrant glial cells, in the degenerating spinal cord, extending lifespan and improving motor neuron pathology [Citation285]. Similarly, the compound SAR443820, an inhibitor of receptor-interacting serine/threonine protein kinase 1 (RIPK1), which is involved in neuroinflammation [Citation286], was found to be beneficial in both SOD1G93A mice and Optn−/− mice (Yuan et al., 2019), preventing oligodendrocyte loss, glial inflammation and axonal degeneration of motor neurons [Citation287]. A phase 2 clinical trial is currently ongoing (NCT05237284).
Pridopidine, a sigma-1 receptor (S1R) agonist, in addition to promoting autophagy, would be able to compensate for TFEB nucleoplasmic transport deficiency in C9orf72-associated pathology [Citation288]. It reduces SOD1 aggregation and ameliorates muscle fiber atrophy in the SOD1G93A mouse model [Citation289], making it a pharmacological compound of choice, now in clinical study (NCT06069934).
5.2. ASOs and other gene therapy approaches
Mouse and rat models have played a decisive role in preclinical efforts to develop new therapeutic strategies based on gene therapy, particularly ASO knockdown of SOD1 [Citation290,Citation291] and C9orf72 [Citation292,Citation293]. Among these studies, preclinical data obtained in SOD1G93A mouse and rat models provide a strong rationale for SOD1 reduction as a therapeutic approach. Indeed, novel SOD1 ASOs reduced mRNA and protein, extended survival and preserved neuromuscular innervation in treated animals [Citation291]. Following a clinical trial [Citation12], the antisense oligonucleotide tofersen was approved by the FDA in 2023 for the treatment of ALS patients with SOD1 mutations. Encouraged by this success, ASO ION-363 is currently being evaluated in a phase III trial (NCT04768972), for instance for FUS mutations, based on its efficacy in the mouse model [Citation294].
TDP-43 plays a key role in RNA metabolism and other cellular functions. Targeting TDP-43 toxicity as a therapeutic approach is increasingly being explored, including in translational studies using the mouse model. A very recent study shows that genetic expression of a fragment of RGNEF (NF242) or injection of AAV9/NF242 into a TDP-43 overexpressing fruit fly ALS model and a TDP-43 mouse model (rNLS8), respectively, improved lifespan and motor phenotype and reduced neuroinflammation markers. These are promising results for a protein that directly interacts with TDP-43 and acts as a modifier of TDP-43 toxicity in vivo [Citation295].
Additionally, depletion of ataxin-2 in animal models, via genetic approaches, reduces TDP-43 toxicity. For instance, inhibition of ataxin-2 in a mouse TARDBP model using ASOs improves motor function and prolongs survival [Citation296]. Recently, it was reported that targeting ataxin-2 using the CRISPR-Cas13 system in a mouse model of TDP-43 proteinopathy significantly improved motor deficits and neuronal survival, increased survival and reduced abnormal TDP-43 pathology [Citation297]. Consequently, ataxin-2 has received much attention as a potential therapeutic target for treating ALS. Currently, BIIB105, an ASO targeting ATAXIN-2 gene to lower its protein levels, is in a phase I clinical trial for ALS (NCT04494256).
Viral vectors derived from adeno-associated virus (AAV) has emerged as the lead vector for CNS gene therapy delivery tool. However, to address the limitations of mRNA- and RNA-based drugs in practice, including those associated with safety, efficacy, and bioavailability, various non-viral vectors are being investigated for drug delivery in ALS therapies [Citation298]. This research focuses on improving the pharmacokinetic profiles of individual drugs, using physical or chemical systems such as nanoparticles, to overcome the clinical failure of some ALS therapies. For instance, Chen and colleagues developed an innovative way of delivering ASO encapsulated in calcium phosphate (CaP) lipid nanoparticles [Citation299]. This alternative approach improves the delivery of the gene construct compared with the free form of ASO and reduces SOD1 levels in HEK293 cells. In vivo, the lipid encapsulation improves bioavailability in the zebrafish bloodstream, for better diffusion into the brain and spinal cord after direct injection. These encapsulation approaches could offer a considerable improvement in gene therapy for ALS in the future.
5.3. Antibody-based therapy approached
The use of antibodies targeting misfolded proteins is a promising approach for the treatment of ALS. Among the large number of antibody-based interventions tested for fALS (see [Citation300] for review), a few stand out. For instance, Minamiyama and colleagues designed a new monoclonal antibody targeting misfolded SOD1 (D3–1) [Citation301] which delayed disease onset and extended lifespan in treated-SOD1H46R rats. Others stand out for their anti-protein aggregation mechanism. One example is the AP-101 antibody (NI005) directed against SOD1 protein, currently in Phase IIa clinical trials (NCT05039099). It has been found to attenuate motor symptoms and increase overall survival in mouse models (Neurimmune).
Tegoprubart (AT-1501) is an antibody that targets the CD40 ligand (CD40L), previously shown to be upregulated in the blood of ALS patients. Inhibition of CD40L delayed paralysis and prolonged survival in the SOD1G93A mouse model [Citation302]. Tegoprubart is now in Phase II clinical trial (NCT04322149). Finally, a Phase IIa study is underway evaluating ANX005 (NCT04569435), a recombinant anti-C1q antibody capable of preserving NMJs in SOD1G93A ALS mice [Citation303].
6. Conclusion
As ALS research continues to evolve, the model organisms presented here are valuable assets as versatile genetic models to study the pathogenesis of ALS and to identify new disease-modifying drugs to treat this devastating neurodegenerative disease. There is currently no sufficiently effective treatment to slow the progression of the disease or to compensate for the pathophysiological defects that have been identified and are yet to be discovered.
7. Expert opinion
Over the past decade, there has been a marked increase in interest in using simple organisms (C. elegans, Drosophila and zebrafish) to model ALS. Due to the key advantages of the simple models (), such as their relatively short lifespan, low cost, ease of genetic manipulation and assay throughput, they have provided important insights into the understanding of ALS pathogenesis. Simple models stood out, offering the possibility of developing new tools for translational research. Their ease of use, small size and, in some cases, transparency have led to the development of new techniques for in vivo imaging, automated low to high-throughput screening of small molecules and genetic screening for human mutations. These developments have been instrumental in therapeutic discovery with the identification of lead compounds such as pimozide, α-lipoic acid and HA-114, which are currently being tested in ALS clinics.
Figure 3. Advantages and limitations of simple models (zebrafish, C. elegans and Drosophila) and rodent models for the translational research of new therapeutics for ALS.
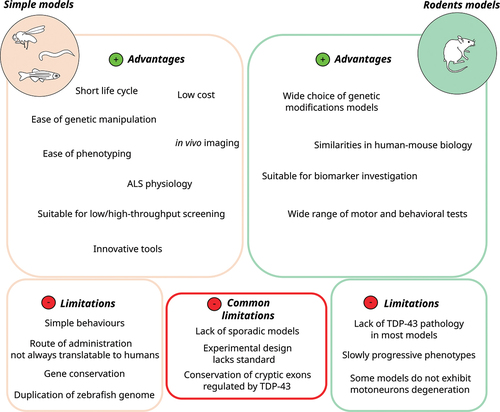
Admittedly, the motor behavior and organization of the neuromuscular system are simplified in these animals. However, this is a real advantage in quickly and easily identifying new therapeutic targets before confirmation in larger animal models or cellular models. Translational studies in simple models have so far focused on the identification of small molecules, particularly as part of a drug repositioning strategy. However, we expect to see emerging proposals for new gene therapies. In zebrafish, ASOs can be administered to the embryo by microinjection or immersion in embryonic water or directly to the brain in adult fish. Moreover, CRISPR-based and cytosine base editing [Citation153], is a powerful method for efficient and rapid insertion of ALS-associated mutations. Compared with traditional genome-editing methods, it can faithfully reproduce the exact point mutations found in ALS patients and could improve model reliability without double-strand DNA cleavage or a donor DNA template.
Likewise, several mouse models have been developed to study the different genetic forms of ALS, and they have provided key insights into pathological pathways implicated in ALS. They have also allowed the development of therapeutic strategies for ALS. The strength of the mouse model lies in its ability to provide multiple therapeutic strategies, ranging from small molecules to antibodies, gene therapy approaches and ASOs. Unlike simple models, the rodent model offers a wide range of behavioral and cognitive tests that allow for a more detailed assessment of the safety and efficacy of a therapeutic intervention.
However, preclinical studies using mouse models face a number of difficulties due to the cost of raising animals and the difficulty of obtaining animal models reflecting critical ALS pathophysiology [Citation304]. As a result, most of the compounds identified in these models have unfortunately failed in the clinic [Citation305]. Some translability issues seem inherent to the model. For example, we can question the robustness of the SOD1G93A mouse model or other commonly used preclinical mouse models that do not exhibit TDP-43 pathology.
But also, the design of preclinical experimental therapy, which now lacks sufficient protocol uniformity, requires improvement. It is crucial to clarify if the experimental drug or therapy was given during the pre-symptomatic or symptomatic stage. The development of symptoms in mouse and rat models varies significantly, with a delayed and progressive pattern. This variability must be carefully taken into consideration for all in vivo models. This allows for the distinction between prospective disease-modifying therapies and those that will have a preventative impact.
Some therapeutic compounds have failed in clinical trials likely due to their inability to cross the blood-brain barrier (BBB), as well as their poor pharmacokinetic profiles (including absorption, bioavailability and metabolism). Research efforts are aimed at overcoming these challenges, notably by developing new strategies for delivery of therapeutics using nanotechnologies [Citation298]. For example, the encapsulation of a therapeutic cocktail composed of Leptin and Pioglitazone (already successfully tested in mice [Citation277–279]) in a mesoporous silica nanoparticles (MSNs) has been explored to optimize the cocktail’s in vivo delivery [Citation306]. Moreover, recently, the formulation of intranasal edaravone-loaded poly(lactic-co-glycolic acid)-poly(ethylene glycol) PLGA-PEG polymeric nanoparticles improved the biodistribution of the preexisting pharmacological compound [Citation307]. In addition, chronic treatment of SOD1G93A mice with the retinoid adapalene encapsulated in similar PLA-PEG nanoparticles has a neuroprotective effect, improving motor performance and prolonging lifespan [Citation308].
Liposomes have the potential to cross the BBB due to their lipophilic properties, and guarantee good bioavailability in the CNS. They are an excellent ally for neurodegenerative diseases. One study showed that they improved the therapeutic potential of steroid compounds methylprednisolone. When intravenously injected in SOD1G93A mice, the anti-inflammatory compound encapsulated in PEGylated liposomes (2B3–201) significantly improves histopathological outcomes in brainstem motor nuclei, resulting in reduced gliosis and reduced neuronal loss [Citation309]. Similarly, the anti-inflammatory agent minocycline, when contained in modified lipopolysaccharide liposomes, has better bioavailability and can ameliorate microglia-dependent neuroinflammation [Citation310]. Thus, the development of new delivery and administration options offers a promising prospect to address the inefficiency of some chemicals in the clinic. Importantly, animal ALS models are essential for testing novel drug delivery approaches in preclinical studies.
Furthermore, it is important to highlight the significant contribution of animal models in the identification of biomarkers for ALS [Citation179]. The exploration of novel biomarkers is widely encouraged. This might enhance our comprehension of the heterogeneity of the disease. Effective biomarkers can also function as surrogate endpoints in clinical trials, offering early indicators of the efficacy of a treatment.
A common constraint in the ALS field is that research is focused on developing animal models of the familial form of the disease, omitting most cases which are sporadic. Developing non-genetic animal models of ALS is one of the major current challenges in the field. Our current knowledge of environmental and genetic ALS risk factors is limited [Citation311]. The disease exhibits significant genetic heterogeneity, with over 150 genes that have been identified as either linked or associated with ALS [Citation8]. This diversity is also reflected in the diversity of pathophysiological mechanisms, for which we have yet to identify the main cellular pathway responsible for the progression, severity and onset of the disease. In this sense, much of the fundamental biology of ALS is still not understood. Consequently, many pathophysiological pathways remain unknown, and we are probably overlooking many therapeutic targets. It is therefore important to focus efforts on a convergent pathway, which remains one of the biggest challenges to be overcome by the community toward developing an effective treatment.
As with many of the examples discussed earlier in this review, the strategy of combining in vivo and in vitro models has been very successful in moving promising compounds along the translational pathway. The modeling of ALS motor neuron phenotypes using human induced pluripotent stem cells (iPSCs) is rapidly expanding [Citation312], and has the advantage of covering both major familial and sporadic forms [Citation313] of the disease. The iPSC motor neuron model, for example, is proving very effective in combination with simple organisms for identifying new compounds and new genetic targets in high-throughput screening studies. We believe that the combination of these two models could be a real asset in overcoming the challenges that remain for the translatability of therapeutic interventions for ALS.
Article highlights
Simple models C. elegans, Drosophila and zebrafish are powerful genetic models for the ALS disease.
Simple and more complex in vivo models provide new insights into ALS pathogenesis.
The synergistic potential of combining multiple ALS models has proven highly effective in identifying new therapeutic targets currently in the clinic.
The use of multiple simple animal models in conjunction with more patient-relevant models has an immense potential in providing insights in ALS pathogenesis and therapeutic targets.
Translational research using animal and other pathology relevant ALS models has enabled the discovery of new therapeutics addressing different pathophysiological aspects of the disease.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207
- Mejzini R, Flynn LL, Pitout IL, et al. ALS genetics, mechanisms, and therapeutics: where are we Now? Front Neurosci. 2019;13:1310. doi: 10.3389/fnins.2019.01310
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018
- Brown RH, Al-Chalabi A, Longo DL. Amyotrophic lateral sclerosis. Longo DL, editor. N Engl J Med. 2017;377(2):162–172. doi: 10.1056/NEJMra1603471
- Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology. 2013;80(17):1590–1597. doi: 10.1212/WNL.0b013e31828f18ac
- Abrahams S. Neuropsychological impairment in amyotrophic lateral sclerosis–frontotemporal spectrum disorder. Nat Rev Neurol. 2023;19(11):655–667. doi: 10.1038/s41582-023-00878-z
- Zou Z-Y, Zhou Z-R, Che C-H, et al. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–549. doi: 10.1136/jnnp-2016-315018
- Wang H, Guan L, Deng M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front Neurosci. 2023;17:1170996.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole study group. N Engl J Med. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901
- Abe K, Itoyama Y, Sobue G, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):610–617. doi: 10.3109/21678421.2014.959024
- Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919–930. doi: 10.1056/NEJMoa1916945
- Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099–1110. doi: 10.1056/NEJMoa2204705
- Benatar M, Wuu J, Andersen PM, et al. Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics. 2022;19(4):1248–1258. doi: 10.1007/s13311-022-01237-4
- Van Daele SH, Masrori P, Van Damme P, et al. The sense of antisense therapies in ALS. Trends Mol Med. 2024;S1471491423002836(3):252–262. doi: 10.1016/j.molmed.2023.12.003
- Mathis S, Goizet C, Soulages A, et al. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. 2019;399:217–226. doi: 10.1016/j.jns.2019.02.030
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011
- van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34(2):363–373. doi: 10.1002/humu.22244
- Gendron TF, Belzil VV, Zhang Y-J, et al. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127:359–376. doi: 10.1007/s00401-013-1237-z
- Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124
- Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN rab-GEFs. Bioinformatics. 2013;29(4):499–503. doi: 10.1093/bioinformatics/bts725
- Zhang D, Iyer LM, He F, et al. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet. 2012;3:283. doi: 10.3389/fgene.2012.00283
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0
- Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258
- Niwa J, Yamada S, Ishigaki S, et al. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J Biol Chem. 2007;282(38):28087–28095. doi: 10.1074/jbc.M704465200
- Crow JP, Ye YZ, Strong M, et al. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997;69(5):1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x
- Higgins CMJ, Jung C, Ding H, et al. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22(6):RC215–RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002
- Spreux-Varoquaux O, Bensimon G, Lacomblez L, et al. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002;193(2):73–78. doi: 10.1016/S0022-510X(01)00661-X
- Ligon LA, LaMonte BH, Wallace KE, et al. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport. 2005;16(6):533–536. doi: 10.1097/00001756-200504250-00002
- Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584
- Ayala YM, Pantano S, D’Ambrogio A, et al. Human, drosophila, and C.Elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348(3):575–588. doi: 10.1016/j.jmb.2005.02.038
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–R64. doi: 10.1093/hmg/ddq137
- Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108
- Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942
- Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066
- Bosco DA, Lemay N, Ko HK, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19(21):4160–4175. doi: 10.1093/hmg/ddq335
- Rademakers R, Stewart H, Dejesus-Hernandez M, et al. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. 2010;42(2):170–176. doi: 10.1002/mus.21665
- Lagier-Tourenne C, Polymenidou M, Hutt KR, et al. Divergent roles of als-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. doi: 10.1038/nn.3230
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34(6):812–826. doi: 10.1002/humu.22319
- Qiu H, Lee S, Shang Y, et al. Als-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124(3):981–999. doi: 10.1172/JCI72723
- Naumann M, Pal A, Goswami A, et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun. 2018;9(1):335. doi: 10.1038/s41467-017-02299-1
- Ward S, Thomson N, White JG, et al. Electron microscopical reconstruction of the anterior sensory anatomy of the nematode caenorhabditis elegans.?2UU. J Comp Neurol. 1975;160(3):313–337. doi: 10.1002/cne.901600305
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56(1):110–156. doi: 10.1016/0012-1606(77)90158-0
- White JG, Southgate E, Thomson JN, et al. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340.
- Mulcahy B, Witvliet D, Holmyard D, et al. A pipeline for volume electron microscopy of the Caenorhabditis elegans nervous system. Front Neural Circuits. 2018;12:94. doi: 10.3389/fncir.2018.00094
- Shaye DD, Greenwald I, Iijima KM. OrthoList: a compendium of C. elegans genes with human orthologs. PLOS ONE. 2011;6(5):e20085. doi: 10.1371/journal.pone.0020085
- Wu Y, Chen Y, Yu X, et al. Towards understanding neurodegenerative diseases: insights from Caenorhabditis elegans. Int J Mol Sci. 2023;25(1):443. doi: 10.3390/ijms25010443
- Baskoylu SN, Yersak J, O’Hern P, et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLOS Genet. 2018;14(10):e1007682. doi: 10.1371/journal.pgen.1007682
- Gidalevitz T, Krupinski T, Garcia S, et al. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. Orr H, editor. PLOS Genet. 2009;5(3):e1000399. doi: 10.1371/journal.pgen.1000399
- Wang J, Farr GW, Hall DH, et al. An ALS-Linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. Cox GA, editor. PLOS Genet. 2009;5(1):e1000350. doi: 10.1371/journal.pgen.1000350
- Boccitto M, Lamitina T, Kalb RG, et al. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. Blagosklonny MV, editor. PLOS ONE. 2012;7(3):e33494. doi: 10.1371/journal.pone.0033494
- Li J, Huang K, Le W. Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol Sin. 2013;34(5):644–650. doi: 10.1038/aps.2012.190
- Li J, Li T, Zhang X, et al. Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol Aging. 2014;35(4):837–846. doi: 10.1016/j.neurobiolaging.2013.09.003
- Xu H, Jia C, Cheng C, et al. Activation of autophagy attenuates motor deficits and extends lifespan in a C. elegans model of ALS. Free Radic Biol Med. 2022;181:52–61. doi: 10.1016/j.freeradbiomed.2022.01.030
- Doonan R, McElwee JJ, Matthijssens F, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22(23):3236–3241. doi: 10.1101/gad.504808
- Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci. 2010;30(48):16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010
- Zhang T, Mullane PC, Periz G, et al. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet. 2011;20(10):1952–1965. doi: 10.1093/hmg/ddr076
- Liachko NF, Saxton AD, McMillan PJ, et al. The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol. 2016;132(4):545–561. doi: 10.1007/s00401-016-1600-y
- Ash PEA, Zhang Y-J, Roberts CM, et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19(16):3206–3218. doi: 10.1093/hmg/ddq230
- Zhang T, Hwang H-Y, Hao H, et al. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J Biol Chem. 2012;287(11):8371–8382. doi: 10.1074/jbc.M111.311977
- Saldi TK, Ash PE, Wilson G, et al. TDP -1, the C aenorhabditis elegans ortholog of TDP -43, limits the accumulation of double-stranded RNA. Embo J. 2014;33(24):2947–2966. doi: 10.15252/embj.201488740.
- Saldi TK, Gonzales P, Garrido-Lecca A, et al. The Caenorhabditis elegans ortholog of TDP-43 regulates the chromatin localization of the heterochromatin protein 1 homolog HPL-2. Mol Cell Biol. 2018;38(15):e00668–17. doi: 10.1128/MCB.00668-17
- Vaccaro A, Patten SA, Ciura S, et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. Le W, editor. PLOS ONE. 2012;7(7):e42117. doi: 10.1371/journal.pone.0042117
- Vaccaro A, Tauffenberger A, Aggad D, et al. Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. Petrucelli L, editor. PLOS ONE. 2012;7(2):e31321. doi: 10.1371/journal.pone.0031321
- Vaccaro A, Patten SA, Aggad D, et al. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol Dis. 2013;55:64–75. doi: 10.1016/j.nbd.2013.03.015
- Murakami T, Yang S-P, Xie L, et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet. 2012;21(1):1–9. doi: 10.1093/hmg/ddr417
- Markert SM, Skoruppa M, Yu B, et al. Overexpression of an als-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol Open. 2020:bio.055129. doi: 10.1242/bio.055129
- Labarre A, Tossing G, Maios C, et al. A single copy transgenic mutant FUS strain reproduces age-dependent ALS phenotypes in C. elegans. MicroPubl Biol. 2021;2021. doi: 10.17912/micropub.biology.000473
- Baskoylu SN, Chapkis N, Unsal B, et al. Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 2022;38(4):110195. doi: 10.1016/j.celrep.2021.110195
- Therrien M, Rouleau GA, Dion PA, et al. FET proteins regulate lifespan and neuronal integrity. Sci Rep. 2016;6(1):25159. doi: 10.1038/srep25159
- Therrien M, Rouleau GA, Dion PA, et al. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLOS ONE. 2013;8(12):e83450. doi: 10.1371/journal.pone.0083450
- Corrionero A, Horvitz HR. A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr Biol. 2018;28(10):1522–1535.e5. doi: 10.1016/j.cub.2018.03.063
- Amick J, Roczniak-Ferguson A, Ferguson SM, et al. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27(20):3040–3051. doi: 10.1091/mbc.e16-01-0003
- Sonobe Y, Aburas J, Krishnan G, et al. A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nat Commun. 2021;12(1):6025. doi: 10.1038/s41467-021-26303-x
- Lamitina T. Length-dependent RNA foci formation and repeat associated non-aug dependent translation in a C. elegans G 4 C 2 model. Micro Publ Biol. 2022;2022. doi: 10.17912/micropub.biology.000600
- Rudich P, Snoznik C, Watkins SC, et al. Nuclear localized C9orf72-associated arginine-containing dipeptides exhibit age-dependent toxicity in C. elegans. Hum Mol Genet. 2017;26(24):4916–4928. doi: 10.1093/hmg/ddx372
- Ashburner M, Bergman CM. Drosophila melanogaster: a case study of a model genomic sequence and its consequences: fi gu re 1. Genome Res. 2005;15(12):1661–1667. doi: 10.1101/gr.3726705
- Hales KG, Korey CA, Larracuente AM, et al. Genetics on the fly: a primer on the Drosophila model system. Genetics. 2015;201(3):815–842. doi: 10.1534/genetics.115.183392
- Adams MD, Celniker SE, Holt RA, et al. The genome sequence of drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185
- Myers EW, Sutton GG, Delcher AL, et al. A whole-genome assembly of drosophila. Science. 2000;287(5461):2196–2204. doi: 10.1126/science.287.5461.2196
- Reiter LT, Potocki L, Chien S, et al. A systematic analysis of human disease-associated gene sequences in drosophila melanogaster. Genome Res. 2001;11(6):1114–1125. doi: 10.1101/gr.169101
- Pandey UB, Nichols CD, Barker EL. Human disease models in drosophila melanogaster and the role of the fly in therapeutic drug discovery. Barker EL, editor. Pharmacol Rev. 2011;63(2):411–436. doi: 10.1124/pr.110.003293
- Lloyd TE, Verstreken P, Ostrin EJ, et al. A genome-wide search for synaptic vesicle cycle proteins in drosophila. Neuron. 2000;26(1):45–50. doi: 10.1016/S0896-6273(00)81136-8
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401
- McGuire SE, Mao Z, Davis RL. Spatiotemporal gene expression targeting with the TARGET and Gene-switch systems in drosophila. Sci STKE [Internet]. 2004 [cited 2024 Feb 5]. Available from: https://www.science.org/doi/10.1126/stke.2202004pl6
- Dietzl G, Chen D, Schnorrer F, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in drosophila. Nature. 2007;448(7150):151–156. doi: 10.1038/nature05954
- Casci I, Pandey UB. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 2015;1607:47–74. doi: 10.1016/j.brainres.2014.09.064
- Layalle S, They L, Ourghani S, et al. Amyotrophic lateral sclerosis genes in drosophila melanogaster. IJMS. 2021;22(2):904. doi: 10.3390/ijms22020904
- Phillips JP, Campbell SD, Michaud D, et al. Null mutation of copper/zinc superoxide dismutase in drosophila confers hypersensitivity to paraquat and reduced longevity. Proc Natl Acad Sci USA. 1989;86(8):2761–2765. doi: 10.1073/pnas.86.8.2761
- Parkes TL, Elia AJ, Dickinson D, et al. Extension of drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19(2):171–174. doi: 10.1038/534
- Watson MR, Lagow RD, Xu K, et al. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283(36):24972–24981. doi: 10.1074/jbc.M804817200
- Islam R, Kumimoto EL, Bao H, et al. Als-linked SOD1 in glial cells enhances ß-N-Methylamino L-Alanine (bmaa)-induced toxicity in drosophila. F1000Res. 2012;1:47. doi: 10.12688/f1000research.1-47.v1
- Kumimoto EL, Fore TR, Zhang B. Transcriptome profiling following neuronal and glial expression of ALS-Linked SOD1 in drosophila. G3 (Bethesda). 2013;3(4):695–708. doi: 10.1534/g3.113.005850
- Gallart-Palau X, Ng C-H, Ribera J, et al. Drosophila expressing human SOD1 successfully recapitulates mitochondrial phenotypic features of familial amyotrophic lateral sclerosis. Neurosci Lett. 2016;624:47–52. doi: 10.1016/j.neulet.2016.05.006
- Şahin A, Held A, Bredvik K, et al. Human SOD1 ALS mutations in a drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics. 2017;205(2):707–723. doi: 10.1534/genetics.116.190850
- Held A, Major P, Sahin A, et al. Circuit dysfunction in SOD1-ALS model first detected in sensory feedback prior to motor neuron degeneration is alleviated by BMP signaling. J Neurosci. 2019;39(12):2347–2364. doi: 10.1523/JNEUROSCI.1771-18.2019
- Feiguin F, Godena VK, Romano G, et al. Depletion of TDP‐43 affects drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583(10):1586–1592. doi: 10.1016/j.febslet.2009.04.019
- Lu Y, Ferris J, Gao F-B. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol Brain. 2009;2(1):30. doi: 10.1186/1756-6606-2-30
- Lin M-J, Cheng C-W, Shen C-K, et al. Neuronal function and dysfunction of drosophila dTDP. Marion-poll F, editor. PLOS ONE. 2011;6(6):e20371. doi: 10.1371/journal.pone.0020371
- Diaper DC, Adachi Y, Sutcliffe B, et al. Loss and gain of drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet. 2013;22(8):1539–1557. doi: 10.1093/hmg/ddt005
- Chang J-C, Hazelett DJ, Stewart JA, et al. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a drosophila, TDP-43 loss of function model of ALS. Brain Res. 2014;1584:39–51. doi: 10.1016/j.brainres.2013.11.019
- Donde A, Sun M, Ling JP, et al. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019;138(5):813–826. doi: 10.1007/s00401-019-02042-8
- Voigt A, Herholz D, Fiesel FC, et al. TDP-43-Mediated neuron loss in vivo requires RNA-Binding activity. Feany MB, editor. PLOS ONE. 2010;5(8):e12247. doi: 10.1371/journal.pone.0012247
- Ritson GP, Custer SK, Freibaum BD, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30(22):7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010
- Li Y, Ray P, Rao EJ, et al. A drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci USA. 2010;107(7):3169–3174. doi: 10.1073/pnas.0913602107
- Ihara R, Matsukawa K, Nagata Y, et al. RNA binding mediates neurotoxicity in the transgenic drosophila model of TDP-43 proteinopathy. Hum Mol Genet. 2013;22(22):4474–4484. doi: 10.1093/hmg/ddt296
- Estes PS, Daniel SG, Mccallum AP, et al. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a drosophila model of amyotrophic lateral sclerosis. Disease Model Mechanisms. 2013:dmm.010710. doi: 10.1242/dmm.010710
- Romano G, Klima R, Buratti E, et al. Chronological requirements of TDP-43 function in synaptic organization and locomotive control. Neurobiol Dis. 2014;71:95–109. doi: 10.1016/j.nbd.2014.07.007
- Sreedharan J, Neukomm LJ, Brown RH, et al. Age-dependent TDP-43-Mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr Biol. 2015;25(16):2130–2136. doi: 10.1016/j.cub.2015.06.045
- Chen Y, Yang M, Deng J, et al. Expression of human FUS protein in drosophila leads to progressive neurodegeneration. Protein Cell. 2011;2(6):477–486. doi: 10.1007/s13238-011-1065-7
- Lanson NA, Maltare A, King H, et al. A drosophila model of fus-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20(13):2510–2523. doi: 10.1093/hmg/ddr150
- Wang J-W, Brent JR, Tomlinson A, et al. The als-associated proteins FUS and TDP-43 function together to affect drosophila locomotion and life span. J Clin Invest. 2011;121(10):4118–4126. doi: 10.1172/JCI57883
- Xia R, Liu Y, Yang L, et al. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol Neurodegener. 2012;7(1):10. doi: 10.1186/1750-1326-7-10
- Bogaert E, Boeynaems S, Kato M, et al. Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 2018;24(3):529–537.e4. doi: 10.1016/j.celrep.2018.06.070
- Machamer JB, Collins SE, Lloyd TE. The ALS gene FUS regulates synaptic transmission at the drosophila neuromuscular junction. Hum Mol Genet. 2014;23(14):3810–3822. doi: 10.1093/hmg/ddu094
- Shahidullah M, Le Marchand SJ, Fei H, et al. Defects in synapse structure and function precede motor neuron degeneration in drosophila models of FUS-Related ALS. J Neurosci. 2013;33(50):19590–19598. doi: 10.1523/JNEUROSCI.3396-13.2013
- Deng J, Yang M, Chen Y, et al. FUS interacts with HSP60 to promote mitochondrial damage. Lu B,editor. PLOS Genet. 2015;11(9):e1005357. doi: 10.1371/journal.pgen.1005357
- Altanbyek V, Cha S-J, Kang G-U, et al. Imbalance of mitochondrial dynamics in drosophila models of amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2016;481(3–4):259–264. doi: 10.1016/j.bbrc.2016.10.134
- Sasayama H, Shimamura M, Tokuda T, et al. Knockdown of the drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLOS ONE. 2012;7(6):e39483. doi: 10.1371/journal.pone.0039483
- Shimamura M, Kyotani A, Azuma Y, et al. Genetic link between cabeza, a Drosophila homologue of fused in sarcoma (FUS), and the EGFR signaling pathway. Exp Cell Res. 2014;326(1):36–45. doi: 10.1016/j.yexcr.2014.06.004
- Frickenhaus M, Wagner M, Mallik M, et al. Highly efficient cell-type-specific gene inactivation reveals a key function for the drosophila FUS homolog cabeza in neurons. Sci Rep. 2015;5(1):9107. doi: 10.1038/srep09107
- Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110(19):7778–7783. doi: 10.1073/pnas.1219643110
- Burguete AS, Almeida S, Gao F-B, et al. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife. 2015;4:e08881. doi: 10.7554/eLife.08881
- Tran H, Almeida S, Moore J, et al. Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron. 2015;87(6):1207–1214. doi: 10.1016/j.neuron.2015.09.015
- Sharpe JL, Harper NS, Garner DR, et al. Modeling C9orf72-related frontotemporal dementia and amyotrophic lateral sclerosis in drosophila. Front Cell Neurosci. 2021;15:770937. doi: 10.3389/fncel.2021.770937
- Mizielinska S, Grönke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–1194. doi: 10.1126/science.1256800
- Wen X, Tan W, Westergard T, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–1225. doi: 10.1016/j.neuron.2014.12.010
- Yang D, Abdallah A, Li Z, et al. Ftd/als-associated poly(gr) protein impairs the notch pathway and is recruited by poly(ga) into cytoplasmic inclusions. Acta Neuropathol. 2015;130(4):525–535. doi: 10.1007/s00401-015-1448-6
- Lee K-H, Zhang P, Kim HJ, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167(3):774–788.e17. doi: 10.1016/j.cell.2016.10.002
- Morón-Oset J, Supèr T, Esser J, et al. Glycine-alanine dipeptide repeats spread rapidly in a repeat length- and age-dependent manner in the fly brain. Acta Neuropathol Commun. 2019;7(1):209. doi: 10.1186/s40478-019-0860-x
- Goodman LD, Prudencio M, Kramer NJ, et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci. 2019;22(6):863–874. doi: 10.1038/s41593-019-0396-1
- Moens TG, Niccoli T, Wilson KM, et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019;137(3):487–500. doi: 10.1007/s00401-018-1946-4
- Atilano ML, Grönke S, Niccoli T, et al. Enhanced insulin signalling ameliorates C9orf72 hexanucleotide repeat expansion toxicity in drosophila. Elife. 2021;10:e58565. doi: 10.7554/eLife.58565
- West RJH, Sharpe JL, Voelzmann A, et al. Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in drosophila. Acta Neuropathol Commun. 2020;8(1):158. doi: 10.1186/s40478-020-01028-y
- Freibaum BD, Lu Y, Lopez-Gonzalez R, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133. doi: 10.1038/nature14974
- Perry S, Han Y, Das A, et al. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing als-related degeneration. Hum Mol Genet. 2017;26(21):4153–4167. doi: 10.1093/hmg/ddx304
- Li S, Wu Z, Li Y, et al. Altered MICOS morphology and mitochondrial ion homeostasis contribute to Poly(GR) toxicity associated with C9-ALS/FTD. Cell Rep. 2020;32(5):107989. doi: 10.1016/j.celrep.2020.107989
- Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4(1):35–44. doi: 10.1038/nrd1606
- Patton EE, Zon LI, Langenau DM. Zebrafish disease models in drug discovery: from preclinical modelling to clinical trials. Nat Rev Drug Discov. 2021;20(8):611–628. doi: 10.1038/s41573-021-00210-8
- Singh J, Patten SA. Modeling neuromuscular diseases in zebrafish. Front Mol Neurosci. 2022;15:1054573. doi: 10.3389/fnmol.2022.1054573
- Lescouzères L, Hassen-Khodja C, Baudot A, et al. A multilevel screening pipeline in zebrafish identifies therapeutic drugs for GAN. EMBO Mol Med. 2023;15(7):e16267. doi: 10.15252/emmm.202216267
- Barbazuk WB, Korf I, Kadavi C. The syntenic relationship of the zebrafish and human genomes. Genome Res. 2000;10(9):1351–1358. doi: 10.1101/gr.144700
- Howe K, Clark MD, Torroja CF, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. doi: 10.1038/nature12111
- Lescouzères L, Bordignon B, Bomont P. Development of a high-throughput tailored imaging method in zebrafish to understand and treat neuromuscular diseases. Front Mol Neurosci. 2022;15:956582. doi: 10.3389/fnmol.2022.956582
- Singh J, Pan YE, Patten SA, et al. NMJ analyser: a novel method to quantify neuromuscular junction morphology in zebrafish. Martelli PL, editor. Bioinformatics. 2023;39(12):btad720. doi: 10.1093/bioinformatics/btad720
- Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135(10):1735–1743. doi: 10.1242/dev.001115
- Schmid B, Haass C. Genomic editing opens new avenues for zebrafish as a model for neurodegeneration. J Neurochem. 2013;127(4):461–470. doi: 10.1111/jnc.12460
- Kawakami K, Asakawa K, Hibi M, et al. Gal4 driver transgenic zebrafish. Advances in genetics [Internet]. Elsevier; 2016 [cited 2024 Feb 26]. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0065266016300207
- Hwang WY, Fu Y, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–229. doi: 10.1038/nbt.2501
- Armstrong GAB, Liao M, You Z, et al. Homology directed knockin of point mutations in the zebrafish tardbp and fus genes in ALS using the CRISPR/Cas9 system. PLOS ONE. 2016;11(3):e0150188. doi: 10.1371/journal.pone.0150188
- Albadri S, Del Bene F, Revenu C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods. 2017;121–122:77–85. doi: 10.1016/j.ymeth.2017.03.005
- Rosello M, Serafini M, Concordet J-P, et al. Precise mutagenesis in zebrafish using cytosine base editors. Nat Protoc. 2023;18(9):2794–2813. doi: 10.1038/s41596-023-00854-3
- Ramesh T, Lyon AN, Pineda RH, et al. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis Model Mech. 2010;3(9–10):652–662. doi: 10.1242/dmm.005538
- Sakowski SA, Lunn JS, Busta AS, et al. Neuromuscular effects of G93A-SOD1 expression in zebrafish. Mol Neurodegener. 2012;7(1):44. doi: 10.1186/1750-1326-7-44
- Lemmens R, Van Hoecke A, Hersmus N, et al. Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum Mol Genet. 2007;16(19):2359–2365. doi: 10.1093/hmg/ddm193
- Da Costa MMJ, Allen CE, Higginbottom A, et al. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis Model Mech. 2014;7:73–81. doi: 10.1242/dmm.012013
- McGown A, McDearmid JR, Panagiotaki N, et al. Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann Neurol. 2013;73(2):246–258. doi: 10.1002/ana.23780
- Laird AS, Van Hoecke A, De Muynck L, et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLOS ONE. 2010;5(10):e13368. doi: 10.1371/journal.pone.0013368
- Kabashi E, Lin L, Tradewell ML, et al. Gain and loss of function of als-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19(4):671–683. doi: 10.1093/hmg/ddp534
- Schmid B, Hruscha A, Hogl S, et al. Loss of als-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc Natl Acad Sci USA. 2013;110(13):4986–4991. doi: 10.1073/pnas.1218311110
- Hewamadduma CAA, Grierson AJ, Ma TP, et al. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum Mol Genet. 2013;22(12):2376–2386. doi: 10.1093/hmg/ddt082
- Dormann D, Rodde R, Edbauer D, et al. Als-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. Embo J. 2010;29(16):2841–2857. doi: 10.1038/emboj.2010.143
- Acosta JR, Goldsbury C, Winnick C, et al. Mutant human FUS is ubiquitously mislocalized and generates persistent stress granules in primary cultured transgenic zebrafish cells. Coulson EJ, editor. PLOS ONE. 2014;9(6):e90572. doi: 10.1371/journal.pone.0090572
- Kabashi E, Bercier V, Lissouba A, et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLOS Genet. 2011;7(8):e1002214. doi: 10.1371/journal.pgen.1002214
- Armstrong GAB, Drapeau P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet. 2013;22(21):4282–4292. doi: 10.1093/hmg/ddt278
- Lebedeva S, de Jesus Domingues AM, Butter F, et al. Characterization of genetic loss-of-function of fus in zebrafish. RNA Biol. 2017;14(1):29–35. doi: 10.1080/15476286.2016.1256532
- Bourefis A-R, Campanari M-L, Buee-Scherrer V, et al. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiol Dis. 2020;142:104935. doi: 10.1016/j.nbd.2020.104935
- Fortier G, Butti Z, Patten SA. Modelling C9orf72-related amyotrophic lateral sclerosis in zebrafish. Biomedicines. 2020;8(10):440. doi: 10.3390/biomedicines8100440
- Lee Y-B, Chen H-J, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. doi: 10.1016/j.celrep.2013.10.049
- Swinnen B, Bento-Abreu A, Gendron TF, et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018;135(3):427–443. doi: 10.1007/s00401-017-1796-5
- Ohki Y, Wenninger-Weinzierl A, Hruscha A, et al. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol Neurodegener. 2017;12(1):6. doi: 10.1186/s13024-016-0146-8
- Swaminathan A, Bouffard M, Liao M, et al. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum Mol Genet. 2018;27(10):1754–1762. doi: 10.1093/hmg/ddy083
- Shaw MP, Higginbottom A, McGown A, et al. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol Commun. 2018;6(1):125. doi: 10.1186/s40478-018-0629-7
- Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74(2):180–187. doi: 10.1002/ana.23946
- Yeh T-H, Liu H-F, Li Y-W, et al. C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp Neurol. 2018;304:114–124. doi: 10.1016/j.expneurol.2018.03.002
- Butti Z, Pan YE, Giacomotto J, et al. Reduced C9orf72 function leads to defective synaptic vesicle release and neuromuscular dysfunction in zebrafish. Commun Biol. 2021;4(1):792. doi: 10.1038/s42003-021-02302-y
- Jaroszynska N, Salzinger A, Tsarouchas TM, et al. C9ORF72 deficiency results in degeneration of the zebrafish retina in vivo [Internet]. Neuroscience. 2023 [cited 2024 Feb 8]. Available from: http://biorxiv.org/lookup/doi/10.1101/2023.10.19.563041
- Picher-Martel V, Valdmanis PN, Gould PV, et al. From animal models to human disease: a genetic approach for personalized medicine in ALS. acta neuropathol commun. Acta Neuropathol Commun. 2016;4(1):70. doi: 10.1186/s40478-016-0340-5
- Kang SH, Li Y, Fukaya M, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16(5):571–579. doi: 10.1038/nn.3357
- Miller SJ, Zhang P, Glatzer J, et al. Astroglial transcriptome dysregulation in early disease of an ALS mutant SOD1 mouse model. J Neurogenet. 2017;31(1–2):37–48. doi: 10.1080/01677063.2016.1260128
- Maniatis S, Äijö T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364(6435):89–93. doi: 10.1126/science.aav9776
- MacLean M, López-Díez R, Vasquez C, et al. Neuronal–glial communication perturbations in murine SOD1G93A spinal cord. Commun Biol. 2022;5(1):177. doi: 10.1038/s42003-022-03128-y
- Filipi T, Matusova Z, Abaffy P, et al. Cortical glia in SOD1(G93A) mice are subtly affected by als-like pathology. Sci Rep. 2023;13(1):6538. doi: 10.1038/s41598-023-33608-y
- Canto MCD, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol. 1997;93(6):537–550. doi: 10.1007/s004010050650
- Alexander GM, Erwin KL, Byers N, et al. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res. 2004;130(1–2):7–15. doi: 10.1016/j.molbrainres.2004.07.002
- Deitch JS, Alexander GM, Bensinger A, et al. Phenotype of transgenic mice carrying a very low copy number of the mutant human G93A superoxide dismutase-1 gene associated with amyotrophic lateral sclerosis. PLOS ONE. 2014;9(6):e99879. doi: 10.1371/journal.pone.0099879
- Bruijn LI, Becher MW, Lee MK, et al. ALS-Linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/S0896-6273(00)80272-X
- Wong PC, Pardo CA, Borchelt DR, et al. An adverse property of a familial als-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105–1116. doi: 10.1016/0896-6273(95)90259-7
- Ripps ME, Huntley GW, Hof PR, et al. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92(3):689–693. doi: 10.1073/pnas.92.3.689
- Jonsson PA, Graffmo KS, Brännström T, et al. Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol. 2006;65(12):1126–1136. doi: 10.1097/01.jnen.0000248545.36046.3c
- Chang-Hong R, Wada M, Koyama S, et al. Neuroprotective effect of oxidized galectin-1 in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;194(1):203–211. doi: 10.1016/j.expneurol.2005.02.011
- Joyce PI, Mcgoldrick P, Saccon RA, et al. A novel SOD1-ALS mutation separates central and peripheral effects of mutant SOD1 toxicity. Hum Mol Genet. 2015;24(7):1883–1897. doi: 10.1093/hmg/ddu605
- AndrewG R, Elliott JL, Hoffman EK, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–47. doi: 10.1038/ng0596-43
- Fischer LR, Li Y, Asress SA, et al. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp Neurol. 2012;233(1):163–171. doi: 10.1016/j.expneurol.2011.09.020
- Muller FL, Song W, Liu Y, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radical Biol Med. 2006;40(11):1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036
- Nagai M, Aoki M, Miyoshi I, et al. Rats expressing human cytosolic copper–zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21(23):9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001
- Howland DS, Liu J, She Y, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002;99(3):1604–1609. doi: 10.1073/pnas.032539299
- Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2009;106(44):18809–18814. doi: 10.1073/pnas.0908767106
- Stallings NR, Puttaparthi K, Luther CM, et al. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40(2):404–414. doi: 10.1016/j.nbd.2010.06.017
- Xu Y-F, Gendron TF, Zhang Y-J, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30(32):10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
- Arnold ES, Ling S-C, Huelga SC, et al. Als-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110(8):E736–745. doi: 10.1073/pnas.1222809110
- Swarup V, Phaneuf D, Bareil C, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(9):2610–2626. doi: 10.1093/brain/awr159
- Zhou H, Huang C, Chen H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. Cox GA, editor. PLOS Genet. 2010;6(3):e1000887. doi: 10.1371/journal.pgen.1000887
- Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010;107(8):3858–3863. doi: 10.1073/pnas.0912417107
- Shan X, Chiang P-M, Price DL, et al. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci USA. 2010;107(37):16325–16330. doi: 10.1073/pnas.1003459107
- Janssens J, Wils H, Kleinberger G, et al. Overexpression of ALS-Associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol. 2013;48(1):22–35. doi: 10.1007/s12035-013-8427-5
- Tsai K-J, Yang C-H, Fang Y-H, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207(8):1661–1673. doi: 10.1084/jem.20092164
- Cannon A, Yang B, Knight J, et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol. 2012;123(6):807–823. doi: 10.1007/s00401-012-0979-3
- Igaz LM, Kwong LK, Lee EB, et al. Dysregulation of the als-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726–738. doi: 10.1172/JCI44867
- Walker AK, Spiller KJ, Ge G, et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015;130(5):643–660. doi: 10.1007/s00401-015-1460-x
- Huang C, Tong J, Bi F, et al. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–118. doi: 10.1172/JCI59130
- Wu L-S, Cheng W-C, Shen C-K. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem. 2012;287(33):27335–27344. doi: 10.1074/jbc.M112.359000
- Iguchi Y, Katsuno M, Niwa J, et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain. 2013;136(5):1371–1382. doi: 10.1093/brain/awt029
- Fratta P, Sivakumar P, Humphrey J, et al. Mice with endogenous TDP ‐43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. Embo J. 2018;37(11):e98684. doi: 10.15252/embj.201798684
- White MA, Kim E, Duffy A, et al. TDP-43 gains function due to perturbed autoregulation in a tardbp knock-in mouse model of ALS-FTD. Nat Neurosci. 2018;21(4):552–563. doi: 10.1038/s41593-018-0113-5
- Ebstein SY, Yagudayeva I, Shneider NA. Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep. 2019;26(2):364–373.e4. doi: 10.1016/j.celrep.2018.12.045
- Huang S-L, Wu L-S, Lee M, et al. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol Commun. 2020;8(1):3. doi: 10.1186/s40478-020-0881-5
- Krus KL, Strickland A, Yamada Y, et al. Loss of stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep. 2022;39(13):111001. doi: 10.1016/j.celrep.2022.111001
- Baughn MW, Melamed Z, López-Erauskin J, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science. 2023;379(6637):1140–1149. doi: 10.1126/science.abq5622
- Nolan M, Talbot K, Ansorge O. Pathogenesis of fus-associated ALS and FTD: insights from rodent models. acta neuropathol commun. Acta Neuropathol Commun. 2016;4(1):99. doi: 10.1186/s40478-016-0358-8
- Sharma A, Lyashchenko AK, Lu L, et al. Als-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7(1):10465. doi: 10.1038/ncomms10465
- Scekic‐Zahirovic J, Sendscheid O, El Oussini H, et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. Embo J. 2016;35(10):1077–1097. doi: 10.15252/embj.201592559
- Scekic-Zahirovic J, Oussini HE, Mersmann S, et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of fus-associated amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133(6):887–906. doi: 10.1007/s00401-017-1687-9
- Devoy A, Kalmar B, Stewart M, et al. Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in ‘FUSDelta14’ knockin mice. Brain. 2017;140(11):2797–2805. doi: 10.1093/brain/awx248
- Mitchell JC, McGoldrick P, Vance C, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125(2):273–288. doi: 10.1007/s00401-012-1043-z
- Shelkovnikova TA, Peters OM, Deykin AV, et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 2013;288(35):25266–25274. doi: 10.1074/jbc.M113.492017
- Huang C, Zhou H, Tong J, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Cox GA, editor. PLOS Genet. 2011;7(3):e1002011. doi: 10.1371/journal.pgen.1002011
- Kino Y, Washizu C, Kurosawa M, et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2015;3(1):24. doi: 10.1186/s40478-015-0202-6
- Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA [Internet]. 2013 [cited 2024 Feb 19]. Available from: https://pnas.org/doi/full/10.1073/pnas.1318835110
- O’Rourke JG, Bogdanik L, Muhammad AKMG, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901. doi: 10.1016/j.neuron.2015.10.027
- Koppers M, Blokhuis AM, Westeneng H, et al. C 9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78(3):426–438. doi: 10.1002/ana.24453
- O’Rourke JG, Bogdanik L, Yáñez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–1329. doi: 10.1126/science.aaf1064
- Atanasio A, Decman V, White D, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci Rep. 2016;6(1):23204. doi: 10.1038/srep23204
- Burberry A, Suzuki N, Wang J-Y, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med [Internet]. 2016 [cited 2024 Feb 19]. Available from: https://www.science.org/doi/10.1126/scitranslmed.aaf6038
- Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-Linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-Containing RNAs. Neuron. 2016;90(3):535–550. doi: 10.1016/j.neuron.2016.04.006
- Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances Autophagic activity via deregulated mTOR and TFEB signaling. Cox GA, editor. PLOS Genet. 2016;12(11):e1006443. doi: 10.1371/journal.pgen.1006443
- Lopez-Herdoiza M-B, Bauché S, Wilmet B, et al. C9ORF72 knockdown triggers ftd-like symptoms and cell pathology in mice. Front Cell Neurosci. 2023;17:1155929. doi: 10.3389/fncel.2023.1155929
- Shao Q, Liang C, Chang Q, et al. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. acta neuropathol commun. Acta Neuropathol Commun. 2019;7(1):32. doi: 10.1186/s40478-019-0685-7
- Zhu Q, Jiang J, Gendron TF, et al. Reduced C9ORF72 function exacerbates gain of toxicity from als/ftd-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–624. doi: 10.1038/s41593-020-0619-5
- Dong W, Ma Y, Guan F, et al. Ablation of C9orf72 together with excitotoxicity induces ALS in rats. FEBS J. 2021;288(5):1712–1723. doi: 10.1111/febs.15501
- Chew J, Gendron TF, Prudencio M, et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348(6239):1151–1154. doi: 10.1126/science.aaa9344
- Chew J, Cook C, Gendron TF, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14(1):9. doi: 10.1186/s13024-019-0310-z
- Herranz-Martin S, Chandran J, Lewis K, et al. Viral delivery of C9ORF72 hexanucleotide repeat expansions in mice lead to repeat length dependent neuropathology and behavioral deficits. Disease Model Mechanisms. 2017:dmm.029892. doi: 10.1242/dmm.029892
- Riemslagh FW, Van Der Toorn EC, Verhagen RFM, et al. Inducible expression of human C9ORF72 36× G4C2 hexanucleotide repeats is sufficient to cause RAN translation and rapid muscular atrophy in mice. Disease Model Mechanisms. 2021;14(2):dmm044842. doi: 10.1242/dmm.044842
- O’Reilly LP, Luke CJ, Perlmutter DH, et al. C. elegans in high-throughput drug discovery. Adv Drug Deliv Rev. 2014;69–70:247–253. doi: 10.1016/j.addr.2013.12.001
- Dibaj P, Zschüntzsch J, Steffens H, et al. Influence of methylene blue on microglia-induced inflammation and motor neuron degeneration in the SOD1G93A model for ALS. Block ML, editor. PLOS ONE. 2012;7(8):e43963. doi: 10.1371/journal.pone.0043963
- Patten SA, Aggad D, Martinez J, et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight. 2017;2(22). doi: 10.1172/jci.insight.97152
- Bose P, Tremblay E, Maios C, et al. The novel small molecule TRVA242 stabilizes neuromuscular junction defects in multiple animal models of amyotrophic lateral sclerosis. Neurotherapeutics. 2019;16(4):1149–1166. doi: 10.1007/s13311-019-00765-w
- Wong SQ, Pontifex MG, Phelan MM, et al. α-methyl-α-phenylsuccinimide ameliorates neurodegeneration in a C. elegans model of TDP-43 proteinopathy. Neurobiol Dis. 2018;118:40–54. doi: 10.1016/j.nbd.2018.06.013
- Labarre A, Guitard E, Tossing G, et al. Fatty acids derived from the probiotic Lacticaseibacillus rhamnosus HA-114 suppress age-dependent neurodegeneration. Commun Biol. 2022;5(1):1340. doi: 10.1038/s42003-022-04295-8
- Tossing G, Livernoche R, Maios C, et al. Genetic and pharmacological PARP inhibition reduces axonal degeneration in C. elegans models of ALS. Hum Mol Genet. 2022;31(19):3313–3324. doi: 10.1093/hmg/ddac116
- Silva MC, Fox S, Beam M, et al. A genetic screening strategy identifies novel regulators of the proteostasis network. Serio TR, editor. PLOS Genet. 2011;7(12):e1002438. doi: 10.1371/journal.pgen.1002438
- Boyd JD, Lee-Armandt JP, Feiler MS, et al. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. SLAS Discov. 2014;19(1):44–56. doi: 10.1177/1087057113501553
- Tseng Y-L, Lu P-C, Lee C-C, et al. Degradation of neurodegenerative disease-associated TDP-43 aggregates and oligomers via a proteolysis-targeting chimera. J Biomed Sci. 2023;30(1):27. doi: 10.1186/s12929-023-00921-7
- Ikenaka K, Kawai K, Katsuno M, et al. Dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. Pandey U, editor. PLOS ONE. 2013;8(2):e54511. doi: 10.1371/journal.pone.0054511
- Ikenaka K, Tsukada Y, Giles AC, et al. A behavior-based drug screening system using a Caenorhabditis elegans model of motor neuron disease. Sci Rep. 2019;9(1):10104. doi: 10.1038/s41598-019-46642-6
- McGown A, Shaw DPJ, Ramesh T. Znstress: a high-throughput drug screening protocol for identification of compounds modulating neuronal stress in the transgenic mutant sod1G93R zebrafish model of amyotrophic lateral sclerosis. Mol Neurodegener. 2016;11(1):56. doi: 10.1186/s13024-016-0122-3
- Joardar A, Menzl J, Podolsky TC, et al. PPAR gamma activation is neuroprotective in a drosophila model of ALS based on TDP-43. Hum Mol Genet. 2015;24(6):1741–1754. doi: 10.1093/hmg/ddu587
- Dupuis L, Dengler R, Heneka MT, et al. A randomized, Double Blind, placebo-controlled trial of Pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. Sensi SL, editor. PLOS ONE. 2012;7(6):e37885. doi: 10.1371/journal.pone.0037885
- Wang T, Cheng J, Wang S, et al. α-Lipoic acid attenuates oxidative stress and neurotoxicity via the ERK/Akt-dependent pathway in the mutant hSOD1 related drosophila model and the NSC34 cell line of amyotrophic lateral sclerosis. Brain Res Bull. 2018;140:299–310. doi: 10.1016/j.brainresbull.2018.05.019
- Lee D, Jeong HC, Kim SY, et al. A comparison study of pathological features and drug efficacy between drosophila models of C9orf72 ALS/FTD. Mol Cells. 2024;47(1):100005. doi: 10.1016/j.mocell.2023.12.003
- Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi: 10.1038/nature14973
- Maragakis NJ, de Carvalho M, Weiss MD. Therapeutic targeting of ALS pathways: refocusing an incomplete picture. Ann Clin Transl Neurol. 2023;10(11):1948–1971. doi: 10.1002/acn3.51887
- Gurney ME, Cutting FB, Zhai P, et al. Benefit of vitamin E, riluzole, and gababapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39(2):147–157. doi: 10.1002/ana.410390203
- Desnuelle C, Dib M, Garrel C, et al. A double-blind, placebo-controlled randomized clinical trial of α-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord. 2001;2(1):9–18. doi: 10.1080/146608201300079364
- Klivenyi P, Ferrante RJ, Matthews RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5(3):347–350. doi: 10.1038/6568
- Shefner JM, Cudkowicz ME, Schoenfeld D, et al. A clinical trial of creatine in ALS. Neurology. 2004;63(9):1656–1661. doi: 10.1212/01.WNL.0000142992.81995.F0
- Ikeda K, Iwasaki Y, Kaji R. Neuroprotective effect of ultra-high dose methylcobalamin in wobbler mouse model of amyotrophic lateral sclerosis. J Neurol Sci. 2015;354(1–2):70–74. doi: 10.1016/j.jns.2015.04.052
- Oki R, Izumi Y, Fujita K, et al. Efficacy and safety of ultrahigh-dose methylcobalamin in early-stage amyotrophic lateral sclerosis: a randomized clinical trial. JAMA Neurol. 2022;79(6):575. doi: 10.1001/jamaneurol.2022.0901
- Gurney ME, Fleck TJ, Himes CS, et al. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology. 1998;50(1):62–66. doi: 10.1212/WNL.50.1.62
- Ito H, Wate R, Zhang J, et al. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp Neurol. 2008;213(2):448–455. doi: 10.1016/j.expneurol.2008.07.017
- Aoki M, Warita H, Mizuno H, et al. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011;1382:321–325. doi: 10.1016/j.brainres.2011.01.058
- Yan J, Wang YM, Hellwig A, et al. TwinF interface inhibitor FP802 stops loss of motor neurons and mitigates disease progression in a mouse model of ALS. Cell Rep Med. 2024;5(2):101413. doi: 10.1016/j.xcrm.2024.101413
- Wang I-F, Guo B-S, Liu Y-C, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci USA. 2012;109(37):15024–15029. doi: 10.1073/pnas.1206362109
- Crisafulli SG, Brajkovic S, Cipolat Mis MS, et al. Therapeutic strategies under development targeting inflammatory mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol. 2018;55(4):2789–2813. doi: 10.1007/s12035-017-0532-4
- Schütz B, Reimann J, Dumitrescu-Ozimek L, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25(34):7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005
- Kiaei M, Kipiani K, Chen J, et al. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191(2):331–336. doi: 10.1016/j.expneurol.2004.10.007
- Shibata N, Kawaguchi‐Niida M, Yamamoto T, et al. Effects of the PPARγ activator pioglitazone on p38 MAP kinase and IκBα in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology. 2008;28(4):387–398. doi: 10.1111/j.1440-1789.2008.00890.x
- Pelaez MC, Desmeules A, Gelon PA, et al. Neuronal dysfunction caused by FUSR521G promotes als-associated phenotypes that are attenuated by nf-κB inhibition. Acta Neuropathol Commun. 2023;11(1):182. doi: 10.1186/s40478-023-01671-1
- Chen Y, Wang H, Ying Z, et al. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through tfeb-mediated autophagy and lysosomal biogenesis: the new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem Biophys Res Commun. 2020;526(1):231–238. doi: 10.1016/j.bbrc.2020.03.051
- López-Blanch R, Salvador-Palmer R, Oriol-Caballo M, et al. Nicotinamide riboside, pterostilbene and ibudilast protect motor neurons and extend survival in ALS mice. Neurotherapeutics. 2024;21(1):e00301. doi: 10.1016/j.neurot.2023.10.011
- Wu C, Watts ME, Rubin LL. MAP4K4 activation mediates motor neuron degeneration in amyotrophic lateral sclerosis. Cell Rep. 2019;26(5):1143–1156.e5. doi: 10.1016/j.celrep.2019.01.019
- Liu M-L, Ma S, Tai W, et al. Screens in aging-relevant human als-motor neurons identify MAP4Ks as therapeutic targets for the disease. Cell Death Dis. 2024;15(1):4. doi: 10.1038/s41419-023-06395-7
- Trias E, Ibarburu S, Barreto-Núñez R, et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13(1):177. doi: 10.1186/s12974-016-0620-9
- Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20(1):19–33. doi: 10.1038/s41583-018-0093-1
- Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353(6299):603–608. doi: 10.1126/science.aaf6803
- Wang S-M, Wu H-E, Yasui Y, et al. Nucleoporin POM121 signals tfeb-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy. 2023;19(1):126–151. doi: 10.1080/15548627.2022.2063003
- Ionescu A, Gradus T, Altman T, et al. Targeting the sigma-1 receptor via pridopidine ameliorates central features of ALS pathology in a SOD1G93A Model. Cell Death Dis. 2019;10(3):210. doi: 10.1038/s41419-019-1451-2
- Smith RA. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Investigation. 2006;116(8):2290–2296. doi: 10.1172/JCI25424
- McCampbell A, Cole T, Wegener AJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Investigation. 2018;128(8):3558–3567. doi: 10.1172/JCI99081
- Liu Y, Dodart J-C, Tran H, et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. 2021;12(1):847. doi: 10.1038/s41467-021-21112-8
- Tran H, Moazami MP, Yang H, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117–124. doi: 10.1038/s41591-021-01557-6
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104–116. doi: 10.1038/s41591-021-01615-z
- Droppelmann CA, Campos-Melo D, Noches V, et al. Mitigation of TDP-43 toxic phenotype by an RGNEF fragment in amyotrophic lateral sclerosis models. Brain. 2024;147(6):2053–2068. doi: 10.1093/brain/awae078
- Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–371. doi: 10.1038/nature22038
- Ma ZC, Moore HJ, Smith TJ, et al. Mitigating a TDP-43 proteinopathy by targeting ataxin-2 using RNA-targeting CRISPR effector proteins. Nat Commun. 2023;14(1):6492. doi: 10.1038/s41467-023-42147-z
- Ediriweera GR, Chen L, Yerbury JJ, et al. Non-viral vector-mediated gene therapy for ALS: challenges and future perspectives. Mol Pharm. 2021;18(6):2142–2160. doi: 10.1021/acs.molpharmaceut.1c00297
- Chen L, Watson C, Morsch M, et al. Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front Neurosci. 2017;11:476. doi: 10.3389/fnins.2017.00476
- Poulin-Brière A, Rezaei E, Pozzi S. Antibody-based therapeutic interventions for amyotrophic lateral sclerosis: a systematic literature review. Front Neurosci. 2021;15:790114. doi: 10.3389/fnins.2021.790114
- Minamiyama S, Sakai M, Yamaguchi Y, et al. Efficacy of oligodendrocyte precursor cells as delivery vehicles for single-chain variable fragment to misfolded SOD1 in ALS rat model. Mol Ther - Methods Clin Devel. 2023;28:312–329. doi: 10.1016/j.omtm.2023.01.008
- Lincecum JM, Vieira FG, Wang MZ, et al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet. 2010;42(5):392–399. doi: 10.1038/ng.557
- Andrews-Zwilling Y, Mathur V, Kuhn L, et al. Inhibiting C1q improves compound muscle action potential and reduces neuronal damage in the SOD1G93A mouse Model (P1-13.004). Neurology. 2022;98(18_supplement). doi: 10.1212/WNL.98.18_supplement.3302
- Fisher EMC, Greensmith L, Malaspina A, et al. Opinion: more mouse models and more translation needed for ALS. Mol Neurodegener. 2023;18(1):30. doi: 10.1186/s13024-023-00619-2
- Wong C, Stavrou M, Elliott E, et al. Clinical trials in amyotrophic lateral sclerosis: a systematic review and perspective. Brain Commun. 2021;3(4):fcab242. doi: 10.1093/braincomms/fcab242
- Díaz-García D, Ferrer-Donato Á, Méndez-Arriaga JM, et al. Design of mesoporous silica nanoparticles for the treatment of amyotrophic lateral sclerosis (ALS) with a therapeutic cocktail based on leptin and pioglitazone. ACS Biomater Sci Eng. 2022;8(11):4838–4849. doi: 10.1021/acsbiomaterials.2c00865
- Lu Y, Wang J-W, Li N, et al. Intranasal administration of edaravone nanoparticles improves its stability and brain bioavailability. J Control Release. 2023;359:257–267. doi: 10.1016/j.jconrel.2023.06.001
- Medina DX, Chung EP, Teague CD, et al. Intravenously administered, retinoid activating nanoparticles increase lifespan and reduce neurodegeneration in the SOD1G93A mouse model of ALS. Front Bioeng Biotechnol. 2020;8:224. doi: 10.3389/fbioe.2020.00224
- Evans MC, Gaillard PJ, De Boer M, et al. Cns-targeted glucocorticoid reduces pathology in mouse model of amyotrophic lateral sclerosis. acta neuropathol commun. Acta Neuropathol Commun. 2014;2(1):66. doi: 10.1186/2051-5960-2-66
- Wiley NJ, Madhankumar AB, Mitchell RM, et al. Lipopolysaccharide modified liposomes for amyotropic lateral sclerosis therapy: efficacy in SOD1 mouse model. ANP. 2012;1(3):44–53. doi: 10.4236/anp.2012.13007
- Vasta R, Chia R, Traynor BJ, et al. Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine. 2022;75:103795. doi: 10.1016/j.ebiom.2021.103795
- Hawrot J, Imhof S, Wainger BJ. Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol Dis. 2020;134:104680. doi: 10.1016/j.nbd.2019.104680
- Fujimori K, Ishikawa M, Otomo A, et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018;24(10):1579–1589. doi: 10.1038/s41591-018-0140-5