
ABSTRACT
The physiological properties of the native, endogenous prion protein (PrPC) is a matter of concern, due to its pleiotropic functions and links to neurodegenerative disorders and cancer. In line with our hypothesis that the basic function of PrPC is to serve as a cell surface scaffold for the assembly of signaling modules, multiple interactions have been identified of PrPC with signaling molecules, including neurotransmitter receptors. We recently reported evidence that PrPC may modulate monoaminergic neurotransmission, as well as depressive-like behavior in mice. Here, we discuss how those results, together with a number of other studies, including our previous demonstration that both inflammatory and behavioral stress modulate PrPC content in neutrophils, suggest a distributed role of PrPC in clinical depression and inflammation associated with neurodegenerative diseases. An overarching understanding of the multiple interventions of PrPC upon physiological events may both shed light on the pathogenesis of, as well as help the identification of novel therapeutic targets for clinical depression, Prion and Alzheimer's Diseases.
INTRODUCTION
Whereas the main focus in the field of prion biology has been the conformational conversion of the prion protein (PrPC) into abnormal, disease-related forms, there is growing interest in physiological roles of PrPC.Citation1-3 A wide range of functions have now been claimed for PrPC at the molecular, cellular and system levels,Citation2 but such an embarrassment of riches has done little for the understanding of the basic biological function of the prion protein. We have, therefore, proposed that PrPC serves as a cell surface scaffold for the assembly of signaling modules.Citation2,4 Accordingly, dynamic interactions with cell type- and context-dependent ligands may explain its pleiotropic regulation of signaling pathways, which translates into wide-range consequences upon both physiology and behavior.Citation2
The prion protein abounds in synapses,Citation5,6 where contributions to neurotransmission may strongly affect both neural function and dysfunction. Modulation by PrPC of several neurotransmitter systems has been reported, albeit in each case the strength of the evidence for direct interaction is somewhat variable Citation7-14 A phage display screen conducted in our laboratory identified group I metabotropic glutamate receptors (mGluR1/5) as a putative binding partner of PrPC, which was originally validated in the context of signal transduction triggered by the interaction of PrPC with laminin,Citation15 and subsequently by PrPC-dependent effects of oligomers of the Aβ peptide,Citation16 the latter of which fuel the ongoing debate over the role of PrPC in the pathogenesis of Alzheimer Disease.Citation17
Novel hits of our phage display indicated both the serotonergic receptor 5HT5A and the SERT serotonin transporter as putative binding partners of the prion protein (T. A. Americo, M. H. Magdesian and R. Linden, unpublished), and recent results are consistent with the hypothesis of an interaction of PrPC with monoaminergic systems.Citation18 Thus, binding of PrPC to 5HT5A, SERT, as well as to the dopamine receptor D1R, but not to D4R, was detected in overlay assays, and co-localized immunolabeling of both 5HT5A and D1 with PrPC was found in confocal photomicrographs of the cerebral cortex of wildtype mice. In addition, differing responses were found between wildtype and PrPC-null cerebrocortical tissue probed with either serotonin or dopamine, but not to noradrenaline, which were at least partly conveyed by 5HT5A and D1R, respectively. These effects were accompanied in Prnp-null tissue by differential contents of both the 5HT5A (but not 5HT1A) receptor and of dopamine, the latter likely associated with an increased content of tyrosine hydroxylase (TH), the rate-limiting enzyme in the synthesis of dopamine. Concurring behavioral tests characterized depressive-like behavior in the Prnp-null mice,Citation18 in line with an earlier report from other investigators, where such behavioral changes were counteracted by either imipramine, a tricyclic antidepressant, or the NMDA receptor antagonist MK-801.Citation19
The interpretation of our findings as evidence for an interaction of PrPC with monoaminergic systems has been challenged on the basis of the flanking gene problem, that is the persistence in congenic knockout mice of allelotypes of bystander genes closed linked to the targeted gene.Citation20 Thus, a given polymorphism of one such gene, present in the embryonic stem cells used for the production of the original transgenic animals, is expected to remain linked for many generations, to the adjacent targeted locus in mixed strains used for propagation of the knockout genotype. In the present case, the issue was specifically referred to a previous demonstrationCitation21 that hyperphagocytosis of apoptotic bodies by macrophages derived from Prnp-null mice, originally reported by our group,Citation22 was traced to the Prnp-flanking gene Sirpa, that encodes Signal Regulatory Protein α (SIRPα). This protein affects immune responses inclusive of phagocytosis by macrophages,Citation23 and a specific polymorphism of the Sirpa gene (henceforth designated 129Sirpa) is present in both the 129Sv and 129Ola mouse strains that originated the knockout of Prnp, but not in the mouse strains employed for propagation of such genotype, namely C57/BL6 or C57/Bl10SnJ, which led, respectively, to current colonies of mixed B6.129Sv and B10.129Ola mice.Citation21
However, the inferenceCitation20 that the 129Sirpa polymorphism is responsible for the depressive-like phenotype identified in our Prnp-null mice is unwarranted.Citation24 First, a study invoked as precedent for the role of Sirpa in depressive-like behavior,Citation25 indeed showed that mice expressing a form of SIRPα that lacks most of its cytoplasmic region manifest impaired responses in the forced swim test (FST), but there was no effect upon the Tail Suspension test (TST).Citation25 In contrast, our Prnp-null mice differed from wildtype not only in FST, but also in TST, as well as in the additional Novelty Supressed Feeding test, all of which are taken as indicators of depressive-like behavior in rodents. Also differing from our results,Citation18 the engineered SIRPα produced no change in dopamine levels.Citation25 Moreover, the same truncated SIRPα reportedly leads to robust neuroprotection against ischemic damage, similar to deletion of its major ligand CD47,Citation26,27 whereas ischemic damage is aggravated in Prnp-null mice,Citation28 thus corroborating the evidence that effects of either Prnp deletion or truncated Sirpa do not overlap. In addition, other studies in our lab, using the same B10.129Ola strain, failed to show immune phenotypes expected to appear in either neutrophils or microglia of Prnp-null mice as a consequence of the 129Sirpa polymorphism.Citation29,30 The possibility that our colony may have lost the differential effect of the Sirpa polymorphism derived from the 129Ola background is currently under investigation.
Together with the evidence that PrPC may indeed bind to both serotonergic and dopaminergic receptors, the functional changes detected in our studies are consistent with the hypothesis that the prion protein plays a role in the activity of monoaminergic systems and upon depressive-like behavior.Citation18 Here we discuss a framework for investigating the extended hypothesis that PrPC may be involved in major depression associated with neurodegenerative conditions, with focus on the Transmissible Spongiform Encephalopathies (TSEs, or Prion Diseases) and Alzheimer's Disease (AlzD).
THE PRION PROTEIN AND MONOAMINERGIC SYSTEMS
Evidence that PrPC modulates serotonergic function was first described in 1C11 murine neuroectodermal cells, when these were induced to express at least the 5-HT1B/D and 5-HT2A serotonergic receptors, and synthesize, store and transport serotonin. Antibody binding to PrPC at the surface of 1C11 serotonergic cells led to recruitment to lipid rafts of PrPC, caveolin and soluble tyrosine kinases, as well as serotonergic receptors, and prevented signal transduction through the latter.Citation9,31-33 Interestingly, 1C11 cells infected with scrapie (PrPSc) produced oxidized derivates from serotonin or catecholamines, leading to monoaminergic dysfunction.Citation34
Interaction with the dopaminergic system was first tested by overexpressing PrPC in a PC12 pheochromocytoma cell line, which resulted in increased content of monoamine oxidase, followed by a reduction in the release of dopamine and increased levels of the dopamine metabolite 3,4-dihydroxyphenylacetic acid.Citation35 Recently, it was reported that Prnp-null mice showed increased spontaneous climbing, a motor dysfunction related to dopaminergic transmission, whereas in the striatum, PrPC was found in dopaminergic neurons and levels of D1 receptor were reduced, but with no change in dopamine content. Direct interaction was also reported between the C-terminal domain of PrPC and the N-terminal domain of TH, together with evidence that PrPC helped the internalization of the enzyme. Conversely, TH modulated the content of PrPC and its localization in the plasma membrane.Citation13 However, differing from both this and a previous study,Citation19 we found no motor deficits in our Prnp-null mice.Citation18 Notably, the lack of motor symptoms in 8-12 week-old mice, as used in our study, concurs with previous evidence that motor deficits in Prnp-null are restricted to aging animals.Citation36-39
MONOAMINERGIC DYSFUNCTION IN PRION AND ALZHEIMER'S DISEASES
Monoaminergic dysfunction is often found in animals infected with PrPSc, as well as in TSEs. Levels of serotonin, dopamine, norepinephrine and their metabolites, as well as the activity of receptors and related enzymes were reportedly changed in scrapie infected animals.Citation40,41 Among other modifications, hyperactivity of tryptophan hydroxylase and monoamine oxidase A, increased levels of the metabolite 5-HIAA, and increased turnover of serotonin were reported in TSEs.Citation42 Further, decreased levels of TH were described in early stages of the conversion of PrPC, associated with a substantial decrease in dopamine and norepinephrine levels in the brain of mice infected with various scrapie strains.Citation43-45
In turn, in the context of AlzD, Prnp-null mice carrying familial AD transgenes encoding APPswe and PSen1ΔE9, failed to present the extensive serotonin axonal degeneration in the cerebral cortex typical of their Prnp+/+ counterparts, thus suggesting that AβO-PrPC binding is involved in the serotonergic dysfunction presented in AD.Citation46
CLINICAL DEPRESSION ASSOCIATED WITH PRION AND ALZHEIMER'S DISEASES
Despite the current consensus that major depression involves far more than the classical monoamine hypothesis,Citation47 evidence of interaction of PrPC with monoaminergic systems warrants the question of whether and how the prion protein may be involved in clinical depression associated with neurodegenerative diseases. Mood disorders are frequent in prion diseases,Citation48 and depletion of both monoaminergic cells and markers was found in the brains of such patients.Citation49-52 Interestingly, tricyclic antidepressants such as imipramine, and antipsychotic drugs such as chlorpromazine, which showed some effect upon psychiatric symptoms in TSE patients,Citation48,53 also inhibited prion replication in vitro.Citation54,55
Monoaminergic systems are also involved in behavioral and psychological symptoms of dementia,Citation56,57 and the prevalence of depression has been reported at up to 50% in AlzD.Citation58,59 Both morphological and functional changes were described in the monoaminergic system of AD patients' brains,Citation60,61 often associated with worsening clinical symptoms, but it is still uncertain whether these changes are compensatory, adaptive mechanisms, or direct consequences of AD progression. Notably, however, dopaminergic transmission has been hypothesized as a new player in AD pathophysiology,Citation62 and AD patients treated with dopaminergic agonists showed some positive results in restoring LTP-like cortical plasticity.Citation63 Studies in animal models of AD showed that monoaminergic drugs prevented intraneuronal amyloid deposition,Citation64 and either selective serotonin reuptake inhibitors (SSRI) or a high tryptophan diet reduced Aβ production and accumu-lation.Citation65,66
INFLAMMATION AND DEPRESSION IN NEURODEGENERATIVE DISEASES
Both systemic inflammation and neuroinflammation are associated with the progression of neurodegenerative diseases, and may play a major role in their pathogenesis.Citation67-70 A number of co-morbidities/risk factors may contribute to neurodegeneration specifically through modulation of inflammatory reponses.Citation70,71 Within the brain, microglial activation, reactive astrocytosis, and the release of a variety of cytokines have been reported in both experimental models and in patients of both TSEs and AlzD.Citation68,72-75
Microglia and/or microglial dysfunction have been identified as a major factor in a chronic inflammatory state associated with both experimental and human clinical prion diseases,Citation67,76,77 as well as in AlzD.Citation70 Recent studies along this line have challenged the previously proposed roles of NLRP3, ASC, and TREM2 in the pathogenesis of experimental prion infections,Citation78,79 but evidence persists of an involvement in microglial proliferation,Citation80 the fractalkine pathway,Citation81 and reactive oxygen species produced by NOX2.Citation82
Microglia has also been directly implicated in major depressive disorder.Citation83,84 Classical antidepressants reportedly inhibit the production of proinflammatory factor such as TNF-α and nitric oxide, and suppress the activation of microglia.Citation85,86 Injection of Aβ oligomers into the mouse brain caused both memory impairment and depressive-like behavior, along with the release of pro-inflammatory cytokines and microglial activation, which were prevented by treatment with a selective serotonin reuptake inhibitor.Citation87 Thus, microglia dysfunction may play a major role in linking depression with AlzD through neuroinflammation.Citation88 Still, despite the evidence for both similarities as well as differences between the neuroinflammatory components of either TSEs or AlzD,Citation68,89 neither a possible role of microglia in linking depression with prion diseases, nor the role of PrPC in Aβ oligomer-induced depressive-like behavior have been specifically examined to date.
Notably, the role of PrPC upon microglial biology is still unclear. Differential properties were reported as a function of PrPC content in a microglial cell line,Citation90,91 as well as in primary microglial cultures from the brains of either wildtype and Prnp-null B6.129Sv mice.Citation76 In contrast, no difference was found between primary cultures of brain microglial cells from either Prnp-null or wildtype mice of our B10.129Ola colony with regard to cell morphology, expression of the microglial marker Iba1, translocation of NF-κB to the nucleus, cytokine production, levels of iNOS or, notably, either rates of phagocytosis or migration, even following activation.Citation30
Besides microglia, however, PrPC is widely expressed in the immune system, including human T and B lymphocytes, monocytes, dendritic cells, platelets, and neutrophils.Citation2 In particular, despite long-standing recognition that neutrophils are involved in the interplay of immune responses with clinical depression,Citation92,93 these cells have been largely neglected in the context of the contribution of systemic inflammation to neurodegenerative diseases.Citation77,94,95 Early work indicated that the scrapie agent affects neutrophil biology,Citation96-99 whereas conflicting data have been reported regarding an association of neutrophil dysfunction with AlzD.Citation99-105 Nevertheless, recent studies warrant an examination of the hypothesis that neutrophils may play a role in the association of depression with neurodegenerative diseases.
A recent study of two transgenic mouse models, as well as of the brains of human AD patients, provided evidence that neutrophils adhere to blood vessels and invade the brain parenchima, release neutrophil extracellular traps (NETs) and interleukin-17, and gather around areas with Aβ amyloid deposits. This chain of events depends on the LFA-1 integrin, blockade of which, as well as depletion of neutrophils, reduced AlzD-like pathological hallmarks and ameliorated cognitive dysfunction in the transgenic mice. Remarkably, neutrophil depletion or inhibition dramatically reduced microglial activation, showing for the first time a microglia-neutrophil crosstalk in AD pathogenesis.Citation106 This elegant study suggests that neutrophils are major players in the pathogenesis of AlzD, and strongly interact with microglia in the inflammatory responses associated with neurodegeneration. In turn, neutrophils were also implicated in the depressive-like behavior of mice subject to peripheral inflammatory stress,Citation107,108 which is known to worsen the course of neurodegeneration in mouse models of both TSEs and AlzD.Citation109,110 Thus, polymorphonuclear cells may be instrumental for the relationship among inflammation, depression and neurodegeneration.
Evidence that PrPC may be involved in this context stems from our demonstration that inflammatory stress induced overexpression of Prnp, and an increase in the content of PrPC at the surface of mouse neutrophils. This effect is mediated by a combination of TGF-β and glucocorticoid, and results in enhanced cytotoxicity of neutrophils toward vascular endothelial cells.Citation29 It is tempting to speculate that, upon peripheral inflammatory conditions, Prnp-overexpressing neutrophils may help disrupt the blood-brain barrier,Citation111 and invade the central nervous system, thus leading to an impact in both depressive-like behavior, as well as aggravation of neurodegeneration.Citation106,108 Our data showing that non-inflammatory restraint stress produced similar effects as inflammatory stressCitation29 may also be relevant for the suggested, though still controversial link of chronic emotional stress with the course of neurodegenerative conditions.Citation112-115
CONCLUSION
Beyond the purported effects of misfolded conformers of the prion protein upon transmissible spongiform encephalopathies, interest in unraveling the functional properties of the normal conformer of PrPC has now extended from TSEs to AlzD. Our hypothesis that the biological function of PrPC is to provide a cell surface scaffold for the assembly of signaling modules implies that selective interactions with components of signaling systems explain its pleiotropic functions in intercellular interactions both among nerve cells and between the nervous system and other cell types.
Current studies have focused on the triad inflammation-depression-neurodegeneration, as a major determinant of the course of both TSEs and AlzD. This paradigm suggests that the examination of interactions of PrPC with monoaminergic pathways, on the one hand, and PrPC content and function in effectors of inflammatory reactions, such as microglia and neutrophils, are crucial for an informed approach to the understanding of the pathogenesis of neurodegenerative conditions. Various studies reviewed above are indeed consistent with intervention of PrPC in both monoaminergic function and inflammatory events.
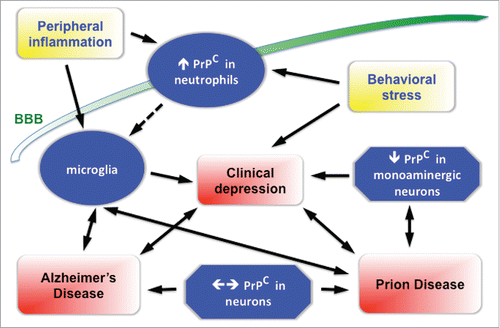
Much of the evidence for such roles of PrPC is still indirect, and at times circumstantial. Nevertheless, the available data converge upon a framework illustrated in , a concerted approach to which may allow the identificartion of novel therapeutic targets for Prion and/or Alzheimer's diseases.
FIGURE 1. Summary diagram of the purported relationship of the prion protein to depression associated with either Prion or Alzheimer's Diseases. The scheme is based on a comprehensive interpretation of results reported by various research groups, although the strength of the evidence varies among the various components of this framework. Ancillary events are coded in yellow, neuropathological outcomes are shown in red boxes. Immune cells are depicted as oval, neural cells as poligonal boxes. An increase or a decrease in the content of PrPC is shown as an upward or downward arrow, whereas engagement of PrPC either by oligomers of β-amyloid, or by its own conformational conversion and aggregation are shown by a horizontal double arrow, related respectively to either Alzheimer's or Prion Diseases. BBB = blood-brain barrier.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
The authors' research is supported by grants and fellowships from the Brazilian Council of Scientific and Technological Development (CNPq) and the Foundation for Research Support of the State of Rio de Janeiro (FAPERJ).
REFERENCES
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439-77; PMID:18558863; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673-728; PMID:18391177; http://dx.doi.org/10.1152/physrev.00007.2007
- Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci 2012; 35:92-103; PMID:22137337; http://dx.doi.org/10.1016/j.tins.2011.10.002
- Linden R, Cordeiro Y, Lima LM. Allosteric function and dysfunction of the prion protein. Cell Mol Life Sci 2012; 69:1105-24; PMID:21984610; http://dx.doi.org/10.1007/s00018-011-0847-7
- Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schurmann P, Windl O, Brose N, Kretzschmar H. Evidence of presynaptic location and function of the prion protein. J Neurosci 1999; 19:8866-75; PMID:10516306
- Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci 2003; 23:7183-93; PMID:12904479
- Kannenberg K, Groschup MH, Sigel E. Cellular prion protein and GABAA receptors: no physical association? Neuroreport 1995; 7:77-80; PMID:8742421
- Rangel A, Madronal N, Gruart i Masso A, Gavin R, Llorens F, Sumoy L, Torres JM, Delgado-Garcia JM, Del Rio JA. Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS One 2009; 4:e7592; PMID:19855845; http://dx.doi.org/10.1371/journal.pone.0007592
- Mouillet-Richard S, Pietri M, Schneider B, Vidal C, Mutel V, Launay JM, Kellermann O. Modulation of serotonergic receptor signaling and cross-talk by prion protein. J Biol Chem 2005; 280:4592-601; PMID:15590675; http://dx.doi.org/10.1074/jbc.M406199200
- Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem 2010; 285:36542-50; PMID:20837487; http://dx.doi.org/10.1074/jbc.M110.157263
- Carulla P, Bribian A, Rangel A, Gavin R, Ferrer I, Caelles C, Del Rio JA, Llorens F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol Biol Cell 2011; 22:3041-54; PMID:21757544; http://dx.doi.org/10.1091/mbc.E11-04-0321
- Black SA, Stys PK, Zamponi GW, Tsutsui S. Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Frontiers Cell Dev Biol 2014; 2:45; http://dx.doi.org/10.3389/fcell.2014.00045
- Rial D, Pamplona FA, Moreira EL, Moreira KM, Hipolide D, Rodrigues DI, Dombrowski PA, Da Cunha C, Agostinho P, Takahashi RN, Walz R, Cunha RA, Prediger RD. Cellular prion protein is present in dopaminergic neurons and modulates the dopaminergic system. Eur J Neurosci 2014; 40:2479-86; PMID:24766164; http://dx.doi.org/10.1111/ejn.12600
- Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 2015; 22:772-84; PMID:25490055; http://dx.doi.org/10.1089/ars.2014.6032
- Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, Lee KS, Magalhaes AC, Caetano FA, Mancini GL, et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. Faseb J 2011; 25:265-79; PMID:20876210; http://dx.doi.org/10.1096/fj.10-161653
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013; 79:887-902; PMID:24012003; http://dx.doi.org/10.1016/j.neuron.2013.06.036
- Westaway D, Jhamandas JH. The P's and Q's of cellular PrP-Abeta interactions. Prion 2012; 6:359-63; PMID:22874673; http://dx.doi.org/10.4161/pri.20675
- Beckman D, Santos LE, Americo TA, Ledo JH, de Mello FG, Linden R. Prion Protein Modulates Monoaminergic Systems and Depressive-like Behavior in Mice. J Biol Chem 2015; 290:20488-98; PMID:26152722; http://dx.doi.org/10.1074/jbc.M115.666156
- Gadotti VM, Bonfield SP, Zamponi GW. Depressive-like behaviour of mice lacking cellular prion protein. Behavioural Brain Res 2012; 227:319-23; http://dx.doi.org/10.1016/j.bbr.2011.03.012
- Nuvolone M, Aguzzi A. Altered Monoaminergic Systems and Depressive-like Behavior in Congenic Prion Protein Knock-out Mice. J Biol Chem 2015; 290:26350; PMID:26500295; http://dx.doi.org/10.1074/jbc.L115.689117
- Nuvolone M, Kana V, Hutter G, Sakata D, Mortin-Toth SM, Russo G, Danska JS, Aguzzi A. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J Exp Med 2013; 210:2539-52; PMID:24145514; http://dx.doi.org/10.1084/jem.20131274
- de Almeida CJ, Chiarini LB, da Silva JP, PM ES, Martins MA, Linden R. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukoc Biol 2005; 77:238-46; PMID:15539455; http://dx.doi.org/10.1189/jlb.1103531
- Murata Y, Kotani T, Ohnishi H, Matozaki T. The CD47-SIRPalpha signalling system: its physiological roles and therapeutic application. J Biochem 2014; 155:335-44; PMID:24627525; http://dx.doi.org/10.1093/jb/mvu017
- Beckman D, Santos LE, Americo TA, Ledo JH, de Mello FG, Linden R. Reply to Altered Monoaminergic Systems and Depressive-like Behavior in Congenic Prion Protein Knock-out Mice. J Biol Chem 2015; 290:26351; PMID:26500296; http://dx.doi.org/10.1074/jbc.L115.689232
- Ohnishi H, Murata T, Kusakari S, Hayashi Y, Takao K, Maruyama T, Ago Y, Koda K, Jin FJ, Okawa K, et al. Stress-evoked tyrosine phosphorylation of signal regulatory protein α regulates behavioral immobility in the forced swim test. J Neurosci 2010; 30:10472-83; PMID:20685990; http://dx.doi.org/10.1523/JNEUROSCI.0257-10.2010
- Jin G, Tsuji K, Xing C, Yang YG, Wang X, Lo EH. CD47 gene knockout protects against transient focal cerebral ischemia in mice. Exp Neurol 2009; 217:165-70; PMID:19233173; http://dx.doi.org/10.1016/j.expneurol.2009.02.004
- Wang L, Lu Y, Deng S, Zhang Y, Yang L, Guan Y, Matozaki T, Ohnishi H, Jiang H, Li H. SHPS-1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J Neurochem 2012; 122:834-43; PMID:22671569; http://dx.doi.org/10.1111/j.1471-4159.2012.07818.x
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis 2005; 20:442-9; PMID:15893468; http://dx.doi.org/10.1016/j.nbd.2005.04.002
- Mariante RM, Nobrega A, Martins RA, Areal RB, Bellio M, Linden R. Neuroimmunoendocrine regulation of the prion protein in neutrophils. J Biol Chem 2012; 287:35506-15; PMID:22910907; http://dx.doi.org/10.1074/jbc.M112.394924
- Pinheiro LP, Linden R, Mariante RM. Activation and function of murine primary microglia in the absence of the prion protein. J Neuroimmunol 2015; 286:25-32; PMID:26298321; http://dx.doi.org/10.1016/j.jneuroim.2015.07.002
- Mouillet-Richard S, Mutel V, Loric S, Tournois C, Launay JM, Kellermann O. Regulation by neurotransmitter receptors of serotonergic or catecholaminergic neuronal cell differentiation. J Biol Chem 2000; 275:9186-92; PMID:10734054; http://dx.doi.org/10.1074/jbc.275.13.9186
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925-8; PMID:10988071; http://dx.doi.org/10.1126/science.289.5486.1925
- Mouillet-Richard S, Schneider B, Pradines E, Pietri M, Ermonval M, Grassi J, Richards JG, Mutel V, Launay JM, Kellermann O. Cellular prion protein signaling in serotonergic neuronal cells. Annals New York Acad Sci 2007; 1096:106-19
- Mouillet-Richard S, Nishida N, Pradines E, Laude H, Schneider B, Feraudet C, Grassi J, Launay JM, Lehmann S, Kellermann O. Prions impair bioaminergic functions through serotonin- or catecholamine-derived neurotoxins in neuronal cells. J Biol Chem 2008; 283:23782-90; PMID:18617522; http://dx.doi.org/10.1074/jbc.M802433200
- Lee HG, Park SJ, Choi EK, Carp RI, Kim YS. Increased expression of prion protein is associated with changes in dopamine metabolism and MAO activity in PC12 cells. J Mol Neurosci 1999; 13:121-6; PMID:10691299; http://dx.doi.org/10.1385/JMN:13:1-2:121
- Coitinho AS, Roesler R, Martins VR, Brentani RR, Izquierdo I. Cellular prion protein ablation impairs behavior as a function of age. Neuroreport 2003; 14:1375-9; PMID:12876477
- Nazor KE, Seward T, Telling GC. Motor behavioral and neuropathological deficits in mice deficient for normal prion protein expression. Biochim Biophys Acta 2007; 1772:645-53; PMID:17531449; http://dx.doi.org/10.1016/j.bbadis.2007.04.004
- Gasperini L, Legname G. Prion protein and aging. Frontiers Cell Dev Biol 2014; 2:44; http://dx.doi.org/10.3389/fcell.2014.00044
- Massimino ML, Peggion C, Loro F, Stella R, Megighian A, Scorzeto M, Blaauw B, Toniolo L, Sorgato MC, Reggiani C, et al. Age-dependent neuromuscular impairment in prion protein knock-out mice. Muscle Nerve 2016; 53(2):269-79; PMID:25989742; http://dx.doi.org/10.1002/mus.24708
- Cross AJ, Kimberlin RH, Crow TJ, Johnson JA, Walker CA. Neurotransmitter metabolites, enzymes and receptors in experimental scrapie. J Neurological Sci 1985; 70:231-41; http://dx.doi.org/10.1016/0022-510X(85)90090-5
- Bassant MH, Picard M, Olichon D, Cathala F, Court L. Changes in the serotonergic, noradrenergic and dopaminergic levels in the brain of scrapie-infected rats. Brain Res 1986; 367:360-3; PMID:2421836; http://dx.doi.org/10.1016/0006-8993(86)91619-7
- Ledoux JM. Effects on the serotoninergic system in sub-acute transmissible spongiform encephalopathies: current data, hypotheses, suggestions for experimentation. Med Hypotheses 2005; 64:910-8; PMID:15780484; http://dx.doi.org/10.1016/j.mehy.2004.11.020
- Durand-Gorde JM, Bert J, Nieoullon A. Changes in tyrosine hydroxylase, glutamic acid decarboxylase and choline acetyltransferase after local microinoculation of scrapie agent into the nigrostriatal system of the golden hamster. Brain Res 1985; 341:243-51; PMID:2864098; http://dx.doi.org/10.1016/0006-8993(85)91063-7
- Yun SW, Choi EK, Ju WK, Ahn MS, Carp RI, Wisniewski HM, Kim YS. Extensive degeneration of catecholaminergic neurons to scrapie agent 87V in the brains of IM mice. Mol Chem Neuropathol 1998; 34:121-32; PMID:10327412; http://dx.doi.org/10.1007/BF02815074
- Gunapala KM, Chang D, Hsu CT, Manaye K, Drenan RM, Switzer RC, Steele AD. Striatal pathology underlies prion infection-mediated hyperactivity in mice. Prion 2010; 4:302-15; PMID:20948312; http://dx.doi.org/10.4161/pri.4.4.13721
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367-74; PMID:20445063; http://dx.doi.org/10.1523/JNEUROSCI.0395-10.2010
- Rosenblat JD, McIntyre RS, Alves GS, Fountoulakis KN, Carvalho AF. Beyond Monoamines-Novel Targets for Treatment-Resistant Depression: A Comprehensive Review. Curr Neuropharmacol 2015; 13:636-55; PMID:26467412; http://dx.doi.org/10.2174/1570159X13666150630175044
- Thompson A, MacKay A, Rudge P, Lukic A, Porter MC, Lowe J, Collinge J, Mead S. Behavioral and psychiatric symptoms in prion disease. Am J Psychiatry 2014; 171:265-74; PMID:24585329; http://dx.doi.org/10.1176/appi.ajp.2013.12111460
- Wanschitz J, Kloppel S, Jarius C, Birner P, Flicker H, Hainfellner JA, Gambetti P, Guentchev M, Budka H. Alteration of the serotonergic nervous system in fatal familial insomnia. Ann Neurol 2000; 48:788-91; PMID:11079543; http://dx.doi.org/10.1002/1531-8249(200011)48:5<788::AID-ANA13>3.0.CO;2-5
- Kloppel S, Pirker W, Brucke T, Kovacs GG, Almer G. Beta-CIT SPECT demonstrates reduced availability of serotonin transporters in patients with Fatal Familial Insomnia. J Neural Transmission 2002; 109:1105-10; http://dx.doi.org/10.1007/s007020200093
- Ragno M, Scarcella MG, Cacchio G, Capellari S, Di Marzio F, Parchi P, Trojano L. Striatal [123I] FP-CIT SPECT demonstrates dopaminergic deficit in a sporadic case of Creutzfeldt-Jakob disease. Acta neurologica Scandinavica 2009; 119:131-4; PMID:18638039; http://dx.doi.org/10.1111/j.1600-0404.2008.01075.x
- Vital A, Fernagut PO, Canron MH, Joux J, Bezard E, Martin-Negrier ML, Vital C, Tison F. The nigrostriatal pathway in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 2009; 68:809-15; PMID:19535991; http://dx.doi.org/10.1097/NEN.0b013e3181abdae8
- Benito-Leon J. Combined quinacrine and chlorpromazine therapy in fatal familial insomnia. Clin Neuropharmacol 2004; 27:201-3; PMID:15319710; http://dx.doi.org/10.1097/01.wnf.0000134853.36429.0e
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A 2001; 98:9836-41; PMID:11504948; http://dx.doi.org/10.1073/pnas.161274798
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241-65; PMID:16816391; http://dx.doi.org/10.1093/brain/awl150
- Lanctot KL, Herrmann N, Mazzotta P. Role of serotonin in the behavioral and psychological symptoms of dementia. J Neuropsychiatry Clin Neurosci 2001; 13:5-21; PMID:11207325; http://dx.doi.org/10.1176/jnp.13.1.5
- Rodriguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer's disease. Prog Neurobiol 2012; 99:15-41; PMID:22766041; http://dx.doi.org/10.1016/j.pneurobio.2012.06.010
- Lee HB, Lyketsos CG. Depression in Alzheimer's disease: heterogeneity and related issues. Biological Psychiatry 2003; 54:353-62; PMID:12893110; http://dx.doi.org/10.1016/S0006-3223(03)00543-2
- Zubenko GS, Zubenko WN, McPherson S, Spoor E, Marin DB, Farlow MR, Smith GE, Geda YE, Cummings JL, Petersen RC, et al. A collaborative study of the emergence and clinical features of the major depressive syndrome of Alzheimer's disease. Am J Psychiatry 2003; 160:857-66; PMID:12727688; http://dx.doi.org/10.1176/appi.ajp.160.5.857
- Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, Dang V, Sanchez MM, De Miguel Z, Ashford JW, Salehi A. Ascending monoaminergic systems alterations in Alzheimer's disease. translating basic science into clinical care. Neurosci Biobehavioral Rev 2013; 37:1363-79; http://dx.doi.org/10.1016/j.neubiorev.2013.05.008
- Palmer AM, Wilcock GK, Esiri MM, Francis PT, Bowen DM. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer's disease. Brain Res 1987; 401:231-8; PMID:2434191; http://dx.doi.org/10.1016/0006-8993(87)91408-9
- Martorana A, Mori F, Esposito Z, Kusayanagi H, Monteleone F, Codeca C, Sancesario G, Bernardi G, Koch G. Dopamine modulates cholinergic cortical excitability in Alzheimer's disease patients. Neuropsychopharmacol 2009; 34:2323-8; http://dx.doi.org/10.1038/npp.2009.60
- Koch G, Di Lorenzo F, Bonni S, Giacobbe V, Bozzali M, Caltagirone C, Martorana A. Dopaminergic modulation of cortical plasticity in Alzheimer's disease patients. Neuropsychopharmacol 2014; 39:2654-61; http://dx.doi.org/10.1038/npp.2014.119
- Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T, et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann Neurol 2011; 69:248-56; PMID:21387370; http://dx.doi.org/10.1002/ana.22319
- Cirrito JR, Disabato BM, Restivo JL, Verges DK, Goebel WD, Sathyan A, Hayreh D, D'Angelo G, Benzinger T, Yoon H, et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A 2011; 108:14968-73; PMID:21873225; http://dx.doi.org/10.1073/pnas.1107411108
- Noristani HN, Verkhratsky A, Rodriguez JJ. High tryptophan diet reduces CA1 intraneuronal β-amyloid in the triple transgenic mouse model of Alzheimer's disease. Aging Cell 2012; 11:810-22; PMID:22702392; http://dx.doi.org/10.1111/j.1474-9726.2012.00845.x
- Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol 2002; 15:349-54; PMID:12045736; http://dx.doi.org/10.1097/00019052-200206000-00020
- Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R, Williams A. Neuroinflammation in Alzheimer's disease and prion disease. Glia 2002; 40:232-9; PMID:12379910; http://dx.doi.org/10.1002/glia.10146
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun 2004; 18:407-13; PMID:15265532; http://dx.doi.org/10.1016/j.bbi.2004.01.004
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015; 16:358-72; PMID:25991443; http://dx.doi.org/10.1038/nrn3880
- Sandu RE, Buga AM, Uzoni A, Petcu EB, Popa-Wagner A. Neuroinflammation and comorbidities are frequently ignored factors in CNS pathology. Neural Regen Res 2015; 10:1349-55; PMID:26604877; http://dx.doi.org/10.4103/1673-5374.165208
- Brown DR. Microglia and prion disease. Microscopy Res Technique 2001; 54:71-80; http://dx.doi.org/10.1002/jemt.1122
- Rozemuller AJ, Jansen C, Carrano A, van Haastert ES, Hondius D, van der Vies SM, Hoozemans JJ. Neuroinflammation and common mechanism in Alzheimer's disease and prion amyloidosis: amyloid-associated proteins, neuroinflammation and neurofibrillary degeneration. Neurodegener Dis 2012; 10:301-4; PMID:22398730; http://dx.doi.org/10.1159/000335380
- Wojtera M, Sobow T, Kloszewska I, Liberski PP, Brown DR, Sikorska B. Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer's disease: morphometric study and review of the literature. Folia Neuropathol 2012; 50:74-84; PMID:22505366
- Llorens F, Lopez-Gonzalez I, Thune K, Carmona M, Zafar S, Andreoletti O, Zerr I, Ferrer I. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Frontiers Aging Neurosci 2014; 6:198; http://dx.doi.org/10.3389/fnagi.2014.00198
- Brown DR, Besinger A, Herms JW, Kretzschmar HA. Microglial expression of the prion protein. Neuroreport 1998; 9:1425-9; PMID:9631441; http://dx.doi.org/10.1097/00001756-199805110-00032
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases–an update. Immunol 2014; 142:151-66; http://dx.doi.org/10.1111/imm.12233
- Nuvolone M, Sorce S, Schwarz P, Aguzzi A. Prion pathogenesis in the absence of NLRP3/ASC inflammasomes. PLoS One 2015; 10:e0117208; PMID:25671600; http://dx.doi.org/10.1371/journal.pone.0117208
- Zhu C, Herrmann US, Li B, Abakumova I, Moos R, Schwarz P, Rushing EJ, Colonna M, Aguzzi A. Triggering receptor expressed on myeloid cells-2 is involved in prion-induced microglial activation but does not contribute to prion pathogenesis in mouse brains. Neurobiol Aging 2015; 36:1994-2003; PMID:25816748; http://dx.doi.org/10.1016/j.neurobiolaging.2015.02.019
- Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 2013; 33:2481-93; PMID:23392676; http://dx.doi.org/10.1523/JNEUROSCI.4440-12.2013
- Grizenkova J, Akhtar S, Brandner S, Collinge J, Lloyd SE. Microglial Cx3cr1 knockout reduces prion disease incubation time in mice. BMC Neurosci 2014; 15:44; PMID:24655482; http://dx.doi.org/10.1186/1471-2202-15-44
- Sorce S, Nuvolone M, Keller A, Falsig J, Varol A, Schwarz P, Bieri M, Budka H, Aguzzi A. The role of the NADPH oxidase NOX2 in prion pathogenesis. PLoS Pathog 2014; 10:e1004531; PMID:25502554; http://dx.doi.org/10.1371/journal.ppat.1004531
- Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, Maier SF, Yirmiya R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 2014; 19:699-709; PMID:24342992; http://dx.doi.org/10.1038/mp.2013.155
- Yirmiya R, Rimmerman N, Reshef R. Depression as a Microglial Disease. Trends Neurosci 2015; 38:637-58; PMID:26442697; http://dx.doi.org/10.1016/j.tins.2015.08.001
- Walker FR. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacol 2013; 67:304-17; http://dx.doi.org/10.1016/j.neuropharm.2012.10.002
- Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, Kanba S. Antidepressants inhibit interferon-gamma-induced microglial production of IL-6 and nitric oxide. Exp Neurol 2007; 206:33-42; PMID:17481608; http://dx.doi.org/10.1016/j.expneurol.2007.03.022
- Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST. Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry 2013; 18:1053-4; PMID:23183490; http://dx.doi.org/10.1038/mp.2012.168
- Santos LE, Beckman D, Ferreira ST. Microglial dysfunction connects depression and Alzheimer's disease. Brain Behav Immun 2015; S0889-1591(15):30056-8
- Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci 1999; 19:928-39; PMID:9920656
- Ding T, Zhou X, Kouadir M, Shi F, Yang Y, Liu J, Wang M, Yin X, Yang L, Zhao D. Cellular prion protein participates in the regulation of inflammatory response and apoptosis in BV2 microglia during infection with Mycobacterium bovis. J Mol Neurosci 2013; 51:118-26; PMID:23345082; http://dx.doi.org/10.1007/s12031-013-9962-2
- Shi F, Yang L, Kouadir M, Yang Y, Ding T, Wang J, Zhou X, Yin X, Zhao D. Prion protein participates in the regulation of classical and alternative activation of BV2 microglia. J Neurochem 2012; 124(2):168-74; PMID:23061439; http://dx.doi.org/10.1111/jnc.12053
- Maes M. Evidence for an immune response in major depression: a review and hypothesis. Progress Neuro-Psychopharmacology Biol Psychiatry 1995; 19:11-38
- Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K. The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 2001; 15:199-226; PMID:11566046; http://dx.doi.org/10.1006/brbi.2000.0597
- Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol 2010; 120:277-86; PMID:20644946; http://dx.doi.org/10.1007/s00401-010-0722-x
- Cunningham C, Hennessy E. Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther 2015; 7:33; PMID:25802557; http://dx.doi.org/10.1186/s13195-015-0117-2
- Licursi PC, Merz PA, Merz GS, Carp RI. Scrapie-induced changes in the percentage of polymorphonuclear neutrophils in mouse peripheral blood. Infect Immun 1972; 6:370-6; PMID:4118048
- Carp RI, Merz PA, Licursi PC, Merz GS. Replication of the factor in scrapie material that causes a decrease in polymorphonuclear neutrophils. J Infect Dis 1973; 128:256-8; PMID:4737373; http://dx.doi.org/10.1093/infdis/128.2.256
- Miragliotta G, Fumarulo R, Fumarola D. Inhibition of neutrophil functions by scrapie prion protein: description of some inhibitory properties. Acta Virol 1990; 34:517-22; PMID:1983177
- Scali C, Prosperi C, Bracco L, Piccini C, Baronti R, Ginestroni A, Sorbi S, Pepeu G, Casamenti F. Neutrophils CD11b and fibroblasts PGE(2) are elevated in Alzheimer's disease. Neurobiol Aging 2002; 23:523-30; PMID:12009501; http://dx.doi.org/10.1016/S0197-4580(01)00346-3
- Licastro F, Morini MC, Davis LJ, Malpassi P, Cucinotta D, Parente R, Melotti C, Savorani G. Increased chemiluminescence response of neutrophils from the peripheral blood of patients with senile dementia of the Alzheimer's type. J Neuroimmunol 1994; 51:21-6; PMID:8157733; http://dx.doi.org/10.1016/0165-5728(94)90124-4
- Davydova TV, Fomina VG, Voskresenskaya NI, Doronina OA. Phagocytic activity and state of bactericidal systems in polymorphonuclear leukocytes from patients with Alzheimer's disease. Bull Exp Biol Med 2003; 136:355-7; PMID:14714081; http://dx.doi.org/10.1023/B:BEBM.0000010950.53560.e2
- Garlind A, Nilsson E, Palmblad J. Calcium ion transients in neutrophils from patients with sporadic Alzheimer's disease. Neurosci Lett 1998; 255:95-8; PMID:9835223; http://dx.doi.org/10.1016/S0304-3940(98)00716-2
- Baik SH, Cha MY, Hyun YM, Cho H, Hamza B, Kim DK, Han SH, Choi H, Kim KH, Moon M, et al. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging 2014; 35:1286-92; PMID:24485508; http://dx.doi.org/10.1016/j.neurobiolaging.2014.01.003
- Achilli C, Ciana A, Minetti G. Amyloid-β (25-35) peptide induces the release of pro-matrix metalloprotease 9 (pro-MMP-9) from human neutrophils. Mol Cell Biochem 2014; 397:117-23; PMID:25087121; http://dx.doi.org/10.1007/s11010-014-2178-0
- Gabbita SP, Johnson MF, Kobritz N, Eslami P, Poteshkina A, Varadarajan S, Turman J, Zemlan F, Harris-White ME. Oral TNFalpha Modulation Alters Neutrophil Infiltration, Improves Cognition and Diminishes Tau and Amyloid Pathology in the 3xTgAD Mouse Model. PLoS One 2015; 10:e0137305; PMID:26436670; http://dx.doi.org/10.1371/journal.pone.0137305
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 2015; 21:880-6; PMID:26214837; http://dx.doi.org/10.1038/nm.3913
- Rummel C, Inoue W, Poole S, Luheshi GN. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol Psychiatry 2010; 15:523-34; PMID:19773811; http://dx.doi.org/10.1038/mp.2009.98
- Aguilar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry 2014; 19:599-606; PMID:24126927; http://dx.doi.org/10.1038/mp.2013.137
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005; 25:9275-84; PMID:16207887; http://dx.doi.org/10.1523/JNEUROSCI.2614-05.2005
- Joshi YB, Giannopoulos PF, Chu J, Pratico D. Modulation of lipopolysaccharide-induced memory insult, gamma-secretase, and neuroinflammation in triple transgenic mice by 5-lipoxygenase. Neurobiol Aging 2014; 35:1024-31; PMID:24332986; http://dx.doi.org/10.1016/j.neurobiolaging.2013.11.016
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011; 12:723-38; PMID:22048062
- Alkadhi KA. Chronic psychosocial stress exposes Alzheimer's disease phenotype in a novel at-risk model. Frontiers Biosci 2012; 4:214-29; http://dx.doi.org/10.2741/E371
- Machado A, Herrera AJ, de Pablos RM, Espinosa-Oliva AM, Sarmiento M, Ayala A, Venero JL, Santiago M, Villaran RF, Delgado-Cortes MJ, et al. Chronic stress as a risk factor for Alzheimer's disease. Rev Neurosci 2014; 25:785-804; PMID:25178904; http://dx.doi.org/10.1515/revneuro-2014-0035
- Djamshidian A, Lees AJ. Can stress trigger Parkinson's disease? J Neurol Neurosurg Psychiatry 2014; 85:878-81; PMID:24259593; http://dx.doi.org/10.1136/jnnp-2013-305911
- Greenberg MS, Tanev K, Marin MF, Pitman RK. Stress, PTSD, and dementia. Alzheimer's Dementia 2014; 10:S155-65; PMID:24924667; http://dx.doi.org/10.1016/j.jalz.2014.04.008