
ABSTRACT
Background
Decades of research have transformed hemophilia from severely limiting children’s lives to a manageable disorder compatible with a full, active life, for many in high-income countries. The direction of future research will determine whether exciting developments truly advance health equity for all people with hemophilia (PWH). National Hemophilia Foundation (NHF) and American Thrombosis and Hemostasis Network conducted extensive inclusive all-stakeholder consultations to identify the priorities of people with inherited bleeding disorders and those who care for them.
Research design and methods
Working group (WG) 1 of the NHF State of the Science Research Summit distilled the community-identified priorities for hemophilia A and B into concrete research questions and scored their feasibility, impact, and risk.
Results
WG1 defined 63 top priority research questions concerning arthropathy/pain/bone health, inhibitors, diagnostics, gene therapy, the pediatric to adult transition of care, disparities faced by the community, and cardiovascular disease. This research has the potential to empower PWH to thrive despite lifelong comorbidities and achieve new standards of wellbeing, including psychosocial.
Conclusions
Collaborative research and care delivery will be key to capitalizing on current and horizon treatments and harnessing technical advances to improve diagnostics and testing, to advance health equity for all PWH.
Plain Language Summary
Hemophilia is the best known of the inherited bleeding disorders (BD). This is a rare condition that causes disproportionate bleeding, often into joints and vital organs. Factor replacement, injecting recombinant or plasma-based clotting factor products directly into the vein, became commonplace to control the disorder in the 1990s and 2000s. Prophylaxis, or injecting replacement factor every few days into people with hemophilia (PWH), has revolutionized patients’ lives. In the last few years, other advances in new therapies have entered this space, such as non-factor replacement therapies and gene therapy. With many more research advances on the horizon, the National Hemophilia Foundation (NHF) initiated a State of the Science Research Summit in 2020. This event was attended by over 880 interested parties to help design an agenda of research priorities for inherited BDs for the next decade, based on community consultations. NHF formed multiple Working Groups (WG), each exploring a theme resulting from the community consultations, and presenting their results at the Summit. Led by 2 hematologists who manage and treat PWH daily, the 21-community member WG1 assigned to hemophilia A and B divided into 7 subgroups to identify and organize research priorities for different topic areas. The outcomes focused on prioritizing patients’ needs, technological advances, and research in the areas of greatest potential for PWH and those who care for them. The results are a roadmap for the future execution of a research plan that truly serves the community.
1. Introduction
1.1. Hemophilia as a lifelong disorder
Decades of research have transformed hemophilia from a disorder that severely limited children’s lives and activities to one that may be managed across a full and very active life, for many in high-income countries. The direction of the research of coming decades will determine whether exciting developments, recent and future, truly advance health equity for all people with hemophilia (PWH).
Historically, research into hemophilia focused heavily on replacing the missing or nonfunctional clotting factor, factor VIII (FVIII) in the case of hemophilia A and factor IX (FIX) in the case of hemophilia B, the deficiency of which is responsible for its characteristic excessive bleeding that disproportionately occurs in joints, muscles, and with surgeries or injury [Citation1,Citation2]. In the 1920s, the life expectancy of a person with severe hemophilia was approximately 11 years [Citation3] with death often resulting from bleeding into vital organs [Citation4]. Many years of collaborative research were needed to obtain safe concentrates of plasma-derived [Citation5] and recombinant [Citation6] FVIII and FIX, with the regular prophylactic infusion of replacement factor becoming the standard of care for severe hemophilia in 1994, and now also for some people with mild or moderate hemophilia [Citation7,Citation8].
Routine prophylaxis, started at a young age and prior to the development of joint disease, dramatically reduced annualized bleeding rates (ABR) and vastly improved joint health, one of the primary morbidities of untreated hemophilia [Citation9], for many children [Citation10,Citation11] and adults [Citation12]. Recent innovations of extended half-life (EHL) recombinant FVIII and FIX, allowing for decreased dosing frequency or increased trough levels [Citation13,Citation14], expanded the treatment landscape [Citation15]. Prophylaxis combined with a patient-centric multidisciplinary team approach to care now means that many PWH in high-income countries enjoy a life expectancy comparable to that of people without a bleeding disorder (BD) [Citation16,Citation17].
Opportunities are boundless for research to improve outcomes in all aspects of the lives of PWH. Many will continue to face important challenges to bone and joint health, with hemophilic arthropathy requiring lifelong management [Citation9]. They will encounter the common concerns of cardiovascular disease and hypertension, with management options complicated by the backdrop of their bleeding tendency [Citation18,Citation19]. The development of antibodies (inhibitors) against exogenously administered FVIII remains the most important complication of the treatment of hemophilia developing in up to 25–30% of previously untreated people with severe hemophilia A, rendering FVIII infusions ineffective [Citation20,Citation21]. Inhibitors to replacement FIX, which may develop in 1.5–3% of people with hemophilia B, are associated with significant morbidity due to the bleeding risk in the absence of effective prophylaxis, as well as frequent occurrence of allergic/anaphylactic reactions and nephrotic syndrome [Citation22]. All these issues profoundly impact the daily quality-of-life (QoL), psychosocial wellbeing, and mental health of PWH [Citation1].
Today, the hemophilia treatment landscape is undergoing more revolutionary changes. The subcutaneously administered non-factor replacement therapy, emicizumab, a humanized monoclonal bispecific antibody that has the ability to rebalance hemostasis by mimicking the function of FVIII, has been approved for prophylaxis in PWH A of any age, with and without FVIII inhibitors [Citation23]. Several other non-factor therapeutics and multiple gene therapies for PWH are in clinical trials, two of which have (at the time of writing) received European or US regulatory approval [Citation24–31]. The current array of effective treatment options combined with emerging novel therapeutic approaches, and perhaps someday even a functional cure for hemophilia, raise the exciting possibility of addressing the lifelong health of PWH in a context of hemostatic stability, and targeting far more ambitious outcomes. The elimination of morbidity, disability, and pain; freedom from the need for hemostatic intervention in the context of trauma or surgery; access to all the therapeutic options available to people without a BD to manage comorbidities, especially with aging; and the opportunity to participate fully in normal family, career, and social activities have migrated from unimaginable aspirations to achievable health equity milestones [Citation1].
1.2. A novel agenda for a novel research era
Hemophilia research has entered an era of new technologies, funding opportunities, and blossoming international interest, providing an unparalleled opportunity to transform the lives of PWH and those who care for them. While the direction of past decades has been driven largely by the interests of researchers and industry, in 2020 the National Hemophilia Foundation (NHF) and the American Thrombosis and Hemostasis Network (ATHN) proposed something different. The people with inherited BDs (PWIBD) develop unique expertise every day of their lives about their disorders, and are truly lived experience experts (LEE) [Citation32], they should set the research agenda [Citation33]. Together, NHF and ATHN initiated a process to set a community-generated national research agenda that seeks to ensure all PWIBD have access to safe, effective, convenient therapeutics, diagnostics, and digital technologies [Citation33,Citation34].
1.3. Community-identified areas of priority research
NHF conducted extensive, inclusive community consultations to learn what matters most to PWIBD [Citation33,Citation34]. The NHF State of the Science Research Summit (SOSRS) Steering Committee structured the community priorities into six focus areas and recruited expert Working Groups (WG) to distill each into concrete research questions. WG1 Research Priorities for Hemophilia A and B were charged with answering the following key question: How can we use new technologies to discover therapies to improve life for PWH while working to deliver a safe and meaningful cure [Citation33,Citation34]? A framework for answering this question is depicted in .
Figure 1. Working Group 1 Hemophilia A and B schematic of community-identified areas for priority research framework POC: point of care, QoL: quality-of-life.
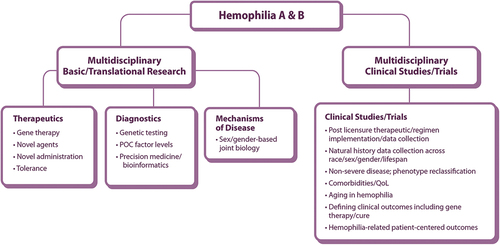
We report WG1ʹs response to this charge, incorporating insights gained through discussions with the inherited BD community at the NHF SOSRS in September 2021. We offer our conclusions to the National Research Blueprint for Inherited Bleeding Disorders so that they may accelerate progress in the areas with the greatest potential to improve the lives of PWH and their families [Citation35].
2. Methods
2.1. Working Group 1 composition
The NHF SOSRS Steering Committee recruited two Co-Chairs, with subsequent recruitment of a diverse stakeholder group (). The group set an agenda to determine the research of greatest impact for individuals and families living with inherited BDs, which requires the expertise of LEEs, medical specialists, allied healthcare professionals (HCP), and industry partners. WG1 brought together adult and pediatric hematologists, physical therapists, a pharmacist, advanced practice providers, a pain researcher, an NHF staff member, an NHF Board member, a Chapter executive director, several LEEs, a social worker, and two members from the pharmaceutical industry. An NHF support person met several times with LEEs individually and in a (virtual) group setting, to ensure that they could contribute their views and expertise. The Co-Chairs proactively invited LEE input upholding a collaborative respectful decorum.
Table 1. Members of Working Group 1 and its research priority subgroups.
2.2. Working group methodology
To accommodate diverse time zones, schedules, and pandemic restrictions, all meetings were held virtually. After several initial whole group meetings, the Co-Chairs divided WG1 into seven community-prioritized research themed subgroups led by members with extensive expertise in the area (). Subgroups met weekly with leads reporting back to the larger WG1 every two weeks to ensure coordination. Membership overlap facilitated exchange between subgroups.
Each subgroup determined hypothesis-based research questions, considered existing data, and defined questions which best encompassed the priorities identified by the community consultations. If existing data were inadequate, they proposed an approach to establish new datasets. Each subgroup generated a list of approximately 10 priority research questions.
It was anticipated that WG1 priorities would overlap with some identified by WG4 Research Priorities for the Health of People Who Have or Had the Potential to Menstruate; WG5 Diversity, Equity, and Inclusion, Health Services Research, and Implementation Science; and WG6 Facilitating Priority Research in the Inherited Bleeding Disorders Community. Periodically, WG Co-Chair meetings facilitated collaborative cross-talk.
2.3. Feasibility-impact-risk scoring
Recognizing that these deliberations would yield many more important research questions than could be considered in the short term, NHF provided the WGs with a set of scoring criteria. This matrix guided evaluation across three dimensions: feasibility, impact, and risk (F-I-R), with pre-defined scores assigned to distinct characteristics (Supplementary Table S1) [Citation33,Citation34,Citation36]. Summing the three scores allowed for a comparison of research initiatives that seek to address very different questions in very different contexts. While a question that appears ‘easy’ to address with the currently available infrastructure and expertise would have a high feasibility score, a question requiring more challenging methodology but with the potential for greater impact and characterized by low risk could yield an equally high sum score.
The standardized F-I-R rating method may generate individual total scores ranging from −10 for a question that has low feasibility, low impact, but high risk to 28 for a question with high feasibility, high impact, and low risk. WG1 added prioritization thresholds to the approach: a question scoring ≤5 was classified as low priority, 6–15 as medium priority, and any question scoring ≥16 was defined as high priority.
Individual subgroup members scored each of their respective research questions, with any grossly disparate results reconciled within the subgroup. Final scores were averaged and the result rounded to the nearest whole number for each question, and discussed by the entire WG1.
2.4. NHF State of the Science Research Summit
The work to generate a blueprint for the next decade of inherited BD research began with community consultations. After six months of intense work with the resulting themes, the WGs presented their prioritized research questions to >880 LEEs, physicians, researchers, multidisciplinary care team professionals, and federal and industry partners at the NHF State of the Science (virtual) Research Summit (SOSRS), September 12–15, 2021 [Citation33,Citation34]. Mark Skinner, JD provided a LEE perspective on living with hemophilia, Norman Fost, MD, MPH, gave a plenary on ethical issues in gene therapy, the WG1 Co-Chairs presented the group’s deliberations and resulting prioritized research questions, and Jill M. Johnsen, MD moderated a live panel discussion engaging the plenary speaker, several WG1 members, and the presenting LEE, with questions and comments from Summit attendees.
Supported by a National Heart Lung and Blood Institute (NHLBI) R13 grant (R13HL158209), NHF offered a Remote Participation Group (RPG) option for PWIBD or people who care for them, LEEs, from underrepresented segments of the community to participate in the SOSRS [Citation33]. An in-person group of 15 Black/African Americans from the Hemophilia of South Carolina NHF Chapter and another (virtual) of 3 men aging with hemophilia from the New York City Hemophilia NHF Chapter, took advantage of this facilitated review of the hemophilia A and B focus content. Reports submitted by their facilitators, along with the content of the SOSRS live panel discussion, informed the Discussion and Conclusions of this paper.
3. Results
3.1. WG1 subject subgroups
The research avenues with the potential to advance the priorities of PWH identified through community consultations were many and diverse (). Based on initial whole group discussions of the framework, the WG1 Co-Chairs identified seven key subject areas in which research advances could best meet their expressed needs:
Arthropathy/pain/bone health
Inhibitors
Diagnostics
Gene therapy
Pediatric to adult transition of care
Disparities
Cardiovascular disease
Multidisciplinary subgroups of WG1 members whose expertise aligned with the subject area () each developed a comprehensive view of the research opportunities, and then identified the specific research questions with the greatest potential to transform the lives of PWH and those who care for them.
3.2. Feasibility-impact-risk scores
The subgroups each scored their questions against the F-I-R matrix based on currently available infrastructure and expertise ( and Suppl Table S2). WG1 emphasizes that all the priority research questions identified are important and should be addressed. It is anticipated that future changes in infrastructure or expertise may increase the feasibility of some questions, and the lists may, therefore, evolve. Some opportunities found to fit more appropriately into the considerations of other WGs were shared with their Co-Chairs and not elaborated further here.
Figure 2. Plot of feasibility, impact, and risk scores of prioritized questions in WG1 subgroup subject areas.Label numbers correspond to those in , segmented circles indicate data points from different subgroups with identical coordinates.
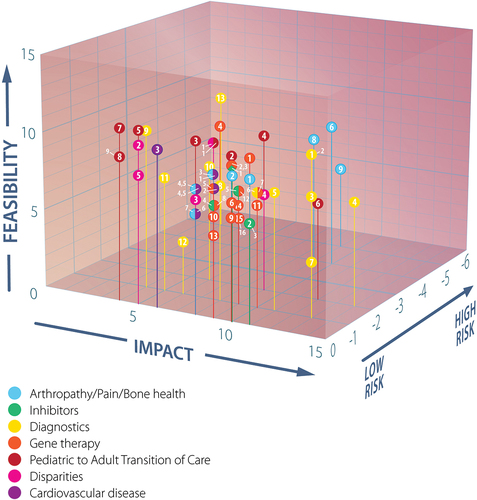
Table 2. Research questions prioritized by the Arthropathy/Pain/Bone Health subgroup
3.3. Arthropathy/pain/bone health
3.3.1. Overview of potential research
The Arthropathy/Pain/Bone Health subgroup mapped out research areas to impact hemophilic arthropathy, both pediatric and adult, across all hemophilia severities (Suppl Fig S1). They discussed chronic and acute pain and its causes identifying five investigational avenues. Regarding preventing the development of hemophilic arthropathy and intervening when it does occur, they focused on optimal management from physical therapy to pharmacologic approaches. The need for basic science investigations to elucidate the pathobiology and progression of hemophilic arthropathy was considered essential. The examination of molecular and multicellular mechanisms to develop targeted therapies to prevent and/or manage pain requires the development of suitable in vivo animal models, ex vivo human tissue models that simulate the hemophilic microenvironment, and in vitro cellular models. Bone mineral density (osteoporosis/osteopenia), and bone health in general (i.e. epiphyseal overgrowth, osteophyte development, cyst and bone bleeding) were recognized as compromised in hemophilia, with large gaps in knowledge. To that end, the subgroup questioned whether traditional bone and joint health outcome measures are sufficient to determine the true extent of disease manifestations impacting patient functionality. The development of diagnostic, prognostic, and response biomarkers, as defined by the U.S. Food and Drug Administration (FDA) and the National Institutes of Health (NIH) [Citation37], specific to hemophilia pain is required to advance early diagnosis and to monitor the potential effectiveness of interventions. Whole patient-important outcomes (e.g. pain, real-world functionality) must be prioritized and validated, and advances in imaging techniques, including magnetic resonance imaging (MRI) and musculoskeletal ultrasound (MSKUS) must be optimized for application in the clinic and/or home of the PWH.
3.3.2. Prioritized research questions
The Arthropathy/Pain/Bone Health subgroup focused this wealth of possibilities into nine research questions and subjected them to the F-I-R scoring process (). Two of the five questions that scored ≥16, the WG1 threshold for high priority, address blood clearance and joint rehabilitation after hemarthrosis. The other three deal with best post-surgical interventions to optimize recovery, the role of thrombin generation for bone health, and effects of bone strengthening agents. The remaining questions address the causes of joint pains, pain quantification and treatment, the incorporation of ultrasound imaging into practice, the role of regenerative medicine, and the type of surgeries or interventions that may mitigate arthropathy. While these four did not score ≥16, the subgroup reiterate their important research potential.
3.4. Inhibitors
3.4.1. Overview of potential research
The Inhibitors subgroup deliberations highlighted the importance of collaborative basic and translational science to better understand the mechanisms involved in inhibitor development and resolution, to be able to predictively model initial inhibitor development and the success of immune tolerance induction (ITI) to eradicate inhibitors, and for an infrastructure that empowers and enables the involvement of the entire community. Their discussions (Suppl Fig S2), included a strong awareness of the national blueprint for future research on FVIII inhibitors developed by the NHLBI State of the Science (SOS) Workshop in 2018 [Citation38–43] and shared many conclusions with this initiative. PWH and inhibitors experience greater morbidity and lower QoL than PWH without inhibitors [Citation44,Citation45], driving a need for research into musculoskeletal health, pain, and psychosocial outcomes.
Observational cohort studies and clinical trials, harnessing the power of all the -omics (genomics, proteomics, transcriptomics, metabolomics, etc.) will be key in advancing inhibitor research [Citation40]. The inherent challenge of enrolling enough participants to adequately power a study of a minority occurrence in a rare disorder underscores the need for national infrastructure that enables every PWH to participate in a cohort [Citation42], something further explored by WG6 Facilitating Priority Research in the Inherited Bleeding Disorders Community [Citation46]. The need for a national infrastructure supporting efficient data and specimen collection, analysis, and communication of results, to maximize learning from each participant [Citation39], has also been discussed by WG3 Research Priorities for Ultra-Rare Inherited Bleeding Disorders [Citation47].
3.4.2. Prioritized research questions
Four of the subgroup’s top research questions scored above the high priority threshold and one fell just below (). Determining the educational needs of PWH, their families, HCPs, and trainees, including the increasingly widespread context of a non-replacement factor therapy, scored the highest, followed by prediction algorithms for inhibitor development and optimal clotting factor exposure and ITI in the non-factor replacement era. The fifth question addresses the prospect of a potential role for gene therapy in ITI. These five clinical questions should complement the NHLBI SOS basic and translational science priorities, together providing a comprehensive road map for future hemophilia inhibitor research.
Table 3. Research questions prioritized by the Inhibitors subgroup
3.5. Diagnostics
3.5.1. Overview of potential research
This subgroup examined research into laboratory diagnostics including clotting factor levels and hemostatic potential of non-factor products, determination of genetic modifiers and biomarkers, and joint health assessment tools (Suppl Fig S3). Advancing accurate and convenient quantitation of clotting factor levels and of hemostatic potential conveyed by non-factor products at the point of care (POC), in the clinic or in the homes of PWH, and in hospitals will be critical in guiding management decisions, including in urgent situations. Improving access to testing is also of paramount importance so all PWH may benefit from these advances.
Examining genotypic-phenotypic patterns for predictors of bleeding tendency/severity, genetic modifiers of hemophilia, biomarkers of specific outcomes, and the application of the various -omics to predicting the risk of inhibitor development were identified as promising research areas. Opportunities for imaging and biomarkers in bone and joint health were discussed. The Diagnostics subgroup also debated whether the results of the most common joint health assessment tools truly correlate with the outcomes of greatest importance to PWH. They propose applying the same scrutiny to common imaging techniques.
3.5.2. Prioritized research questions
The Diagnostics subgroup formulated 13 top priorities as research initiatives to be accomplished, rather than questions (). Six scored above the high priority threshold, three of which seek to exploit advances in imaging technology encompassing POC MSKUS, including PWH self-imaging, and novel MRI sequences for hemosiderin quantification to assess iron clearance. The other three propose rapid and reliable tests for bleed detection and the accurate prediction of bleeding phenotypes. Research priorities of lesser importance were lab tests to universally assess hemostatic potential and measure the risk of bleeding, the relationship between clotting factor levels and bleed propensity, the impact of routine clinical and imaging assessments on outcomes, the occurrence of anti-drug antibodies to non-factor replacement therapies, companion tests, and the impact of differential tissue distribution of FIX products in hemophilia B.
Table 4. Research initiatives prioritized by the Diagnostics subgroup
3.6. Gene therapy
3.6.1. Overview of potential research
With several phase 3 clinical trial programs in hemophilia A and B currently ongoing, two of which have (at the time of writing) lead to European or US regulatory approval [Citation24–31], the Gene Therapy subgroup took a wide view of how advances in this field might benefit as many PWH as possible (Suppl Fig S4). They considered the technical questions defining gene therapy efficacy and safety; how issues of patient education, health literacy, and the PWH experience will shape its impact; as well as determinants of access including eligibility, implementation, and payment details.
The subgroup concurred that the European Haemophilia Safety Surveillance (EUHASS) defined adverse events for the monitoring of hemophilia treatments should center the assessment of gene therapy safety [Citation48]. Efficacy determinations of this novel treatment approach must include patient-important outcomes such as hemarthropathy and health-related QoL as well as clinical outcomes. The potential for gene therapy to be a one-time, life altering treatment makes optimization of patient education and understanding the physical and mental health impacts essential.
The subgroup also considered practical aspects of the implementation of gene therapy including infrastructure, guidelines, and training for delivery and follow-up in diverse healthcare and payment settings. Demonstrating cost-effectiveness, optimizing operational requirements, and studies of multiple alternative reimbursement strategies should contribute to facilitating patient access.
3.6.2. Prioritized research questions
Of the 16 questions prioritized by the Gene Therapy subgroup (), the top six all focus on maximizing the benefit for PWH. The other high scoring questions seek to expand eligibility to wider populations of PWH. The questions addressing safety and comorbidities scored the lowest as they are associated with higher risk (which scores negatively), however this does not diminish their importance. Gene therapy offers an exciting advancement in the treatment of hemophilia, and the subgroup emphasizes the importance of addressing all 16 questions to offer the best care and outcomes to all PWH.
Table 5. Research questions prioritized by the Gene Therapy subgroup
3.7. Pediatric to adult transition of care
3.7.1. Overview of potential research
The Pediatric to Adult Transition of Care subgroup considered that successful transfer of PWH from pediatric to adult care not only incorporates the physical transfer of care but also medical independence for young adults, in the context of the different models of hemophilia treatment center (HTC) care within the US healthcare system (Suppl Fig S5). Defining relevant terms and nomenclature, identifying milestones and graduation requirements, and establishing measures of transition success are key. These specifications should align with the Institute of Health Improvement (IHI) Triple Aim: improving the experience of care, improving the health of populations, and reducing per capita costs of health care [Citation49], as well as the objectives of the Healthy People 2030 initiative of the US Department of Health and Human Services [Citation50].
The subgroup acknowledges that transition tools currently exist but asserts that they require immediate and regular updates. The development and dissemination (including beyond the HTC network since some young adults choose to establish or continue care in practices outside an HTC setting) of updated more inclusive tools constitute a second research focus, addressing different severities, genders, and transitional steps for PWIBD, their families, and HCPs. Flexible educational resources for all stakeholders are also needed. Methods to assess transition readiness and tools, including electronic medical records and/or the ATHN data collection and analysis infrastructure [Citation51], to track and ensure a smooth transition were highlighted. Standardized longitudinal data collection on the transition population and their outcomes in a national database would facilitate characterization of predictors of success and identification of gaps in care. The subgroup remarked that this need is not particular to PWH, but shared across all BDs and refers the reader also to the deliberations of WG2 on the management of von Willebrand disease (VWD), platelet dysfunctions, and other mucocutaneous BDs across the lifespan [Citation52], and that of WG4 on the issues particular to people who have or had the potential to menstruate with BDs [Citation53].
3.7.2. Prioritized research questions
The subgroup formulated nine specific research questions with six reaching or surpassing the F-I-R score high priority threshold (). The top-ranking question recognizes the physical, emotional, and social aspects of transition and seeks to serve the entire target population, while the second underscores medical independence as a critical component. The remainder of the research questions build upon the definition of successful transition and seek to identify keys to this success.
Table 6. Research questions prioritized by the Pediatric to Adult Transition of Care subgroup
3.8. Disparities
3.8.1. Overview of potential research
This subgroup’s discussions fell into two categories: disparities experienced relative to the general population (Suppl Fig S6A) and within the hemophilia community (Suppl Fig S6B). This report develops the latter. Recognizing, characterizing, and actively resolving these disparities is essential to achieving health equity and to the effectiveness of every aspect of diagnosis, treatment, and care.
Understanding disparities in access to HTC resources, where so much of hemophilia management happens in partnership with PWH [Citation17], is key. The impact of geographic proximity, outreach programs, barriers to accessing funds from the US Health Resources & Services Administration 340B Drug Pricing on outcomes constitute one promising line of investigation. Understanding how demographics such as sex/gender, race, and ethnicity impact the experiences of PWH in the healthcare system, and on their outcomes, requires detailed study. Research into different types of health insurance as a source of inequity has the potential to profoundly impact the lives of PWH.
3.8.2. Prioritized research questions
The subgroup formulated five questions investigating disparities specific to the hemophilia community, with one scoring above the threshold of 16 (). Since increasing numbers of females are being recognized and diagnosed with hemophilia [Citation54], the subgroup prioritized determining the differences in the HTC comprehensive care experiences of males and females with similar clotting factor levels. The second-ranked question asks about the determinants considered by providers when selecting a diagnosis of mild hemophilia versus hemophilia carrier, for affected girls and women, a distinction that may impact access to, and insurance coverage of, therapeutics and care. Benefits of two questions regarding the impact of access to physical therapy (PT) and/or MSKUS resources on joint outcomes are expected to extend to other inherited BDs.
Table 7. Research questions prioritized by the Disparities subgroup
Table 8. Research questions prioritized by the Cardiovascular Disease subgroup
The subgroup formulated research questions around the many disparities that also impact people with other inherited BDs, presented in the lower portion of , but did not score them. These questions address clinical trial access, HTC staffing and funding, and inherited BDs that may be associated with specific cultural or ethnic populations. The group also discussed disparities faced by gender diverse people and related to health literacy, more completely addressed by WG4 Research Priorities for the Health of People Who Have or Had the Potential to Menstruate [Citation53] and WG5 Diversity, Equity, and Inclusion; Health Services Research; and Implementation Science [Citation55].
3.9. Cardiovascular disease
3.9.1. Overview of potential research
Understanding cardiovascular disease and its management in people aging with hemophilia is more important than ever as many PWH in the US now enjoy a life expectancy comparable to that of men without a BD [Citation16,Citation17,Citation56]. The evidence base in this field is particularly thin, since aging with hemophilia is a relatively new phenomenon. The subgroup mapped out a multitude of promising research avenues (Suppl Fig S7).
Hypertension has a higher prevalence in PWH compared to the general male population [Citation57–59], and carries a high mortality risk from intracranial hemorrhage [Citation60], which is known to be exacerbated with elevated blood pressure in the general population [Citation61]. Therefore, investigations of its etiology and pathophysiology were considered particularly urgent. To this end, better delineation of cardiovascular risk factors in PWH through large-scale surveys, perhaps querying the ATHNdataset [Citation51], should inform impactful quality improvement (QI) programs. Similarly, it will be important to elucidate the ideal assessment and management of atrial fibrillation and myocardial infarction in PWH, including the potential role for novel non-factor and factor therapies when antiplatelet and anticoagulation treatments are required.
3.9.2. Prioritized research questions
The top-ranking question of six formulated by the subgroup (), seeks to clarify the relationship between hypertension, blood pressure, hemorrhagic stroke, and death. The remaining questions address further elucidation of hypertension and cardiovascular health stratified by hemophilia severity and other risk factors, as well as the role of non-factor replacement therapy for management of cardiovascular conditions (e.g. atrial fibrillation), perhaps affording more optimal antiplatelet and anticoagulant treatments. These questions garnered high feasibility scores and interest from all stakeholders, prompting optimism that they may be addressed in the near future. An expectation that studying the pathophysiology of hypertension in hemophilia will require considerable basic science, including work with animal models, contributed to a lower overall score for these questions. The evolving hemophilia treatment landscape drove the prioritization of research into the potential for safer anticoagulation in PWH to manage cardiovascular disease.
4. Discussion
Over several decades, research and advances in technology and care innovations have transformed hemophilia from a largely pediatric condition [Citation3] into a lifelong disorder with a life expectancy comparable that of people without a BD for many in the US [Citation16,Citation17,Citation56]. At the dawn of this new era, hemophilia research priorities must now go beyond minimizing bleeding rates and increasing survival to transforming the lives of all PWH and those who care for them to an experience of health equity, and a life unencumbered by disease [Citation1,Citation62–64]. In addition to continuing to push toward a cure for hemophilia, research must also further the understanding and management of comorbidities such as inhibitors and joint and cardiovascular disease, and optimize diagnostic laboratory, imaging, and other assessment tools with a new focus on psychosocial and QoL outcomes. Moreover, any research endeavors must be all inclusive, dismantling the barriers to access and care that deny certain populations within the hemophilia community the hope of health equity.
Each WG1 subgroup () generated a list of research questions () through extensive scrutiny of their subject area in response to the community-identified priorities () and sorted them via a feasibility-impact-risk scoring matrix (Suppl Tables S1 and S2) [Citation33,Citation36]. WG1 would, however, like to underscore the importance of all research questions formulated by each subgroup, regardless of ranking order. Requirements for randomized controlled trials, extensive infrastructure, intensive basic lab science including animal models, and/or expertise beyond that which is currently available lowered the feasibility scores of some questions, highlighting the need for investment in the priorities established by WG6 Facilitating Priority Research in the Inherited Bleeding Disorders Community [Citation46]. WG1 hopes that such advances will increase the feasibility of numerous priority questions identified by WGs 1–5, such as those defined by WG4 for the health of people who have or had the potential to menstruate affected by hemophilia [Citation46,Citation47,Citation52,Citation53,Citation55].
4.1. Thriving despite lifelong comorbidities
Research priorities for a lifelong disorder must include optimizing management of its comorbidities [Citation65] and target improvements in outcomes that allow PWH and their families to thrive throughout their lives. Recent advances in technological capabilities, especially pertaining to POC MSKUS, and therapeutics offer new research avenues with the potential to impact health concerns shared with the general aging population such as cardiovascular risk factors like hypertension, inhibitors, hemarthropathy, and reduced mobility. The medical independence of PWH and continuity of care, integrating the management of increasingly present or complex comorbidities with the management of their BD, must be considerations in the transition to and progression of this later life phase.
Though one novel therapeutic currently provides an alternative to factor replacement, the development of antibodies (inhibitors) to exogenously administered FVIII remains the most common treatment side effect occurring in up to 25–30% of people with severe hemophilia A. Inhibitors typically develop in young children during early exposure to clotting factor [Citation66]. PWH and inhibitors experience greater morbidity and lower QoL than PWH without inhibitors [Citation44,Citation45]. While a smaller proportion (1.5–3%) of PWH B develop inhibitors to replacement FIX, they are associated with equally grave outcomes with frequent allergic/anaphylactic reactions and nephrotic syndrome [Citation22]. Eradication of inhibitors is more complex and less often successful in PWH B [Citation67,Citation68]. At the time of writing, no non-factor alternative has been approved for PWH B and inhibitors. The top research priority identified by WG1 in this context is a better understanding of the education needs of PWH and their families, and of trainees and HCPs. A significant number of PWH are at risk for or currently live with inhibitors, or will develop them, and optimizing bidirectional communication and transmission of information regarding inhibitor recognition and diagnosis and management to minimize the negative impact and maximize their ability to live unhindered full lives is of utmost importance. Understanding the risk factors for the development of inhibitors and the optimal initial exposure to exogenous factor in young children in order to avoid inhibitor development, and the best approach to inhibitor eradication, all in the current treatment landscape, including non-factor replacement therapies, will be key to minimizing the negative impacts of inhibitors in the future. Achieving these research priorities will require large-scale collaborative observational cohort studies, clinical trials, and basic and translational science as established by the national blueprint for future research on FVIII inhibitors developed by the NHLBI SOS Workshop in 2018 [Citation38–43].
Pain, reduced mobility, and functional impairment associated with the progressive joint deterioration and hemophilic arthropathy that result from bleeding into joints negatively impact clinical outcomes and the QoL of PWH [Citation69], and were consistently highly prioritized in the community consultations upon which this research agenda is founded [Citation33]. Improving our understanding of how to facilitate clearance of blood/iron from the joint, to prevent/minimize the damage caused by joint bleeds, and to promote regeneration and rehabilitation stands to meet precisely these needs. There is optimism that treatment with EHL recombinant factors, non-factor replacement therapies, and potentially gene therapy, will effectively convert severe hemophilia into mild or moderate hemophilia with a substantial reduction in joint bleeds and damage for many [Citation70]. The effects of EHL and non-factor replacement products on joint preservation are not yet fully characterized, and joint bleeds in people with mild or moderate hemophilia may be less frequent but are not without damaging sequelae. Joint function and pain will continue to be important preoccupations of PWH for at least the near future requiring continuing and evolving support from the multidisciplinary care team [Citation9,Citation70]. Understanding the impact of access to musculoskeletal professionals, such as physical therapists, on outcomes will inform the evolution of the HTC model of care.
The management of hypertension in PWH exemplifies the need for collaborative care that extends beyond the traditionally defined HTC. PWH tend to have higher blood pressure than the general male population. The etiology remains poorly understood and the phenomenon is underrecognized [Citation19,Citation57–59]. Primary care physicians must be made aware of the urgency of controlling high blood pressure in, even young, PWH since it increases the risk for hemorrhagic stroke, a major cause of death in this population [Citation60,Citation61]. The increased incidence of certain risk factors in some populations (e.g. African American PWH), obstacles to accessing or paying for the required medications (e.g. type or lack of insurance), and lack of comprehension of the seriousness of the condition (e.g. younger PWH) necessitate a collaborative management plan between the primary care physician, the HTC multidisciplinary care team, and the PWH. Improving education and awareness with primary care physicians and PWH is even more important for individuals not cared for at an HTC.
4.2. Collaborative research and care delivery
Collaboration will be key to achieving the research priorities with the greatest potential to transform the lives of PWH. From defining biomarkers of joint health to detailing predictive models of inhibitor development to understanding the risk factors for cardiovascular disease in the context of hemophilia, multi-center longitudinal observational cohort studies and clinical trials, coordinated data collection and analysis, and optimization of specimen collection and assay protocols will maximize the contributions from each of the often too few participants. ATHN offers a platform of standardized integrated systems, data, and processes across >140 HTCs upon which novel collaborative inclusive research may be efficiently initiated, decreasing the time and cost required to implement significant projects [Citation71,Citation72]. Engaging expertise beyond that of the inherited BD community will be essential to team science (e.g. addressing the combined concerns of hemophilia and hypertension), informing coordinated comprehensive care for PWH throughout their lives. Collaboration must extend beyond the HTC; this joint effort must not only engage federal partners, but also primary care physicians, hematologists, and PWH whose care is not within the HTC network.
PWIBD and care partners, true LEEs [Citation32], have an invaluable perspective to contribute to every phase of research and care. The deliberations of WG1 were inspired and enriched by the involvement of LEEs and their input impacted the direction and scoring of multiple top priority questions. This was an important first step; continued involvement is essential. Some opportunities include involving LEEs of all ages and diverse populations in developing tools for successful transition or education around gene therapy trials; engaging aging PWH in the design of studies concerning joint health; and proactively recruiting African American PWH, who are less likely to engage with healthcare systems, into care and research initiatives.
Patient organizations, like NHF and Hemophilia Federation of America, are also key drivers of the community outreach, advocacy, and education that underpins research. The NHF initiative, Community Voices in Research [Citation73], facilitates data collection directly from community members regardless of association with an HTC and must be capitalized upon. The wealth of LEE expertise creates an opportunity for NHF to contribute to the communication of results and benefits of research to the community, and to the education of HCPs to inform care.
4.3. Capitalizing upon current and horizon treatments
Non-factor replacement and gene therapies hold the potential to transform many outcomes for PWH by creating a hemostatic balance. This may be particularly helpful for the management of cardiovascular disease. While PWH appear to be at lower risk of thrombosis, they are not exempt from cardiovascular diseases common to aging such as ischemic heart disease, myocardial infarction, and atrial fibrillation [Citation65,Citation74–76]. In the general population thrombotic comorbidities are most often managed with anticoagulants, and/or antiplatelet agents, all of which have historically been contraindicated in clotting factor deficiencies [Citation19,Citation77]. Therapeutics that rebalance hemostasis without employing replacement factor present an exciting new possibility, perhaps creating a sufficiently procoagulant steady-state to allow anticoagulant and/or antiplatelet management for cardiovascular comorbidities. For instance, rebalancing therapeutics may establish safe conditions for stenting, which requires anticoagulants and/or antiplatelet agents.
Gene therapy appears closer than ever on the horizon of the hemophilia treatment landscape [Citation26,Citation30]. The promise of a possible ‘functional cure’ is monumental and while data continue to be collected in clinical trials to ensure the reliable and safe expression of sustained predictable factor levels, the anticipation in the community is palpable [Citation24,Citation25,Citation27,Citation29,Citation78]. The wellbeing of PWH and their families and the principles of health equity must remain paramount in gene therapy. The ultimate goal must be the best possible psychosocial and clinical outcomes for all PWH. This requires optimization of educational interventions and consenting methods especially for different age groups and cultural contexts, empowerment and knowledge, pathways of advocacy, and mental health and psychosocial initiatives. It also requires investigations into operational methodologies that uphold equity, affordability, and fairness for all stakeholders, and into technical aspects that facilitate expansion of eligible populations (e.g. those with AAV antibodies, children, women, PWH and inhibitors). Here too, the expertise of a diversity of LEEs will be invaluable in understanding and meeting the psychosocial, education, and communication needs of this exceptional context.
4.4. Harnessing technical advances to improve diagnostics and testing
More sophisticated, accurate, and convenient assessment and diagnostic tools must accompany advances in therapeutic opportunities and management objectives. Important opportunities exist in the realm of imaging and factor activity/hemostatic potential determinations, as well as biomarkers and functional evaluations.
Accurate detection and characterization of joint bleeding (including subclinical) episodes, and their resolution is key to evaluating the efficacy of therapies and to aligning joint health management with the priorities of PWH. Recent advances in MSKUS techniques (comprehensively reviewed in [Citation79]) have yielded protocols for the assessment of longitudinal joint health, disease activity, and even osteochondral derangement. MSKUS has emerged as a critical adjunct to clinical evaluation of acute bleeding episodes [Citation80,Citation81], due to its ability to detect small volumes of blood in the joint [Citation82], for POC and full diagnostic scanning, and perhaps even for early detection of clinically significant joint disease [Citation79]. Developing and demonstrating the value of MSKUS home self-imaging constitutes an exciting opportunity to harness technological advances to improve the daily QoL and clinical outcomes of PWH [Citation83]. The advent of mobile handheld devices should remove some barriers imposed by equipment costs and technical requirements. To further delineate the clearance of blood products in the joint, especially important with the advent of non-factor treatments or gene therapy, it is particularly exciting to pursue the development of novel MRI sequences for quantitative iron assessment. Current MRI sequences only allow semi-quantitative hemosiderin scoring, which is insufficient to detect dynamic changes over time.
As novel therapies stand poised to potentially revolutionize hemophilia treatment paradigms, the accurate determination of clotting factor activity levels and hemostatic potential in the context of diverse factor and non-factor replacement products, and gene therapy, is critically important [Citation84]. Just as with self-imaging, POC tools for near-patient use should be developed to streamline patient care in a timely and convenient manner.
4.5. Psychosocial and quality-of-life outcomes to define the new standard of care
Mental health, psychosocial outcomes, activities of daily living, and QoL rank among the highest priorities of PWH [Citation33]. The exciting improvements in clinical outcomes promised by current advances must also progress meeting these expressed needs of PWH [Citation1,Citation64]. In addition to clinical advances reducing lifestyle restrictions, WG1 identified numerous such opportunities in transition and health disparities research. Psychosocial and QoL outcomes must be front and center in the objectives of hemophilia research, and not simply an assumed but unconsidered byproduct of the focus on clinical outcomes.
The successful transition from pediatric to adult care is a key milestone in the lives of PWH and those who care for them [Citation85,Citation86]. Defining the success of this transition must include the acquisition of medical independence, the physical transfer of care, but also outcomes of daily QoL and personal wellbeing. All PWH, regardless of severity and gender, must be equally empowered to thrive, pursuing education, career, family, and social aspirations, by the advances sweeping the field, and transition research must contribute to a knowledge base that informs this.
Addressing the many disparities encountered by the community is essential to achieving health equity for all PWH. Hemophilia treatment products are very expensive. The annual cost of prophylaxis with replacement factor products for one individual in the US is in the range of at least US$ 500,000 [Citation87,Citation88]. Hemophilia gene therapies are expected to cost in the order of US$ 2,000,000–3,000,000 and the single non-factor replacement therapy currently approved by regulators, emicizumab, is estimated to cost over US$ 500,000 per year [Citation87,Citation88]. These high costs impact fair and equitable access to appropriate treatment for PWH. As further explored by WG5 Diversity, Equity, and Inclusion; Health Services Research; and Implementation Science, it will be important to investigate how insurance coverage may affect access to care, treatment, and outcomes [Citation55]. Lower costs of all hemophilia treatment products would be expected to increase opportunities for PWH from all socioeconomic contexts to access optimal care [Citation87].
Keen to capitalize upon the promise of novel therapeutic options, PWH often seek guidance on how to safely become more physically active. Research into the impact of differential availability of physical therapy professionals and advanced imaging services in HTCs on functional and pain outcomes, and the institutional and financial barriers that influence who can access these resources, has the potential to inform meaningful change. Similarly, understanding the barriers to diagnosis and care for women and girls with hemophilia has enormous potential to improve their lives. Understanding what might influence a woman or girl’s decision to consult an HCP about potentially abnormal bleeding, how their HTC care experience differs from that of their male counterparts, the implications of being labeled as a carrier versus having mild hemophilia, and the repercussions for insurance and access to therapies and specialized care all merit further study.
The HTC is also well placed to address disparities in inclusion in clinical trials for hemophilia subpopulations. An absence of data about underrepresented populations will deepen existing inequities in benefiting from advances in hemophilia research. The disparity between PWH receiving care through an HTC and those who do not is particularly challenging for HTCs to address, however HTCs must not shy away from this challenge. Together with LEEs, patient organizations, and primary care physicians, they can identify the barriers to equitable care for PWH outside of HTCs and the keys to either bringing them into the HTC sphere of care or extending that sphere out to meet them where they are.
LEE perspective
Being invited to the table to build research priorities as a voice in this patient-centric, transformative project was truly an honor. From the beginning, we were invited to take part in the prioritization of research, to––not just listen––but actively provide input into what questions the research teams were looking to address, while also providing our own research priorities, based on our extensive knowledge about living with bleeding disorders and as representatives of the bleeding disorders community.This inclusive approach allowed for unique perspectives to be brought to the table, so researchers could address priorities that otherwise may have not been on their radar and issues they may not have realized affected the bleeding disorders community. The purpose of our involvement from the beginning was very clear: ‘We want and desire to have your voice heard in the direction of the future research priorities.’ This was not a tokenistic involvement; it was a real chance to provide our input on what is important to the community. As a result, several of the key priorities, were implemented, discovered, and analyzed from the LEE voice.To see the final manuscript, knowing we had a direct hand in the final project outcome, was empowering. The commitment of the research and medical WG members should be noted, a commitment to ensuring the voice of the PEOPLE with bleeding disorders is present as we collectively move research into the future.
5. Conclusions
The fundamental goal of hemophilia research has changed from achieving minimal standards of survivability to health equity for all PWH. This means prioritizing the studies that seek to improve outcomes that matter most to PWH such as living long full lives without pain, with spontaneity, and free from fear, lives that are not restricted by hemophilia. These studies must incorporate PWH of all severities, at all life stages, and from all populations and socioeconomic contexts. Complete inclusivity also means intentionally seeking out, defining, and dismantling the barriers to optimal care and participation in research faced by diverse populations within the inherited BD community. As long as any group is underrepresented in research and has unequal access to care, all the dramatic and exciting advances in hemophilia research fail to achieve health equity.
Hemophilia has set the bar high for multidisciplinary comprehensive care with the PWH as an empowered partner. Similarly, the hemophilia research blueprint for the coming decades must be determined by inclusive multidisciplinary deliberations complete with LEE expertise, and its successful execution will hinge upon coordinated collaboration between all stakeholder groups. WG1 was inspired by the passionate engagement of all its members in the work that led to the NHF SOSRS and this manuscript. Hailing from different professions, areas of expertise, and perspectives within the inherited BD community, all members were united in their commitment to health equity for all PWIBD. Carrying this passionate collaboration forward and expanding it to include ever more stakeholders and contributors promises to fuel the execution of a National Research Blueprint that truly serves the community whose interests lie at its core.
List of abbreviations
Declaration of interests
The authors are integrated members of the inherited bleeding disorders community: people with inherited bleeding disorders, their family members, health care providers and researchers (including physicians, nurses, physical therapists, pharmacists, social workers/psychologists, geneticists/genetic counselors, etc.), industry partners, government officials/regulators, local community organization representatives and others.
DQ Tran Jr. discloses: participation on a Data Safety Monitoring Board or Advisory Board: Bayer, Biomarin, Genentech, HEMA Biologics, Takeda, UniQure. C Benson discloses: all support for the present manuscript: Sanofi; consulting Fees: Annette von Drygalski; patents planned, issued or pending: Sanofi; stock or stock options: Sanofi. J Boice discloses: support for attending meetings and/or travel: Tremeau Pharmaceuticals, Inc.; other financial or non-financial interests: employee of Tremeau Pharmaceuticals, Inc. M Chitlur discloses: grants or contracts from any entity: Genentech, Agios, Inc., Novartis, Inc.; participation on a Data Safety Monitoring Board or Advisory Board: PUMPKIN, NovoNordisk, BPL, Inc., Genentech, Inc., Sanofi, Agios Pharmaceuticals; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: FWGBD. A Dunn discloses: all support for the present manuscript (e.g. funding, provision of study materials, medical writing, article processing charges, etc.): National Hemophilia Foundation; grants or contracts from any entity: Biomarin, Pfizer, Sanofi, Novo Nordisk, Freeline, Genentech, Takeda, ATHN; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Biomarin, France Foundation; support for attending meetings and/or travel: World Federation of Hemophilia, National Hemophilia Foundation; participation on a Data Safety Monitoring Board or Advisory Board: Uniqure; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: National Hemophilia Foundation-MASAC chair, Board member, World Federation of Hemophilia-USA, Board Member. MA Escobar discloses: all support for the present manuscript: National Hemophilia Foundation; grants or contracts from any entity: Takeda, UniQure, Bayer, Sanofi, NovoNordisk; consulting Fees: National Hemophilia Foundation; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Takeda, NovoNordisk, Genentech, Biomarin, Bayer; participation on a Data Safety Monitoring Board or Advisory Board: NovoNordisk, Takeda, Pfizer, Bayer, Biomarin, Genentech, Sanofi, CSL Behring, Hemabiologics/LFB. K Gupta discloses: all support for the present manuscript: ICTS, Univ California, Irvine, (UCI) CA, NIH R01 HL147562, NIH R01 CA263806; grants or contracts from any entity: Novartis, Cyclerion, Yale University. 1910 Genetics; royalties or licenses: 1910 Genetics; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Novartis, CSL Behring; support for attending meetings and/or travel: Novartis; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Member, Diversity and Equity Initiative, American Society of Hematology; receipt of equipment, materials, drugs, medical writing, gifts or other services: Gencore. SD Martin disclosures: leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Board Chair of NHF. S Martin discloses: support for attending meetings and/or travel: Bleeding Disorders Association of South Carolina; participation on a Data Safety Monitoring Board or Advisory Board: LEE with NHF; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Bleeding Disorders Association of South Carolina, SC Advocacy Coalition. S Meeks discloses: grants or contracts from any entity: NIH, HTRS, NHF, Hemophilia of Georgia, Octapharma, Genentech; Consulting fees: Spark, Teraimmune; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: WFH, ISTH, THSNA; participation on a Data Safety Monitoring Board or Advisory Board: Pfizer, Takeda, CSL Behring, Biomarin, Genentech; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: HTRS, MASAC. A Narvaez, Jr. discloses: payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: HFA; support for attending meetings and/or travel: National Hemophilia Foundation. D Quon discloses: consulting fees: Bayer, Biomarin, Genentech, Novo Nordisk, Sanofi, Takeda; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Biomarin, Genentech, Novo Nordisk, Sanofi, Takeda. M Reding discloses: grants or contracts from any entity: Bayer, Biomarin; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Bayer, Biomarin, CSL Behring, Sanofi Genzyme, Takeda; participation on a Data Safety Monitoring Board or Advisory Board: Bayer, Biomarin, CSL Behring, HEMA Biologics, Novo Nordisk, Sanofi Genzyme, Takeda. U Reiss discloses: all support for the present manuscript: ALSAC; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: ATHN. B Savage discloses: consulting fees: Novo Nordisk, Inc; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Novo Nordisk, Inc; support for attending meetings and/or travel: National Hemophilia Foundation; participation on a Data Safety Monitoring Board or Advisory Board: Genentech USA, Inc; leadership or fiduciary role in other board, society, committee, or advocacy group, paid or unpaid: National Hemophilia Foundation. K Schafer discloses: consulting fees: Bayer, Genentech, Novo Nordisk, Takeda, Biomarin; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: HFA, Novo Nordisk, Biomarin. B Steiner discloses: grants or contracts from any entity: Genentech, Bioverativ, Pfizer; Consulting fees: Uniqure; Payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Sanofi. C Thornburg discloses: grants or contracts from any entity: National Hemophilia Foundation, Biomarin, Center for Inherited Blood Disorders (CDC and HRSA funding distributed through regional center), ATHN, Novo Nordisk, Sanofi Genzyme/Alnylam; consulting fees: Biomarin, HemaBiologics, Takeda, Octapharma, CSL Behring, Sanofi Genzyme, Genentech; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Vindico Medical Education, LLC, MED-IQ, Genentech; support for attending meetings and/or travel: National Hemophilia Foundation, ATHN; participation on a Data Safety Monitoring Board or Advisory Board: Bluebird Bio, Cyclerion; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: National Hemophilia Foundation. A von Drygalski discloses: all support for the present manuscript (e.g. funding, provision of study materials, medical writing, article processing charges, etc.): National Hemophilia Foundation; grants or contracts from any entity: Pfizer, Sanofi; Consulting fees: Biomarin, CSL-Behring, Sanofi, Takeda, Regeneron, ASC Therapeutics, Uniqure, Novo Nordisk. LM Volland is an employee of NHF. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
A von Drygalski and DQ Tran led the working group recruitment, organization, discussions, and analysis. They divided WG1 into subgroups led by A von Drygalski, B Steiner, C Thornburg, DQ Tran, DV Quon, SL Meeks, and UM Reiss; and coordinated and synthesized the output of the subgroups. All authors contributed to analysis and deliberations. DQ Tran and A von Drygalski led manuscript preparation, all authors offered input and contributed to revisions of the manuscript, approved the final version to be published, and agree to be accountable for all aspects of the work. SD Martin and S Martin led writing of the Plain Language Summary and LEE Perspective sections.
Presentation of content
DQ Tran and A von Drygalski presented highlights of the deliberations of Working Group 1 at the National Hemophilia Foundation State of the Science (virtual) Research Summit (SOSRS), September 12–15, 2021. Summit discussions informed the Discussion and Conclusions of this paper.
Supplemental Material
Download Zip (5.8 MB)Acknowledgments
The Executive Committee of the National Hemophilia Foundation (NHF) National Research Blueprint initiative were actively engaged in the conception, design, preparation, and oversight of each of the State of the Science manuscripts in this supplement. Maria E. Santaella actively engaged with the lived experience expert (LEE) WG members throughout the process, empowering their inclusion and participation. The Executive Committee consisted of: Kevin Mills, Michael Recht, Michelle L. Witkop, Maria E. Santaella, Donna DiMichele, Keri L. Norris, Esmeralda Vázquez and Brett Spitale. The authors thank Kevin Mills, PhD for his review of the manuscript.
Fiona Robinson, PhD provided professional medical writing support during manuscript development; medical illustrations were created by Matt Evans; both paid by NHF.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17474086.2023.2171981
Additional information
Funding
References
- Skinner MW, Nugent D, Wilton P, et al. Achieving the unimaginable: health equity in haemophilia. Haemophilia. 2020;26(1):17–24.
- Pierce GF. Uncertainty in an era of transformative therapy for haemophilia: addressing the unknowns. Haemophilia. 2021;27(Suppl 3):103–113.
- Larsson SA. Life expectancy of swedish haemophiliacs, 1831-1980. Br J Haematol. 1985;59(4):593–602.
- Jones PK, Ratnoff OD. The changing prognosis of classic hemophilia (factor VIII “deficiency”). Ann Intern Med. 1991;114(8):641–648.
- Farrugia A, Liumbruno GM, Candura F, et al. Factors affecting the quality, safety and marketing approval of clotting factor concentrates for haemophilia. Blood Transfus. 2018;16(6):525–534.
- Raso S, Hermans C. Recombinant factor VIII: past, present and future of treatment of hemophilia A. Drugs Today (Barc). 2018;54(4):269–281.
- Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe haemophilia should be started at an early age but can be individualized. Br J Haematol. 1999;105(4):1109–1113.
- Skolnick AA. Hemophilia foundation recommends prophylactic use of clotting factors. JAMA. 1994;272(15):1153–1154.
- Gualtierotti R, Solimeno LP, Peyvandi F. Hemophilic arthropathy: current knowledge and future perspectives. J Thromb Haemost. 2021;19(9):2112–2121.
- Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544.
- Warren BB, Thornhill D, Stein J, et al. Young adult outcomes of childhood prophylaxis for severe hemophilia A: results of the joint outcome continuation study. Blood Adv. 2020;4(11):2451–2459.
- Manco-Johnson MJ, Lundin B, Funk S, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. J Thromb Haemost. 2017;15(11):2115–2124.
- Mahlangu J, Young G, Hermans C, et al. Defining extended half-life rFVIII-A critical review of the evidence. Haemophilia. 2018;24(3):348–358.
- Peters R, Harris T. Advances and innovations in haemophilia treatment. Nat Rev Drug Discov. 2018;17(7):493–508.
- Mahlangu JN, Blanchette V, Klamroth R. Redefining prophylaxis in the modern era. Haemophilia. 2021;27(Suppl 3):21–27.
- Oldenburg J, Dolan G, Lemm G. Haemophilia care then, now and in the future. Haemophilia. 2009;15(Suppl 1):2–7.
- Valentino L, Baker J, Butler R, et al. Integrated hemophilia patient care via a national network of care centers in the United States: a model for rare coagulation disorders. J Blood Med. 2021;12:897–911.
- Ferraris VA, Boral LI, Cohen AJ, et al. Consensus review of the treatment of cardiovascular disease in people with hemophilia A and B. Cardiol Rev. 2015;23(2):53–68.
- Seaman CD, Apostolova M, Yabes J, et al. Prevalence and risk factors associated with hypertension in hemophilia: cross-sectional analysis of a national discharge register. Clin Appl Thromb Hemost. 2017;23(7):871–875.
- Hay CR, Palmer B, Chalmers E, et al. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. 2011;117(23):6367–6370.
- Peyvandi F, Miri S, Garagiola I. Immune responses to plasma-derived versus recombinant FVIII products. Front Immunol. 2020;11:591878.
- Santoro C, Quintavalle G, Castaman G, et al. Inhibitors in hemophilia B. Semin Thromb Hemost. 2018;44(6):578–589.
- Shima M, Sidonio RF Jr. Substitution therapy. Haemophilia. 2021;27(Suppl 3):53–59.
- Batty P, Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere. 2021;5(3):e540.
- Croteau SE, Wang M, Wheeler AP. clinical trials update: innovations in hemophilia therapy. Am J Hematol. 2021;96(1):128–144.
- European Medicines Agency. First gene therapy to treat severe haemophilia A 2022 [cited 2022 Jul 20]. Available from: https://www.ema.europa.eu/en/news/first-gene-therapy-treat-severe-haemophilia.
- Garrison LP, Kleinermans D. Is the world ready for gene therapy? Haemophilia. 2022;28(Suppl 2):5–8.
- Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N Engl J Med. 2022;386(11):1013–1025.
- Rodriguez-Merchan EC, De Pablo-Moreno JA, Liras A. Gene therapy in hemophilia: recent advances. Int J Mol Sci. 2021;22:14.
- U.S. Food and Drug Administration. FDA approves first gene therapy to treat adults with hemophilia B 2022 [cited 2022 Nov 23]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treat-adults-hemophilia-b.
- U.S. Food and Drug Administration. HEMGENIX Prescribing Information 2022 [cited 2022 Nov 23]. Available from: https://www.fda.gov/media/163467/download.
- Vázquez E, Kim M, Santaella ME. Lived experience experts: a name created by us for us, Expert Rev Hematol, 2023;16(S1):7–11. DOI: 10.1080/17474086.2023.2178410.
- Valentino LA, Witkop ML, Santaella ME, et al. Building the blueprint: formulating a community-generated national plan for future research in inherited bleeding disorders. Haemophilia. 2022;28(5):760–768.
- Rauch A, Valentino LA, Mills K, et al. Big picture initiatives in bleeding disorders. Haemophilia. 2022;28(Suppl 4):53–60.
- Valentino LA, Witkop ML, Santaella ME, et al. The National Hemophilia Foundation State of the Science Research Summit initiative: executive summary, Expert Rev Hematol, 2023;16(S1):129–134. DOI: 10.1080/17474086.2023.2181782.
- Valentino LA, Witkop ML, Santaella ME, et al. The National Hemophilia Foundation’s State of the Science Research Summit Initiative: The Foundation of a National Research Blueprint for Inherited Bleeding Disorders. Expert Rev Hematol. 2023;16(S1):1–5. DOI: 10.1080/17474086.2023.2178412.
- U.S. Food and Drug Administration. BEST (biomarkers, endpoints, and other tools) resource [cited 2022 May 31]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK326791/.
- Johnsen JM, Brown DL. Members of working group 4 of the NHLBI state of the science workshop on factor VIII inhibitors generating a national blueprint for future research. The national blueprint for pregnancy/birth longitudinal cohorts to study factor VIII immunogenicity: NHLBI State of the Science (SOS) Workshop on factor VIII inhibitors. Haemophilia. 2019;25(4):603–609.
- Konkle BA, Recht M. Members of working group 2 of the NHLBI state of the science workshop on factor VIII inhibitors generating a national blueprint for future research. The national blueprint for 21st century data and specimen collection and observational cohort studies: NHLBI State of the Science Workshop on factor VIII inhibitors. Haemophilia. 2019;25(4):590–594.
- Meeks SL, Herzog RW. Members of working group 3 of the NHLBI state of the science workshop on factor VIII inhibitors generating a national blueprint for future research. The national blueprint for future basic and translational research to understand factor VIII immunogenicity: NHLBI State of the Science Workshop on factor VIII inhibitors. Haemophilia. 2019;25(4):595–602.
- Pipe SW, Sabatino DE, Nugent DJ, et al. Executive summary of the NHLBI State of the Science (SOS) Workshop: overview and next steps in generating a national blueprint for future research on factor VIII inhibitors. Haemophilia. 2019;25(4):610–615.
- Ragni MV, George LA. Members of working group 1 of the NHLBI state of the science workshop on factor VIII inhibitors generating a national blueprint for future research. The national blueprint for future factor VIII inhibitor clinical trials: NHLBI State of the Science (SOS) Workshop on factor VIII inhibitors. Haemophilia. 2019;25(4):581–589.
- Sabatino DE, Pipe SW, Nugent DJ, et al. Origins and organization of the NHLBI State of the Science Workshop: generating a national blueprint for future research on factor VIII inhibitors. Haemophilia. 2019;25(4):575–580.
- Lillicrap D, Fijnvandraat K, Young G, et al. Patients with hemophilia A and inhibitors: prevention and evolving treatment paradigms. Expert Rev Hematol. 2020;13(4):313–321.
- Tieu P, Chan A, Matino D. Molecular mechanisms of inhibitor development in hemophilia. Mediterr J Hematol Infect Dis. 2020;12(1):e2020001.
- Ragni MV, Young G, Batsuli G, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: facilitating research through infrastructure, workforce, resources and funding. Expert Rev Hematol. 2023;16(S1):107-127. DOI: 10.1080/17474086.2023.2181781.
- Nugent D, Acharya SS, Baumann KJ, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities for ultra-rare inherited bleeding disorders. Expert Rev Hematol. 2023;16(S1):55–70. DOI: 10.1080/17474086.2023.2175661.
- Makris M, Calizzani G, Fischer K, et al. EUHASS: the european haemophilia safety surveillance system. Thromb Res. 2011;127(Suppl 2):S22–5.
- Berwick DM, Nolan TW, Whittington J. The triple AIM: care, health, and cost. Health Aff (Millwood). 2008;27(3):759–769.
- Leading Health Indicators 2030: Advancing health, equity, and well-being Washington (DC) 2020 [cited 2021 Dec 11]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32200597.
- American Thrombosis and Hemostasis Network. Advancing research with better data through the ATHNdataset [cited 2021 Nov 12]. Available from: https://athn.org/what-we-do/national-projects/athndataset.html.
- Sidonio RF Jr., Bryant PC, Di Paola J, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities for mucocutaneous bleeding disorders. Expert Rev Hematol. 2023;16(S1):39–54. DOI: 10.1080/17474086.2023.2171983.
- Baldwin MK, Ahmadzia HK, Bartlett DL, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research to advance the health of people with inherited bleeding disorders with the potential to menstruate. Expert Rev Hematol. 2023;16(S1):71–86. DOI: 10.1080/17474086.2023.2175660.
- Haley KM, Sidonio RF Jr., Abraham S, et al. A cross-sectional study of women and girls with congenital bleeding disorders: the American thrombosis and hemostasis network cohort. J Womens Health (Larchmt). 2020;29(5):670–676.
- Byams VR, Baker JR, Bailey C, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities in health services; diversity, equity, and inclusion; and implementation science. Expert Rev Hematol. 2023;16(S1):87–106. DOI: 10.1080/17474086.2023.2183836.
- National Hemophilia Foundation. History from 2AD to the present [cited 2021 Nov 12]. Available from: https://www.hemophilia.org/bleeding-disorders-a-z/overview/history.
- Barnes RF, Cramer TJ, Sait AS, et al. The hypertension of hemophilia is not explained by the usual cardiovascular risk factors: results of a cohort study. Int J Hypertens. 2016;2016:2014201.
- Fransen van de Putte DE, Fischer K, Makris M, et al. Increased prevalence of hypertension in haemophilia patients. Thromb Haemost. 2012;108(4):750–755.
- von Drygalski A, Kolaitis NA, Bettencourt R, et al. Prevalence and risk factors for hypertension in hemophilia. Hypertension. 2013;62(1):209–215.
- Zwagemaker AF, Gouw SC, Jansen JS, et al. Incidence and mortality rates of intracranial hemorrhage in hemophilia: a systematic review and meta-analysis. Blood. 2021;138(26):2853–2873.
- Lattanzi S, Silvestrini M. Blood pressure in acute intra-cerebral hemorrhage. Ann Transl Med. 2016;4(16):320.
- Konkle BA, Skinner M, Iorio A. Hemophilia trials in the twenty-first century: defining patient important outcomes. Res Pract Thromb Haemost. 2019;3(2):184–192.
- Manco-Johnson MJ, Warren BB, Buckner TW, et al. Outcome measures in haemophilia: beyond ABR (Annualized Bleeding Rate). Haemophilia. 2021;27(Suppl 3):87–95.
- O’Mahony B, Dolan G, Nugent D, et al. Patient-centred value framework for haemophilia. Haemophilia. 2018;24(6):873–879.
- Marchesini E, Oliovecchio E, Coppola A, et al. Comorbidities in persons with haemophilia aged 60 years or more compared with age-matched people from the general population. Haemophilia. 2018;24(1):e6–e10.
- Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055.
- Chitlur M, Warrier I, Rajpurkar M, et al. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. 2009;15(5):1027–1031.
- Di Michele DM. Immune tolerance induction in haemophilia: evidence and the way forward. J Thromb Haemost. 2011;9(Suppl 1):216–225.
- McLaughlin P, Hermans C, Asghar S, et al. Problem joints and their clinical and humanistic burden in children and adults with moderate and severe hemophiliaa: CHESS paediatrics and CHESS II. 62nd ASH Annual Meeting and Exposition; Atlanta, GA (and virtual) 2021. [cited 2022 Mar 13]. Available from: https://ash.confex.com/ash/2020/webprogram/Paper140306.html
- Lobet S, Timmer M, Konigs C, et al. The role of physiotherapy in the new treatment landscape for haemophilia. J Clin Med. 2021;10:13.
- Aschman DJ, Abshire TC, Shapiro AD, et al. A community-based partnership to promote information infrastructure for bleeding disorders. Am J Prev Med. 2011;41(6 Suppl 4):S332–7.
- American Thrombosis and Hemostasis Network. Why conduct your research sudy with ATHN [cited 2022 May 31]. Available from: https://www.athn.org/what-we-do/for-researchers/why-conduct-your-research-study-with-athn.html.
- National Hemophilia Foundation. What is CVR? [cited 2021 Nov 12]. Available from: https://www.hemophilia.org/research/community-voices-in-research/what-is-cvr.
- Kulkarni R, Soucie JM, Evatt BL. Hemophilia surveillance system project i. Prevalence and risk factors for heart disease among males with hemophilia. Am J Hematol. 2005;79(1):36–42.
- Mannucci PM. Aging with hemophilia: the challenge of appropriate drug prescription. Mediterr J Hematol Infect Dis. 2019;11(1):e2019056.
- Biere-Rafi S, Tuinenburg A, Haak BW, et al. Factor VIII deficiency does not protect against atherosclerosis. J Thromb Haemost. 2012;10(1):30–37.
- Badescu MC, Ciocoiu M, Rezus E, et al. Current therapeutic approach to acute myocardial infarction in patients with congenital hemophilia. Life (Basel). 2021;11:10.
- Thornburg CD. Prepare the way for hemophilia a gene therapy. N Engl J Med. 2022;386(11):1081–1082.
- Bakeer N, Dover S, Babyn P, et al. Musculoskeletal ultrasound in hemophilia: results and recommendations from a global survey and consensus meeting. Res Pract Thromb Haemost. 2021;5(5):e12531.
- Berro M, Elichiry M, Wasen K, et al. Use of ultrasound for evaluation of painful joint episodes perceived as haemarthrosis in adult patients with severe haemophilia. Haemophilia. 2018;24(3):e124–e5.
- Ceponis A, Wong-Sefidan I, Glass CS, et al. Rapid musculoskeletal ultrasound for painful episodes in adult haemophilia patients. Haemophilia. 2013;19(5):790–798.
- Nguyen S, Lu X, Ma Y, et al. Musculoskeletal ultrasound for intra-articular bleed detection: a highly sensitive imaging modality compared with conventional magnetic resonance imaging. J Thromb Haemost. 2018;16(3):490–499.
- von Drygalski A, Pasta G, de la Corte-Rodriguez H. Ultrasound and patient self-imaging in hemophilia. Haemophilia. 2021;27(2):e298–e301.
- Batty P, Lillicrap D. Gene therapy for hemophilia: current status and laboratory consequences. Int J Lab Hematol. 2021;43(Suppl 1):117–123.
- Bhatt N, Boggio L, Simpson ML. Using an educational intervention to assess and improve disease-specific knowledge and health literacy and numeracy in adolescents and young adults with haemophilia A and B. Haemophilia. 2021;27(2):229–236.
- White PH, Cooley WC, Transitions Clinical Report Authoring G, et al. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2018;142:5.
- Institute for clinical and economic review. valoctocogene roxaparvovec and emicizumab for hemophilia a without inhibitors: effectiveness and value 2020 [cited 2022 Dec 29]. Available from: https://icer.org/wp-content/uploads/2020/10/ICER_Hemophilia-A_Final-Report_112020.pdf.
- Institute for clinical and economic review. gene therapy for hemophilia b and an update on gene therapy for hemophilia a: effectiveness and value 2022 [cited 2022 Dec 29]. Available from: https://icer.org/wp-content/uploads/2022/05/ICER_Hemophilia_Final_Report_12222022.pdf.