
ABSTRACT
Mitochondrial dysfunction has been suggested to contribute to neurodegenerative diseases, including Alzheimer and Parkinson disease. Cells respond to changes in the functional state of mitochondria via retrograde signaling pathways from the mitochondria to the nucleus, but little is known about retrograde signaling in the nervous system. We have recently shown that inhibition of retrograde signaling reduces the impact of neuronal mitochondrial dysfunction. We performed a study designed to characterize the mitochondrial retrograde signaling pathway in the Drosophila nervous system. Using several different models we found that neuronal specific mitochondrial dysfunction results in defects in synapse development and neuronal function. Moreover, we identified the Drosophila hypoxia inducible factor α (HIFα) ortholog Sima as a key neuronal transcriptional regulator. Knock-down of sima restores function in several Drosophila models of mitochondrial dysfunction, including models of human disease. Here we discuss these findings and speculate on the potential benefits of inhibition of retrograde signaling. We also describe how our results relate to other studies of mitochondrial retrograde signaling and the potential therapeutic applications of these discoveries.
Mitochondria produce the majority of cellular energy in the form of ATP and play key roles in amino acid synthesis, calcium buffering, iron-sulfur cluster synthesis, generation of reactive oxygen species (ROS) and apoptosis. Dysfunction of this multifunctional organelle is associated with a wide range of disorders, from diabetes to cancer and neurodegeneration.Citation1,2 The high metabolic demand of the nervous system makes it particularly sensitive to mitochondrial deficits. Altered mitochondrial morphology, decreased complex I activity and reduced mitochondrial DNA (mtDNA) levels are observed in the cortex of Alzheimer disease (AD) patients.Citation3-5 Complex I deficiencies and increased production of ROS are also detected in the substantia nigra pars compacta in Parkinson disease patients.Citation6,7 Similarly, complex I deficiency and mtDNA loss are observed in the prefrontal cortex of patients with Parkinson disease dementia.Citation8,9
The mitochondrial retrograde response is a signaling pathway that enables cells to respond to changes in the functional state of mitochondria. Mitochondrial retrograde signaling is a pathway of communication from mitochondria to the nucleus, resulting in changes in nuclear gene expression ().Citation10 This is opposite to anterograde signaling from the nucleus to mitochondria. Retrograde signaling varies according to the context, ranging from cellular metabolic changes to cell cycle arrest and even modification of the systemic integrated stress response.Citation10-12 Retrograde signaling has been well characterized in budding yeast and has been linked to the mechanistic target of rapamycin (mTOR) pathway in this model.Citation13 In yeast, reduced glutamine levels activate the retrograde response,Citation14 resulting in nuclear translocation of the Rtg1/Rtg3 transcriptional heterodimer.Citation15 Activation of retrograde signaling can result from changes in cellular metabolic status, AMP:ATP ratio, ROS levels, Ca2+ concentration, or compromised mitochondrial membrane potential.Citation10,16-18 A number of different mitochondrial retrograde signaling pathways have been identified in mammalian cells. For example, increased cytosolic Ca2+ concentration activates cAMP-responsive element-binding protein (CREB), triggering a transcriptional response.Citation19 Other pathways such as mTOR, NFκB and Sirt1 are also implicated in the mammalian retrograde response.Citation20 These studies have focused on proliferating cells and it is unclear how relevant these pathways are to mitochondrial retrograde signaling in post-mitotic neurons.
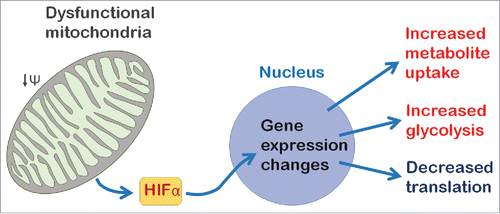
Figure 1. A model for neuronal mitochondrial retrograde signaling in Drosophila. Mitochondrial dysfunction causes reduced mitochondrial membrane potential (ΔΨ) and triggers the retrograde response. In the Drosophila nervous system HIFα (Sima) regulates retrograde response genes to reprogram neurons by changing the metabolic state of the cell and inhibiting protein translation.
To address this we recently developed a model to investigate the mitochondrial retrograde response in the Drosophila central nervous system (CNS). We characterized the transcriptional response to mitochondrial dysfunction caused either by overexpression of mitochondrial transcription factor A (TFAM), or knock-down of ATP synthase coupling factor 6 (ATPsynCF6) in neurons.Citation21 Neuronal mitochondrial dysfunction, induced using either of these tools, causes reduced adult climbing ability and lifespan, decreased redox potential, as well as reduced numbers of presynaptic mitochondria and active zones.Citation21 Using microarray analysis we found that neuronal mitochondrial dysfunction, induced using either TFAM overexpression or knock-down of ATPsynCF6, alters the expression of hundreds of genes in the late third instar larval CNS.Citation21 Approximately 50% of the genes significantly altered by TFAM overexpression were similarly altered by knock-down of ATPsynCF6. This novel model therefore demonstrates that mitochondrial retrograde signaling is active in the Drosophila CNS.
A well-defined role of the mitochondrial retrograde response is to reprogram metabolism to compensate for mitochondrial dysfunction.Citation10 We also found evidence of metabolic adaptation to mitochondrial dysfunction in Drosophila neurons, suggesting a switch to glycolytic metabolism. Increased lactate dehydrogenase expression occurs in our model, in yeast and several other Drosophila models of mitochondrial dysfunction and may thus be a hallmark of the mitochondrial retrograde response.Citation22-25 Increased CNS lactate levels are associated with mitochondrial diseases such as mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and have been observed in Drosophila parkin mutants and some Parkinson disease patients.Citation26,27
Studies in human cellular models of mitochondrial disease have demonstrated that the transcriptional response elicited by retrograde signaling depends on the severity of mitochondrial dysfunction. The A3243G mutation in the mtDNA tRNALeu gene causes MELAS. Studies using cells containing varying copy numbers of the A3243G mtDNA mutation, or completely lacking mtDNA, have shown that the transcriptional response varies according to the severity of mitochondrial dysfunction, even in the same cell type.Citation28,29 In one study, 4 distinct transcriptional phases were observed according to increasing A3243G mutant mtDNA copy number.Citation29 These studies suggest that the complex nature of mitochondrial diseases may be in part due to differences in retrograde signaling.
Activation of the basic helix-loop-helix PAS domain transcription factor hypoxia inducible factor α (HIFα) switches cells to a glycolytic state in hypoxic conditions. The Drosophila HIFα ortholog Similar (Sima), also directly regulates the transcription of several of the retrograde response genes we identified.Citation30,21 To functionally test whether Sima participates in retrograde signaling we performed epistasis experiments. Surprisingly, these experiments showed that neuronal-specific knock-down of sima partially restores the reduced climbing and lifespan phenotypes of TFAM overexpressing flies.Citation21 Similarly, pan-neuronal sima knock-down rescued the defect in neuronal function in a Drosophila model of the mitochondrial disease Leigh syndrome (using Surf1 RNAi).Citation21 These data suggest that HIFα is a key regulator of mitochondrial retrograde signaling in the nervous system (). However, they also indicate that, counterintuitively, the retrograde response mediated by HIFα contributes to the pathology caused by mitochondrial dysfunction.
A number of other studies in a variety of systems have also shown that inhibition of the mitochondrial retrograde response can reverse pathological phenotypes. In Drosophila, inhibition of retrograde signaling can reverse the cell cycle arrest phenotype caused by mutations in mitochondrial genes. In the developing eye, imaginal disc cells that are mutant for the complex I subunit Pdsw, or the complex IV subunit cytochrome c oxidase Va (CoVa) arrest during the G1/S phase transition.Citation11 These mitochondrial mutations trigger retrograde signaling via either FoxO or p53. Inhibition of retrograde signaling in cells that are double mutant for Pdsw and FoxO, or CoVa and p53, prevents the cell cycle arrest caused by activation of retrograde signaling, allowing cells to re-enter the cell cycle.
Mitochondrial dysfunction is also observed in cancerous cells, which accumulate mtDNA mutationsCitation31 and express reduced levels of ETC complexes.Citation32 Work from Avadhani's group using cultured mouse myoblast C2C12 cells has shown that mitochondrial dysfunction, caused by drug-induced mtDNA loss, or uncoupling of the ETC from ATP synthase, causes decreased apoptosis and increased tumor invasiveness in transplant models.Citation16 Loss of mitochondrial membrane potential causes increased cytosolic Ca2+, resulting in activation of calcineurin.Citation33 Calcineurin activates NFκB, which causes nuclear translocation of the REL-p50 heterodimer, where it associates with C/EBPδ, CREB and NFAT in a nuclear enhanceosome to regulate gene expression.Citation34 Thus, mitochondrial retrograde signaling can drive tumorigenesis. In this model inhibition of retrograde signaling (by knock-down of IκBβ) reduces resistance to apoptosis and decreases the invasive phenotype caused by mitochondrial dysfunction.Citation34
The A1555G mutation in the mtDNA encoded 12S rRNA gene causes nonsyndromic and/or aminoglycoside-induced deafness.Citation35 AMP kinase activation is a key retrograde signal in mammalian cells carrying the mtDNA A1555G mutation.Citation36 In this context mitochondrial dysfunction causes ROS-dependent signaling through AMP kinase and the transcription factor E2F1. Mitochondrial retrograde pathway activation of E2F1 in A1555G mutant cells causes increased sensitivity to apoptosis. However, inhibition of the mitochondrial retrograde response, through loss of the transcription factor E2F1, restores hearing function in a mouse deafness model.Citation36 Thus, inhibition of mitochondrial retrograde signaling may restore cellular homeostasis in a variety of cell and tissue types and is therefore a potential avenue for clinical intervention.
The finding that inhibition of retrograde signaling reverses disease phenotypes is somewhat counter-intuitive. We hypothesize that during normal fluctuations in mitochondrial function, resulting from variable oxygen supply or changing nutrient availability, retrograde signaling benefits the cell and organism. The retrograde response promotes the expression of glycolytic genes, which allow cells to compensate for reduced mitochondrial ATP synthesis.Citation21,22,37 Moreover, gene expression analysis in neurons suggests that retrograde signaling inhibits protein translation.Citation21 As translation has the greatest single requirement for ATP, this would also allow cells to cope with mitochondrial dysfunction. By contrast, the permanent mitochondrial damage that occurs in disease models (and potentially in human disease) causes long-term activation of mitochondrial retrograde signaling. We propose that this long-term activation contributes to disease phenotypes. Thus, transient retrograde activation temporally increases cell proliferation, but unchecked activation in cancer cells results in tumorigenesis. In neurons, transient retrograde activation results in a temporary beneficial reduction in translation, but constitutive activation and long-term loss of protein synthesis in neurodegenerative disease contributes to neuronal dysfunction and loss. Similarly, switching to glycolytic metabolism is temporarily beneficial, but long-term dependence on glycolysis and lack of mitochondrial ATP synthesis may contribute to the disease phenotype. This hypothesis may explain the findings from our study and other disease models in which retrograde signaling contributes to the disease phenotype. Furthermore, in our Drosophila model, inhibition of retrograde signaling ameliorates, but does not fully restore neuronal function. We postulate that this is because there is still a mitochondrial deficit, but that the deleterious consequences of constitutive retrograde signaling have been reversed.
We showed that HIFα is a key regulator of mitochondrial dysfunction in Drosophila, but the mechanism by which mitochondria regulate HIFα in neurons is currently unknown. During hypoxia HIFα is regulated by the action of oxygen depended prolyl hydroxylases (PHDs). In the presence of oxygen PHDs hydroxylate HIFα on specific proline residues that act as recognition sites for the von Hippel-Lindau protein (pVHL) E3 ubiquitin ligase complex, resulting in proteosomal degradation of HIFα. During hypoxia PHD activity is reduced, HIFα is stabilised and translocates to the nucleus where it activates gene transcription. Whether mitochondrial dysfunction in the Drosophila CNS results in ‘pseudohypoxia’ resulting in HIFα stabilization, as observed in cancer cells mutant for components of the tricarboxylic acid (TCA) cycle,Citation38 or whether an alternative mechanism causes HIFα stabilization/activation is not known. Moreover, modulation of HIFα may have different effects depending on the tissue or disease context. A recent study using the Ndufs4 mouse model of the mitochondrial disease Leigh syndrome demonstrates a protective effect of hypoxia.Citation39 In this study the reduced lifespan and locomotor phenotypes of Ndufs4 knockout mice were rescued by exposure to hypoxia. By contrast, in another recent study hypoxia promoted neurodegeneration in photoreceptor neurons in mice.Citation40 Degeneration of neurons in this mouse model could also be triggered by genetically inducing HIFα expression and was rescued by genetic ablation of HIFα in retinal pigment epithelium cells. Clearly, HIFα plays a key role in neurological disease and understanding its contribution to pathology in specific cells and tissues could have important therapeutic potential.
In conclusion, the discovery that inhibiting mitochondrial retrograde signaling reverses disease phenotypes suggests that this approach could be used in any number of conditions associated with mitochondrial dysfunction. Further work will be required to elucidate the interplay of these retrograde signals and their exact role in disease. However, the prevalence of mitochondrial dysfunction associated with human disease means that basic studies in Drosophila and other models could have widespread potential applications. It seems that manipulating the retrograde response by blocking the action of key transcription factors may hold potential as a therapeutic strategy to treat disorders caused by mitochondrial dysfunction.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This original work described was funded by the Wellcome Trust (WT089622MA), the BBSRC (BB/I012273/1), the National Institute for Health Research (NIHR), the Biomedical Research Center at King's College London (KCL), the Edmund J. Safra foundation and KCL. This article presents independent research funded by the NIHR. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
References
- Iacovino M, Granycome C, Sembongi H, Bokori-Brown M, Butow RA, Holt IJ, Bateman JM. The conserved translocase Tim17 prevents mitochondrial DNA loss. Hum Mol Genet 2009; 18:65-74; PMID:18826960; http://dx.doi.org/10.1093/hmg/ddn313
- Schapira AH. Mitochondrial disease. Lancet 2006; 368:70-82; PMID:16815381; http://dx.doi.org/10.1016/S0140-6736(06)68970-8
- Baloyannis SJ, Costa V, Michmizos D. Mitochondrial alterations in Alzheimer's disease. Am J Alzheimers Dis Other Demen 2004; 19:89-93; PMID:15106389; http://dx.doi.org/10.1177/153331750401900205
- Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A 2004; 101:10726-31; PMID:15247418; http://dx.doi.org/10.1073/pnas.0403649101
- Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci 2001; 68:2741-50; PMID:11400916; http://dx.doi.org/10.1016/S0024-3205(01)01074-8
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem 1989; 52:381-9; PMID:2911023; http://dx.doi.org/10.1111/j.1471-4159.1989.tb09133.x
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989; 1:1269; PMID:2566813; http://dx.doi.org/10.1016/S0140-6736(89)92366-0
- Gatt AP, Duncan OF, Attems J, Francis PT, Ballard CG, Bateman JM. Dementia in Parkinson's disease is associated with enhanced mitochondrial complex I deficiency. Mov Disord 2016; 31:352-9; PMID:26853899; http://dx.doi.org/10.1002/mds.26513
- Gatt AP, Jones EL, Francis PT, Ballard C, Bateman JM. Association of a polymorphism in mitochondrial transcription factor A (TFAM) with Parkinson's disease dementia but not dementia with Lewy bodies. Neurosci Lett 2013; 557 Pt B:177-80; PMID:24184878; http://dx.doi.org/10.1016/j.neulet.2013.10.045
- Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell 2004; 14:1-15; PMID:15068799; http://dx.doi.org/10.1016/S1097-2765(04)00179-0
- Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 2008; 40:356-61; PMID:18246068; http://dx.doi.org/10.1038/ng.2007.50
- Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci U S A 2015; 112:E6614-23; PMID:26627253; http://dx.doi.org/10.1073/pnas.1515733112
- Komeili A, Wedaman KP, O'Shea EK, Powers T. Mechanism of metabolic control. Target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J Cell Biol 2000; 151:863-78; PMID:11076970; http://dx.doi.org/10.1083/jcb.151.4.863
- Crespo JL, Powers T, Fowler B, Hall MN. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci U S A 2002; 99:6784-9; PMID:11997479; http://dx.doi.org/10.1073/pnas.102687599
- Jia Y, Rothermel B, Thornton J, Butow RA. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol Cell Biol 1997; 17:1110-7; PMID:9032238; http://dx.doi.org/10.1128/MCB.17.3.1110
- Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013; 13:577-91; PMID:24004957; http://dx.doi.org/10.1016/j.mito.2013.08.007
- Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol 2013; 23:311-8; PMID:23489877; http://dx.doi.org/10.1016/j.tcb.2013.02.002
- Jazwinski SM. The retrograde response: when mitochondrial quality control is not enough. Biochim Biophys Acta 2013; 1833:400-9; PMID:22374136; http://dx.doi.org/10.1016/j.bbamcr.2012.02.010
- Arnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, Remacle J, Raes M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J 2002; 21:53-63; PMID:11782425; http://dx.doi.org/10.1093/emboj/21.1.53
- Jones AW, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G. PGC-1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012; 12:86-99; PMID:21983689; http://dx.doi.org/10.1016/j.mito.2011.09.009
- Cagin U, Duncan OF, Gatt AP, Dionne MS, Sweeney ST, Bateman JM. Mitochondrial retrograde signaling regulates neuronal function. Proc Natl Acad Sci U S A 2015; 112:E6000-9; PMID:26489648; http://dx.doi.org/10.1073/pnas.1505036112
- Chelstowska A, Liu Z, Jia Y, Amberg D, Butow RA. Signalling between mitochondria and the nucleus regulates the expression of a new D-lactate dehydrogenase activity in yeast. Yeast 1999; 15:1377-91; PMID:10509019; http://dx.doi.org/10.1002/(SICI)1097-0061(19990930)15:13<1377::AID-YEA473>3.0.CO;2-0
- Fernandez-Ayala DJ, Chen S, Kemppainen E, O'Dell KM, Jacobs HT. Gene expression in a Drosophila model of mitochondrial disease. PLoS One 2010; 5:e8549; PMID:20066047; http://dx.doi.org/10.1371/journal.pone.0008549
- Freije WA, Mandal S, Banerjee U. Expression profiling of attenuated mitochondrial function identifies retrograde signals in Drosophila. G3 (Bethesda) 2012; 2:843-51; PMID:22908033; http://dx.doi.org/full_text
- Tufi R, Gandhi S, de Castro IP, Lehmann S, Angelova PR, Dinsdale D, Deas E, Plun-Favreau H, Nicotera P, Abramov AY, et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson's disease. Nat Cell Biol 2014; 16:157-66; PMID:24441527; http://dx.doi.org/10.1038/ncb2901
- Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann New York Acad Sci 2008; 1142:133-58; PMID:18990125; http://dx.doi.org/10.1196/annals.1444.011
- Vincent A, Briggs L, Chatwin GF, Emery E, Tomlins R, Oswald M, Middleton CA, Evans GJ, Sweeney ST, Elliott CJ. parkin-induced defects in neurophysiology and locomotion are generated by metabolic dysfunction and not oxidative stress. Hum Mol Genet 2012; 21:1760-9; PMID:22215442; http://dx.doi.org/10.1093/hmg/ddr609
- Jahangir Tafrechi RS, Svensson PJ, Janssen GM, Szuhai K, Maassen JA, Raap AK. Distinct nuclear gene expression profiles in cells with mtDNA depletion and homoplasmic A3243G mutation. Mutat Res 2005; 578:43-52; PMID:16202796; http://dx.doi.org/10.1016/j.mrfmmm.2005.02.002
- Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O'Hearn S, Levy S, Potluri P, Lvova M, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A 2014; 111:E4033-42; PMID:25192935; http://dx.doi.org/10.1073/pnas.1414028111
- Lavista-Llanos S, Centanin L, Irisarri M, Russo DM, Gleadle JM, Bocca SN, Muzzopappa M, Ratcliffe PJ, Wappner P. Control of the hypoxic response in Drosophila melanogaster by the basic helix-loop-helix PAS protein similar. Mol Cell Biol 2002; 22:6842-53; PMID:12215541; http://dx.doi.org/10.1128/MCB.22.19.6842-6853.2002
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A 2005; 102:719-24; PMID:15647368; http://dx.doi.org/10.1073/pnas.0408894102
- Putignani L, Raffa S, Pescosolido R, Aimati L, Signore F, Torrisi MR, Grammatico P. Alteration of expression levels of the oxidative phosphorylation system (OXPHOS) in breast cancer cell mitochondria. Breast Cancer Res Treat 2008; 110:439-52; PMID:17899367; http://dx.doi.org/10.1007/s10549-007-9738-x
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J 2001; 20:1910-20; PMID:11296224; http://dx.doi.org/10.1093/emboj/20.8.1910
- Biswas G, Tang W, Sondheimer N, Guha M, Bansal S, Avadhani NG. A distinctive physiological role for IkappaBbeta in the propagation of mitochondrial respiratory stress signaling. J Biol Chem 2008; 283:12586-94; PMID:18272519; http://dx.doi.org/10.1074/jbc.M710481200
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993; 4:289-94; PMID:7689389; http://dx.doi.org/10.1038/ng0793-289
- Raimundo N, Song L, Shutt TE, McKay SE, Cotney J, Guan MX, Gilliland TC, Hohuan D, Santos-Sacchi J, Shadel GS. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 2012; 148:716-26; PMID:22341444; http://dx.doi.org/10.1016/j.cell.2011.12.027
- Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, Uchikado Y, Natsugoe S, Nakagawa H, Avadhani NG. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2015; 35:1585-95; PMID:26148236
- MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 2007; 27:3282-9; PMID:17325041; http://dx.doi.org/10.1128/MCB.01927-06
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. Hypoxia as a therapy for mitochondrial disease. Science (New York, NY) 2016; 352:54-61; PMID:26917594; doi: aad9642
- Kurihara T, Westenskow PD, Gantner ML, Usui Y, Schultz A, Bravo S, Aguilar E, Wittgrove C, Friedlander MS, Paris LP, et al. Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. eLife 2016; 5; PMID:26978795; http://dx.doi.org/10.7554/eLife.14319