
ABSTRACT
Genome editing via the CRISPR/Cas9 RNA-guided nuclease system has opened up exciting possibilities for genetic analysis. However, technical challenges associated with homology-directed repair have proven to be roadblocks for producing changes in the absence of unwanted, secondary mutations commonly known as “scars.” To address these issues, we developed a 2-stage, marker-assisted strategy to facilitate precise, “scarless” edits in Drosophila with a minimal requirement for molecular screening. Using this method, we modified 2 base pairs in a gene of interest without altering the final sequence of the CRISPR cut sites. We executed this 2-stage allele swap using a novel transformation marker that drives expression in the pupal wings, which can be screened for in the presence of common eye-expressing reporters. The tools we developed can be used to make a single change or a series of allelic substitutions in a region of interest in any D. melanogaster genetic background as well as in other Drosophila species.
Introduction
In recent years, genome editing by site-specific nucleases has rapidly increased in accessibility, ease, and efficiency. Most notably, the CRISPR/Cas9 RNA-guided nuclease system has been developed and optimized for a variety of applications from basic gene disruption to knock-ins, endogenous tagging of proteins, and modulation of gene expression.Citation1–5 In the CRISPR/Cas9 system, adapted from Streptococcus pyogenes (henceforth shortened as “CRISPR”), the Cas9 endonuclease complexes with a single-guide RNA (sgRNA), which is designed by the experimenter to match a ∼20bp target sequence, and causes double-strand cleavage 3 nucleotides 5′ of the 3′ end of the target sequence.Citation5 The selection of sgRNA target sites is flexible, but a protospacer-adjacent motif (PAM) (the trinucleotide 5′-NGG-3′) is required immediately 3′ of the 20bp target sequence.Citation5 After the double-strand break is induced at the target site, the cell's native DNA repair machinery can (i) rejoin the 2 ends of the break through non-homologous end joining (NHEJ), an error-prone process that frequently results in small insertions or deletions, or (ii) close the gap using homology-directed repair (HDR), in which a DNA molecule with sequence homologous to the break site is used as a repair template.Citation3 The repair template used for double-strand break repair by HDR can be a homologous chromosome or an exogenous donor molecule with homology to both sides of the break site.
Exogenous repair templates, which are generally used to make one or more nucleotide changes in a sequence of interest, typically come in one of 2 forms: a single-stranded oligonucleotide donor (ssODN)Citation1 or a double-stranded DNA (dsDNA) plasmid.Citation4,6 ssODNs are more quickly synthesized, but most synthesis companies limit their length to ∼200bp, making them useful only when a suitable CRISPR target site is in close proximity to the site of interest ().Citation3,4,6,7 dsDNA plasmids can be much larger than ssODNs, allowing modifications to be made at a greater distance from the cleavage site as well as allowing larger sequences to be inserted at the site of repair.Citation4,6 To modify larger regions of sequence, 2 target sites can be used to cut out the region to be modified. In this case, the dsDNA used for repair contains the desired change(s) flanked by homology arms targeting regions of DNA outside the 2 target sites (). Regardless of which type of repair template is used, the end result of homology-directed repair (HDR) is that the DNA sequence included in the repair template is incorporated into the native locus.
Figure 1. Challenges for single-stage allele replacement strategies using CRISPR. (A) HDR using a ssODN as repair template is shown. Because ssODNs are limited to ∼200bp, the sgRNA target site (shown in green) must be in close proximity to the sequence to be edited, as the ssODN must span the repair site and have homology to both sides. Unless the introduced change disrupts the target site, the edited locus may be re-cleaved by Cas9, potentially leading to error-prone NHEJ repair. (B) HDR using a dsDNA plasmid as the repair template is shown. If the genomic target site sequences (shown in blue and purple) are present in the donor plasmid, Cas9 cleavage may lead to cutting and subsequent degradation of the donor plasmid; successful repair events will also be vulnerable to cleavage (C) A second mutation at the PAM site (NGG → N*G) may be introduced to prevent re-cleavage of the edited sequence, but this additional mutation(s) may have undesired phenotypic effects. (D) Mutating the PAM sites (NGG → N*G) prevents unwanted cleavage of dsDNA donor or repaired genomic sequence, but involves the addition of extra mutations known as “scars” that may affect phenotypes. Abbreviations used in the figure are defined as follows: NGG = protospacer-adjacent motif; CCN = reverse complement (opposite strand) PAM sequence; N*G or C*N = mutated protospacer-adjacent motif; ssODN = single-stranded oligodeoxynucleotide donor; HDR = Homology-directed repair; HA = Homology arm. Scissor symbols represent target sites expected to be cleaved.
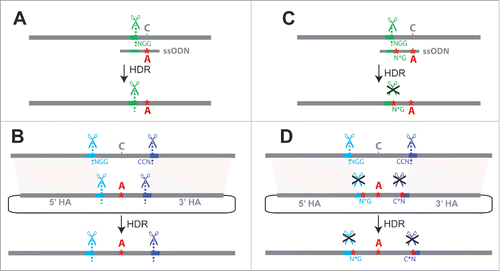
Using CRISPR-induced HDR to edit specific nucleotides is not always straightforward, however. A major challenge results from the fact that the target site sequence recognized by the sgRNA that directs the initial double-stranded break in the genome is also typically present in the repair template (a.k.a. donor DNA) as well as in the genome after editing. These sequences also interact with the sgRNA-Cas9 complex and experience unwanted cleavage, complicating the process of editing nucleotides in a single step of HDR (). This unwanted cleavage can be prevented by introducing one or more secondary changes that ablate the PAM site or alter the target site in the donor DNA in addition to the desired sequence edits, preventing Cas9 from cleaving the donor as well as the genome after it has been edited ().Citation4,8,9 Because these secondary mutations, or “scars,” remain in the genome after editing, care must be taken to minimize their phenotypic effects. When the desired sequence edits are located in (or near) a coding sequence, a synonymous change(s) can be introduced as the secondary change(s) to prevent recutting.Citation4,9,10 Synonymous changes are often assumed to have little impact on protein function, but they do have phenotypic effects in some cases.Citation11–13 When the desired changes are located in a non-coding region far from coding sequence, the PAM site can be ablated with a secondary non-coding change, but the impact of non-coding changes is even more difficult to predict.Citation11 Ideally, HDR should be used to change only the desired nucleotide(s) without introducing any other changes to the final modified genome (i.e. “scarless” editing).
A second challenge when using CRISPR to modify genomes is identifying individuals that have successfully inherited the desired genome alterations. This challenge is especially acute for modifications that require HDR because HDR resolution of double-strand breaks tends to be more rare than non-homologous end joining (NHEJ)Citation14 and imprecise HDR often occurs that results in unwanted changes such as incorporating additional DNA at the edited locus.Citation15 In multicellular species, only genome edits present in germ cells can be transmitted to offspring, and each individual carrying a desired genome modification in its germline (a “founder”) may transmit it to only a small percentage of its progeny.Citation1,6,16 Molecular techniques such as high-resolution melt analysis, Surveyor assays, or even complete sequencing of the targeted region can be used to identify F1 individuals with edited genomes and are especially useful in cultured cells that can be propagated throughout the screening process.Citation4,6,17 Molecular screening methods can be costly and time consuming in a species such as Drosophila, however, where F1 progeny are usually genotyped only after F2 progeny are produced because the F1 individuals must be killed to extract DNA and perform genotyping most reliably. As an alternative, Drosophila researchers often choose to incorporate a visible transformation marker for phenotypic screening.Citation3
When gene deletion, disruption or tagging are desired, a selectable marker may be permanently integrated at the targeted locus, but if the goal is to determine the effects of precise nucleotide changes, any transformation markers used should be removed prior to phenotypic analysis. Recombinase-mediated cassette exchange (RMCE) has been used to remove and replace such a marker gene in DrosophilaCitation18; however, RMCE is not ideal for this purpose because it leaves scars in the form of 2 attR sequences at the site of reporter excision. A related strategy has recently been developed in which the transformation marker is flanked by repeats that are recognized by the PiggyBac transposase (http://flycrispr.molbio.wisc.edu/scarless). PiggyBac-mediated excision leaves a TTAA motif behind, thus this method can be used to remove the transformation marker in a scarless manner when the locus of interest contains an endogenous TTAA motif. An alternative scarless “pop-in/pop-out” strategy was recently described for use in mammalian cells in which a fluorescent reporter gene is inserted along with the desired nucleotide changes in a first transformation step (pop-in), allowing modified cells to be identified using fluorescence-activated cell sorting (FACS), and then removed via a second round of CRISPR editing (pop-out).Citation19,20
Here, we describe an alternative pop-in/pop-out strategy for precisely editing one or more nucleotides in Drosophila. This method requires less molecular screening than single-stage allele replacement strategies and does not result in any unwanted sequence changes in the genome (i.e., it is scarless). It uses a customizable intermediate donor plasmid with a fluorescent reporter gene for easily identifying germline transmission of HDR events that is expressed in the pupal wings, allowing screening for it in the presence of the widely used eye-expressing fluorescent markers. This reporter gene is then cleanly replaced with the desired sequence, resulting in a “scarless” allele swap. We have successfully used this method to introduce 2 single-nucleotide changes into a D. melanogaster genome without inducing any additional modifications. All components used for these reactions (dsDNA repair templates and plasmids encoding Cas9 and sgRNAs) were co-injected into the embryo, making this method suitable for use in any strain of D. melanogaster as well as in other Drosophila species.
Results
The potential of using CRISPR to modify as few as one nucleotide in the genome makes it a powerful tool for testing the phenotypic consequences of changes in DNA sequence ranging from single nucleotide variants to more substantial differences in haplotype. We developed our strategy for making precise nucleotide changes in Drosophila by using CRISPR to modify a strain of D. melanogaster (melDA tan) that carries a Drosophila americana allele of the tan gene in a PiggyBac transgene marked with an eye-expressing green fluorescent protein (GFP). This strain also carries loss-of-function mutations in the endogenous D. melanogaster yellow, white, and tan genes (ywt), with the D. americana tan transgene rescuing the tan mutant phenotype. The D. americana tan allele was inserted into D. melanogaster to study changes in its first intron that contribute to pigmentation divergence between D. americana and its sister species D. novamexicana,Citation21 thus we targeted the first intron of tan for genome modification when developing the tools described below.
First, we identified unique CRISPR target sites flanking our region of interest by using the flyCRISPR Optimal Target Finder tool to rule out target sites with sequence similarity elsewhere in the genome that might cause off-target cleavage.Citation22 We chose to place the 5′ target site within the first exon of the tan transgene so that the pigmentation phenotype could serve as a secondary indicator of successful gene disruption and later repair (). The 3′ target site was located in the first intron of tan. Each of these 2 selected target sequences was then cloned into its own sgRNA expression plasmid (pCFD3).Citation23
Figure 2. Schematic of marker-assisted, 2-stage allele swap within the D. americana tan transgene in D. melanogaster. In stage 1, the 3′ end of the first exon (black rectangle) and a portion of the first intron of D. americana tan were excised by cleavage at the t5 (antisense direction) and t3 sgRNA target sites shown in brown. In the donor plasmid used to repair this region, pGEM-WingGFP-tan, the PAM sites (highlighted in yellow) and 3 PAM-proximal nucleotides at each target site contain sequence from the native target site, but the remaining 17 nucleotides of each sgRNA target sequence have been edited to differ from the D. americana tan sequence. These edited sequences serve as new, unique CRISPR target sites for reporter excision, and are labeled as t5re and t3re (“re” for “reporter excision”). These t5re and t3re target sites are not recognized by the sgRNA-Cas9 complexes targeting sites t5 and t3, thus preventing cleavage of pGEM-WingGFP-tan or the HDR product. When the wing-GFP transformation marker was incorporated into the genome, so were these unique t5re and t3re target sites, which contain restriction sites that double as multiple cloning regions. The donor plasmid used for stage 2, pGEM-tan-edits, contained the region of the D. americana tan sequence amplified with primers shown as arrows labeled A and B, which was cloned into the pGEM T-Easy vector. Changes in the length of 2 homopolymer runs used to confirm genome modification are represented by red asterisks. sgRNAs targeting the t5re and t3re sites flanking the reporter gene were used to remove it, with the D. americana tan sequence restored from pGEM-tan-edits via HDR. Locations of PCR primers used to test flies that lost wing-GFP expression following stage 2 of the allele swap are shown with arrows labeled X and Y. Precise HDR was confirmed by Sanger sequencing the amplicon produced by these primers. For primer sequences and details about screening PCRs, see Table S1 in the supplement. Sanger sequencing chromatograms for all edited sites are shown in Figure S1.
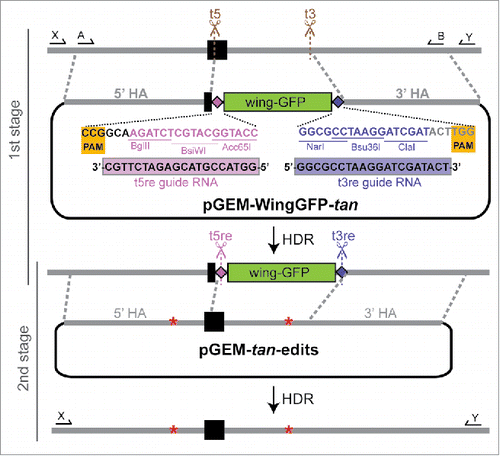
Next, we constructed an intermediate donor plasmid designed to replace our region of interest with a visible transformation marker flanked by unique CRISPR target sites (). The melDA tan strain we sought to modify already contained an eye-expressing red fluorescent protein (RFP) reporter gene marking the attP landing-site used to insert the piggyBac transgene and an eye-expressing GFP marking the transgene, so we chose a transformation marker for CRISPR that expressed a fluorescent protein in a tissue other than the eye: a GFP reporter gene under the control of a ∼1kb enhancer of the D. melanogaster yellow gene that drives robust expression in pupal wings.Citation24 This reporter gene was amplified from genomic DNA of a previously constructed transgenic line using primers with sequences designed to function as unique CRISPR target sites appended at the 5′ and 3′ ends (). These unique CRISPR target sites showed no exact matches (using a NCBI BLAST search) in any sequenced Drosophila genome, making them suitable for use in most, if not all, Drosophila species. They also contained cut sites for restriction enzymes, which can be used to easily remove and replace the wing-expressing GFP reporter gene with a different transformation marker.
To direct this reporter gene to the desired region of the genome, homology arms adjacent to the original CRISPR target sites in the tan transgene were amplified and attached to the ends of this reporter gene flanked by unique CRISPR target sites in the pGEM T-Easy plasmid using Gibson Assembly ().Citation25 The restriction enzyme cut sites in the pGEM T-Easy part of this pGEM-WingGFP-tan donor plasmid can be used in combination with the restriction enzyme cut sites in the unique CRISPR target sequences to easily replace one or both homology arms with different sequences for other studies ().
We injected 1220 melDA tan embryos with a mixture of the pGEM-WingGFP-tan plasmid, both sgRNA expression plasmids targeting the D. americana tan transgene sequence, and a pBS-Hsp70-Cas9 expression plasmid producing Cas9 protein. We crossed 150 of the adult flies that emerged from these injected embryos back to the melDA tan strain and screened their F1 progeny for inheritance of the reporter gene by looking for GFP expression in the developing pupal wings daily under a GFP-enabled stereoscope (). Six of these 150 injected flies produced progeny with GFP expression in pupal wings and were thus considered “founders.” The percentage of progeny expressing the WingGFP reporter construct from each founder ranged from 2.5% to 25.4%. In all, 70 pupae were positive for wing GFP expression, 43 of which were ultimately used to establish lines homozygous for the wing-expressing GFP marker gene. A summary of these statistics is provided in Table S2.
Figure 3. Representative pigmentation and fluorescence phenotypes of flies at each stage of the tan allele swap process. (A) Dorsal pigmentation of adult flies is shown for the melDA tan strain prior to editing (left), the melDA tan strain in which the targeted region of tan has been replaced with the wing-GFP marker (middle), and the melDA tan strain after the wing-GFP marker was replaced with the edited tan sequence (right). The darker pigmentation seen in flies on the left and right is caused by a functional D. americana tan transgene. When this transgene is disrupted (middle), pigmentation is visibly lighter on the dorsal head, thorax, and abdomen. Double-headed black arrows indicate areas in the thorax and abdomen where the change in pigmentation was most readily apparent. (B) GFP fluorescence in late-stage pupae is shown for the melDA tan strain prior to editing (left), the melDA tan strain in which the targeted region of tan has been replaced with the wing-GFP marker (middle), and the melDA tan strain after the wing-GFP marker was replaced with the edited tan sequence (right). The GFP fluorescence in eyes of all 3 flies results from the 3XP3-GFP reporter gene included in the D. americana tan transgene. GFP expression in the developing wings (indicated with arrows) is visible in flies after the first stage of the 2-step allele swap procedure (middle) and lost following the second stage (right). All pupae shown were deemed to be at the same developmental stage based on visible features of wing development, expression of 3XP3-GFP, and lack of pigmentation on the developing wing.
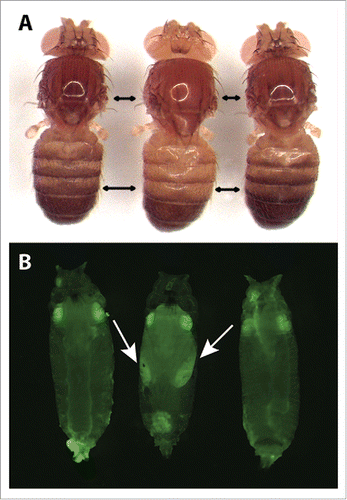
All 43 of these homozygous lines showed lighter body pigmentation than the D. melDA tan parental line, consistent with the marker gene disrupting the D. americana tan transgene in an otherwise tan mutant D. melanogaster genetic background (). The pGEM T-Easy vector backbone was found to have been incorporated along with the marker gene in 28 of these 43 lines, and these lines were excluded from further study. PCR amplifications were then used to check the 5′ and 3′ insertion sites of the WingGFP marker in the remaining 15 lines, 13 of which were found to have incorporated it into the correct genomic location. DNA sequencing subsequently confirmed that the GFP reporter gene, unique CRISPR target sites, and homology regions were as expected in all 13 of these lines. Two of these 13 lines (25.17 and 9.14) were expanded for embryo collection and subsequent injection to excise the GFP reporter gene and replace it with a modified tan sequence.
A second donor plasmid was designed to restore the function of the D. americana tan transgene by replacing the wing-expressing GFP transformation marker with tan sequence excised in the first step. This plasmid, pGEM-tan-edits, was constructed by amplifying the D. americana tan transgene sequence from the beginning of the 5′ homology arm upstream of exon 1 to the end of the 3′ homology arm in the first intron (which includes the original CRISPR target sites) and cloning it into the pGEM T-Easy plasmid (). The specific amplicon chosen to construct this plasmid contained 2 changes in non-coding homopolymer runs (9T->10T in the 5′ homology arm and10T->9T in the intron) that were not expected to affect the function of this sequence (). These changes were included to allow us to confirm that the transformants recovered were not contaminants from the original melDA tan strain. Guide RNAs matching the 2 unique CRISPR target sites introduced with the GFP transformation marker were also cloned into the pCFD3 sgRNA expression plasmid.
We injected 631 embryos from the wing-GFP expressing line 25.17 and 730 from the wing-GFP expressing line 9.14 with a mixture containing the pBS-Hsp70-Cas9 expression plasmid, pCFD3 sgRNA expression plasmids targeting the unique CRISPR target sites, and pGEM-tan-edits. We crossed 179 flies emerging from these injected embryos to the same ywt strain of D. melanogaster that harbored the original D. americana tan transgene. Pupae from these crosses were screened for the absence of wing-expressing GFP along with the presence of eye-expressing GFP and RFP indicating presence of the tan transgene and the attP landing site the transgene was inserted into, respectively. From the 15 crosses found to contain one or more pupae that met these criteria, we collected a total of 32 pupae with this pattern of fluorescence before eclosion. Adult flies emerging from these pupae were crossed to a third chromosome (TM6B) balancer line and their progeny screened a second time for pupal wing GFP expression to make sure that the transient wing fluorescence was not simply missed during the initial screen. Ultimately, 5 founders produced 11 flies whose progeny were verified to have lost wing-expressing GFP. Ten of these 11 progeny were successfully used to establish lines homozygous for the edited transgene (), none of which showed evidence of the pGEM T-Easy backbone being incorporated. One of these 10 lines showed the ∼2.5kb product expected from a PCR spanning the edited region from one homology region to the other and had dark pigmentation consistent with a rescue of D. americana tan function (). The remaining 9 lines failed to produce the expected PCR product and had light pigmentation suggesting that the tan sequence was not successfully restored. Sanger sequencing of the modified region of D. americana tan in the line with dark pigmentation showed precise repair with both homopolymer runs matching the donor plasmid sequence rather than the original transgene sequence (Fig. S1), confirming that we successfully introduced 2 single-nucleotide changes in the D. americana tan transgene in our desired D. melanogaster genetic background. A summary of efficiency at each stage of this second swap is provided in Table S3.
The fact that mutations in both the 5′ homology arm and the intron were incorporated suggests that the HDR was likely initiated from a double-strand break at the t3re site. Because HDR is initiated by one of the two free 3′ ends at the double-strand break, repair from the t5re site could only result in the incorporation of one or the other of these mutations, which are positioned to either side of the t5re site, whereas repair from the t3re site could incorporate both mutations.Citation26 We mention the directionality of repair to illustrate the importance of careful experimental design regarding the position of desired insertions/mutations relative to CRISPR target sites.
All of the reagents used in this work were developed with flexibility for future studies in mind. For example, restriction sites were included in the unique CRISPR target sequences for easy modifications, as described above. We have already used these restriction sites to make an alternative version of the intermediate donor vector (pGEM-WingGFP-tan) in which the pupal wing-expressing GFP reporter gene was replaced with an eye-expressing RFP (pGEM-3XP3.RFP-tan). Fluorescent proteins expressed in the adult eye by the 3XP3 promoter have been shown to function in a wide variety of insect taxa,Citation27,28 making this donor plasmid useful for HDR not only in D. melanogaster, but also in many other insect species. The wing-GFP marker we used allows screening in lines that already carry eye-expressing markers or have eye color that makes the detection of eye-expressing fluorescent markers difficult, but we encourage the use of the 3xP3-RFP intermediate donor when screening for fluorescence in the eyes of white mutant flies is possible because it is less laborious than screening for the gain and loss of expression from the pupal wing-GFP marker. pGEM-3XP3.RFP-tan also contains restriction sites in novel CRISPR target sites introduced to prevent re-cutting and facilitate easy cloning. The homology arms in this plasmid target the D. americana tan gene, but other researchers can replace these homology arms with their own sequences of interest. When preparing reagents for a new locus, it should be noted that the 3 PAM-proximal nucleotides of these unique CRISPR target sequences are specific to each locus, and the sgRNAs should be customized to match the locus targeted by the donor plasmid ().
Discussion
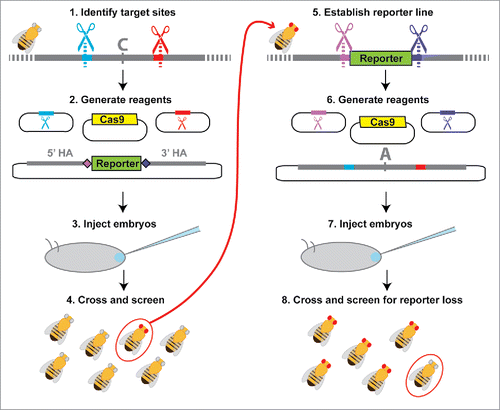
We have developed tools and protocols to implement a 2-step, marker-assisted genome editing strategy suitable for making precise changes at targeted sites in Drosophila with greatly reduced requirements for molecular screening (). Our method adds to the few available techniques that leave no unwanted changes (“scars”) in the genome, such as those that occur when ablating PAM sites or using integrase-mediated excision to remove selectable markers (http://flycrispr.molbio.wisc.edu/scarless).Citation19,20 Our method is also better-suited than these other methods for making a series of allelic changes at the same locus, as the intermediate line containing the marker gene need only be generated once. This is useful, for example, when reintroducing the original, unedited sequence in parallel with an experimental manipulation as a control for side effects of the CRISPR process or when testing a set of allelic variants to identify sites with specific functions.
Figure 4. Workflow for 2-stage marker assisted allele swap. (1) Target sites flanking the area to be edited are identified (red and blue) using online tools to identify optimal target sites and search for potential off-target cleavage sites.Citation22,39 (2) Sequences from the selected target sites are then cloned into sgRNA expression plasmids or used to generate in vitro transcribed sgRNAs. Homology arms flanking the region of interest (recommended length ∼1kb) are cloned into the reporter donor plasmid, which contains unique CRISPR target sites (light and dark purple) in place of the genomic target sites (red and blue). (3) Embryos are injected with the donor plasmid, sgRNAs (expression plasmids or in vitro transcribed RNAs) and a source of Cas9 (expression plasmid, mRNA, or protein) unless any of these components is produced by a transgene already present in the host. (4) Adult flies that develop from the injected embryos are collected as virgins and then crossed back to the parental line. F1 progeny emerging from these crosses are screened for the selectable marker, with flies positive for the selectable marker allowed to produce F2 progeny before extracting their DNA for molecular screening. (5) Individuals with the correct reporter gene insertion are made homozygous and the population is expanded for embryo collections. (6) sgRNAs with sequences matching the unique CRISPR target sites introduced with the reporter gene (light and dark purple) as well as a plasmid containing the original CRISPR target sites and the edited version of the original sequence are prepared. (7) Flies carrying the reporter at the locus of interest are injected for the second allele swap step. (8) Because the selectable marker is dominant, adult flies developing from injected embryos must be crossed back to either the original parental line from step (1) or to a balancer line (if available) for screening. Progeny from this cross that do not show expression of the selectable marker are then crossed and analyzed with molecular tests to determine whether they contain the desired editing events.
With three scarless, 2-stage allele swap methods now described (Xi et al.,Citation20 http://flycrispr.molbio.wisc.edu/scarless, and this study), researchers should consider the differences among these methods when designing their own experiments. First, both our method and the pHD-ScarlessDsRed method (http://flycrispr.molbio.wisc.edu/scarless) are specifically optimized for use in Drosophila, whereas the “pop-in/pop-out” strategy described by Xi et alCitation20 uses reagents designed for use in mammalian cells. Second, the pHD-ScarlessDsRed method requires the presence of a TTAA motif at the target locus for scarless editing, which adds some restriction to target site selection, but circumvents the need for a second round of injections when working with D. melanogaster because flies carrying the reporter can be crossed to existing transgenic lines that express the PiggyBac transposase.Citation29 However, to apply this method in species other than D. melanogaster, a second injection step will still be required in order to introduce the PiggyBac transposase. Third, our method uses a novel transformation marker and uniquely designed target sites in the reporter construct which double as cloning sites for later customization of homology arms or reporter cassette. Finally, with both the Xi et al.Citation20 and pHD-ScarlessDsRed methods, the desired changes are introduced along with a reporter gene during the first step and the reporter gene is excised in the second step. In our method, only the reporter gene is introduced in the first step. In instances where larger genomic regions are being edited, this feature reduces the size of the region that must be inserted initially by HDR, which increases efficiency,Citation30 and creates a stable genotype that can be used to eliminate the first stage CRISPR modification in any future experiments that modify the same locus.
Our method is also particularly well-suited for use in any D. melanogaster genetic background or in any Drosophila species. This feature realizes the great potential of genome editing via site-specific nucleases for making genetic manipulations at a gene's native locus and in its native genomic background. For example, the function of sites that have diverged between 2 Drosophila species can now be tested in their native context rather than in a heterologous species such as D. melanogaster.Citation31–33 However, currently available Cas9- and sgRNA-expression plasmids (including those used in this study) contain promoters derived from D. melanogaster, so injecting purified Cas9 protein or mRNA along with in vitro transcribed sgRNAs instead of using expression plasmids will likely give better results when working with other Drosophila species.Citation17,34,35 We have recently had success using commercially available Cas9 protein and in vitro transcribed sgRNAs to induce NHEJ and/or HDR in Drosophila elegans, Drosophila americana, Drosophila novamexicana, and Drosophila virils (unpublished data).
The two-stage allele swap method reported here provides additional precision and flexibility for allele replacements using CRISPR in Drosophila. Further modifications are likely to increase the efficiency of this method even more, however. For example, if a specific genetic background is not required, one of several lines of D. melanogaster developed to increase CRISPR efficiency can be used, such as lines with Cas9 and/or sgRNA expressed from transgenes integrated into the genomeCitation16,36,37 or lines with reduced lig4 activity that increase the frequency of HDR events by inhibiting the NHEJ pathway.Citation10,38 Similar lines could also be constructed in other Drosophila species to optimize CRISPR-based genome modifications in these hosts. Selection of sgRNA target sites may also be optimized to maximize the likelihood of cleavage according to criteria that have been identified in other studies.Citation39–41 We note, however, that the need to improve CRISPR efficiency is decreased by the use of methods which employ fast and easy phenotypic screening of large populations.
In designing genome editing experiments, the most pertinent strategies and screening techniques will depend on the types of changes desired as well as the resources available to the researcher. For instance, experiments to alter the coding sequence of essential genes would rule out the use of an intermediate stage that disrupts both copies of the gene, which would preclude the use of our 2-stage method as described. Nonetheless, the applicability of our method to many other types of experiments in a wide variety of genetic backgrounds makes it a valuable addition to the existing methods and tools for scarless genome modifications available to the Drosophila research community.
Materials and methods
Fly strains
The D. americana tan transgene was constructed as previously described in Wittkopp et al.Citation21 The transgenic strain of D. melanogaster melDA tan was constructed by integrating this D. americana tan transgene and a 3XP3-GFP transformation marker in a piggyBac plasmid containing an attB sequence into an attP landing site marked with 3XP3-RFP (Flybase ID FBst0024749) at cytological location 86Fb on the third chromosome using phi-C31-mediated integration.Citation42 GenetiVision (Houston, TX) performed the injections that produced the melDA tan transgenic line. Transformant flies carrying the melDA tan transgene were crossed to a line that was mutant for yellow, white, and tan (ywt) to confirm that the D. americana tan transgene rescued the D. melanogaster tan mutant phenotype and to allow easier detection of the 3xP3-GFP and 3xP3-RFP fluorescent markers.
The transgenic D. melanogaster line carrying the wing-expressing GFP reporter gene (referred to as “line 890”) was constructed using D. melanogaster yellow enhancer sub-element “mel_a2” described in Kalay, 2012.Citation24 The reporter gene from line 890 was chosen as a selectable marker for this study because of its clear expression pattern in the developing wings, which can be screened independently of the eye-expressing fluorescent markers present in melDA tan.
To generate lines homozygous for the edited D. americana tan transgene on the third chromosomes, we first constructed a ywt;+;TM6B strain by crossing the TM6B third chromosome balancer (Flybase ID FBst0007197) into the same ywt genetic background used in the construction of melDA tan. This ywt;+;TM6B genotype was crossed with the originally recovered melDA tan flies to produce a stock homozygous for ywt as well as melDA tan. Flies were maintained at ∼25°C on standard cornmeal media except were otherwise specified.
Plasmids
To construct pGEM-WingGFP-tan, the wing-expressing GFP reporter gene sequence from line 890 and homology arms flanking the targeted region of D. americana tan were PCR amplified to generate overlapping regions of homology for cloning into the pGEM T-easy vector via Gibson Assembly.Citation25 The 831bp 5′ homology arm was PCR amplified from D. americana tan using primer pair 5 (Table S1), which appended a region homologous to the pGEM T-easy vector on the 5′ end and added new sequence to form the t5re target site on the 3′ end (). The 966bp 3′ homology arm was amplified using primer pair 6 (Table S1), appending new sequence to form the t3re target site at the 5′ end () and a region homologous to the pGEM T-easy vector to the 5′ end. Both homology arm PCR reactions used melDA tan genomic DNA as template. The wing-expressing GFP reporter was PCR amplified from line 890 genomic DNA using primer pair 7 (Table S1), which appended the t5re target site to the 5′ end and the t3re target site to the 3′end (). These amplicons and the pGEM T-easy vector were assembled using New England Biolabs (NEB) Gibson Assembly Master Mix.
pGEM-tan-edits, the donor plasmid used for the second stage of the allele swap, was generated by PCR amplifying the targeted D. americana tan region along with the flanking homology regions from melDA tan genomic DNA using primer pair 8 (Table S1, see ) and inserting the resulting amplicon into pGEM T-Easy vector via Gibson Assembly.
To construct the eye-expressing RFP donor, pGEM-3XP3.RFP-tan, the 3XP3-RFP reporter was PCR amplified from D. melanogaster genomic DNA containing the M{3XP3-RFP.attP}ZH-51C landing site (Flybase ID FBtp0023088) using primer pair 11, which added Acc65I and Bsu36I restriction sites (Table S1). Before constructing pGEM-3XP3.RFP-tan, we had replaced the 3′ homology arm of pGEM-wingGFP-tan with sequence from another region of D. americana tan using Bsu36I and MluI restriction sites. This new homology arm was amplified from melDA tan genomic DNA using primer pair 10 (Table S1). The 3XP3-RFP reporter amplicon was cloned into this modified pGEM-WingGFP-tan plasmid using the Acc65I and Bsu36I restriction enzyme cut sites.
sgRNA expression plasmids were made by ligating target-site specific annealed oligonucleotide inserts into BbsI-digested pCFD3 (Addgene # 49410) according to the methods described by Port et al.Citation23 The following oligonucleotide pairs were used to generate the cloning inserts for the indicated sgRNA target sites: ‘t5’ – primer pair 12, ‘t3’ – primer pair 13, ‘t5re’ – primer pair 14, ‘t3re’ – primer pair 15 (Table S1). We used the pBS-Hsp70-Cas9 plasmid (Addgene #46294) as a Cas9 source.
Drosophila husbandry and injection
Plasmids for CRISPR were prepared for injection using either Zymo Zyppy Plasmid Maxi Prep kit or Mechery-Nagle Nucleobond Xtra EF Midi Prep kit followed by ethanol precipitation and re-suspension in nuclease-free water. For all injections, plasmid concentrations were as follows: 500ng/µL HDR donor, 100ng/µL each sgRNA plasmid, 250ng/µL pBS-Hsp70-Cas9. After injection, embryos were maintained at 25°C for 3–4 d, at which time larvae were moved to vials with cornmeal media. Embryo injections were performed as described previously.Citation43 For pupal wing reporter screening, flies were moved to 18°C upon entering the wandering larval stage to slow development in an effort to prolong the amount of time the fluorescent marker signal was present.
Fluorescence screening
To screen for the presence of fluorescent markers, we used a Leica MZ6 stereoscope equipped with a Kramer Scientific Quad Fluorescence Illuminator. GFP expression from the wing-GFP reporter gene used in this study becomes easily detectable in the wings after the developing wing is clearly visible, but before 3XP3-GFP signal is visible in the eyes. GFP signal in wings is easily detectable for approximately 2 d at 18°C, with GFP signal fading rapidly at the onset of wing pigmentation.
To screen for the presence or absence of the wing-expressing GFP marker, F1 pupae (progeny of injected parents) were observed daily under the GFP stereoscope at 18°C. After the first stage of the allele swap, when the wing-GFP marker was inserted, pupae with detectable GFP expression in the developing wings were removed from the vial with a wet paintbrush and isolated in a ventilated microcentrifuge tube with food to await future crossing and molecular screening. Surviving wing-GFP positive pupae were crossed to the ywt TM6B balancer line. From these balancer crosses, siblings with both the wing-GFP phenotype and the TM6B bristle phenotype were crossed to form homozygous lines.
Following the second stage of the allele swap (marker excision and replacement), pupae with detectable GFP expression in the developing wings were removed and discarded. Any pupae that remained in the vial until their wings darkened were removed and isolated for future crossing. Crosses were performed as described in the results.
Molecular screening
To test for the presence of unwanted pGEM T-easy vector (plasmid “backbone”) in edited flies, we used PCR reactions with one primer in the vector backbone and the other in either the 5′ or 3′ homology arm, using primer pair 1 and primer pair 2 for the 5′ and 3′ sides, respectively (Table S1), while the donor plasmid was used as a positive control template. Strains that produced a band from either of these PCR reactions were excluded from further study.
To confirm integration of the wing-GFP reporter gene into the correct genomic location, we used a PCR reaction that amplifies DNA sequence from within the reporter gene sequence to outside the homology region on both the 5′ and 3′ sides of the reporter gene, using primer pair 3 to screen the 5′ side and primer pair 4 to screen the 3′ side (Table S1). The amplicons from these PCR reactions were Sanger sequenced to confirm scarless repair at both the target sites and throughout both homology regions. To screen for correct HDR after the second stage of the allele swap, the entire edited locus was amplified via PCR using primers outside the homology regions (see and primer pair 9 in Table S1 for details). This amplicon was Sanger sequenced to confirm the presence of expected sequence edits.
All diagnostic PCRs were performed using genomic DNA extracted from single flies following the Gloor and Engels “squish prep” protocol.Citation44
Imaging
Fly images shown in were captured using a Leica MZFLIII fluorescence stereoscope equipped with a Leica DC480 microscope camera.
Abbreviations
| CRISPR | = | Conserved Regularly Interspaced Short Palindromic Repeats |
| Cas9 | = | CRISPR-associated protein-9 nuclease |
| sgRNA | = | single guide RNA |
| PAM | = | protospacer-adjacent motif |
| ssODN | = | single-stranded oligodeoxynucleotide |
| dsDNA | = | double stranded DNA |
| NHEJ | = | Non-homologous end-joining |
| HDR | = | Homology-directed repair |
| GFP | = | green fluorescent protein |
| RFP | = | red fluorescent protein |
| SNP | = | single nucleotide polymorphism |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KFLY_A_1220463_Supplemental.pdf
Download PDF (1.6 MB)Acknowledgments
We would like to thank Kathy Vaccarro and the Carroll Lab at University of Wisconsin for training and assistance in microinjection techniques, Scott Barolo and Lisa Johnson for assistance optimizing microinjection procedures, and Fabien Duveau, Andrea Hodgins-Davis, Alisha John, Jennifer Lachoweic, and Jonathan Massey for feedback and comments on this manuscript.
Funding
This work was funded by the National Institute of Health (5-R01-GM-089736, P.J.W.), the National Institute of Health Genetics Training Grant (T32GM00754, A.M.L.), and the National Science Foundation Graduate Research Fellowship Program (DGC 1256260 A.M.L.).
Related Research Data
References
- Gratz SJ, Wildonger J, Harrison MM, O'Connor-Giles KM. CRISPR/Cas9-mediated genome engineering and the promise of designer flies on demand. Fly (Austin) 2013; 7:249-55; PMID:24088745; http://dx.doi.org/10.4161/fly.26566
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 2013; 10:977-9; PMID:23892898; http://dx.doi.org/10.1038/nmeth.2598
- Gratz SJ, Rubinstein CD, Harrison MM, Wildonger J, O'Connor-Giles KM. CRISPR-Cas9 genome editing in Drosophila. In: Current protocols in molecular biology. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2015. page 31.2.1-31.2.20
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8:2281-308; PMID:24157548; http://dx.doi.org/10.1038/nprot.2013.143
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337:816-22; PMID:22745249; http://dx.doi.org/10.1126/science.1225829
- Bassett AR, Liu JL. CRISPR/Cas9 and Genome Editing in Drosophila. J Genet Genomics 2014; 41:7-19; PMID:24480743; http://dx.doi.org/10.1016/j.jgg.2013.12.004
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O'Connor-Giles KM. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013; 194:1029-35; PMID:23709638; http://dx.doi.org/10.1534/genetics.113.152710
- Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 2014; 32:819-21; PMID:24880488; http://dx.doi.org/10.1038/nbt.2925
- Inui M, Miyado M, Igarashi M, Tamano M, Kubo A, Yamashita S, Asahara H, Fukami M, Takada S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci Rep 2014; 4:5396; PMID:24953798; http://dx.doi.org/10.1038/srep05396
- Böttcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, Förstemann K. Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res 2014; 42:e89; PMID:24748663; http://dx.doi.org/10.1093/nar/gku289
- Stern DL, Orgogozo V. The loci of evolution: how predictable is genetic evolution? Evolution 2008; 62:2155-77; PMID:18616572; http://dx.doi.org/10.1111/j.1558-5646.2008.00450.x
- Stam LF, Laurie CC. Molecular dissection of a major gene effect on a quantitative trait: The level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics 1996; 144:1559-64; PMID:8978044
- Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human Catechol-O-Methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 2006; 314:1930-3; PMID:17185601; http://dx.doi.org/10.1126/science.1131262
- Miyaoka Y, Berman JR, Cooper SB, Mayerl SJ, Chan AH, Zhang B, Karlin-Neumann GA, Conklin BR. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci Rep 2016; 6:23549; PMID:27030102; http://dx.doi.org/10.1038/srep23549
- Yu Z, Chen H, Liu J, Zhang H, Yan Y, Zhu N, Guo Y, Yang B, Chang Y, Dai F, et al. Various applications of TALEN- and CRISPR/Cas9-mediated homologous recombination to modify the Drosophila genome. Biol Open 2014; 3:271-80; PMID:24659249; http://dx.doi.org/10.1242/bio.20147682
- Sebo ZL, Lee HB, Peng Y, Guo Y. A simplified and efficient germline-specific CRISPR/Cas9 system for Drosophila genomic engineering. Fly (Austin) 2014; 8:52-7; PMID:24141137; http://dx.doi.org/10.4161/fly.26828
- Bassett AR, Tibbit C, Ponting CP, Liu J-L. Highly efficient targeted mutagenesis of drosophila with the CRISPR/Cas9 system. Cell Rep 2013; 4:220-8; PMID:23827738; http://dx.doi.org/10.1016/j.celrep.2013.06.020
- Zhang X, Koolhaas WH, Schnorrer F. A versatile two-step CRISPR- and RMCE-based strategy for efficient genome engineering in Drosophila. G3 (Bethesda) 2014; 4:2409-18; PMID:25324299; http://dx.doi.org/full_text
- Kühn R, Chu VT. Pop in, pop out: a novel gene-targeting strategy for use with CRISPR-Cas9. Genome Biol 2015; 16:244; PMID:26553112; http://dx.doi.org/10.1186/s13059-015-0810-2
- Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol 2015; 16:231; PMID:26553065; http://dx.doi.org/10.1186/s13059-015-0791-1
- Wittkopp PJ, Stewart EE, Arnold LL, Neidert AH, Haerum BK, Thompson EM, Akhras S, Smith-Winberry G, Shefner L. Intraspecific polymorphism to interspecific divergence: Genetics of pigmentation in drosophila. Science 2009; 326:540-4; PMID:19900891; http://dx.doi.org/10.1126/science.1176980
- Gratz SJ, Ukken FP, Rubinstein CD, Thiede G, Donohue LK, Cummings AM, O'Connor-Giles KM. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 2014; 196:961-71; PMID:24478335; http://dx.doi.org/10.1534/genetics.113.160713
- Port F, Chen HM, Lee T, Bullock SL. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci U S A 2014; 111:E2967-76; PMID:25002478; http://dx.doi.org/10.1073/pnas.1405500111
- Kalay G. Rapid evolution of cis-regulatory architecture and activity in the Drosophila yellow gene. [dissertation]. [Ann Arbor]: University of Michigan; 2012. 197 p.
- Gibson DG, Smith HO, Hutchison CA, Venter JC, Merryman C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods 2010; 7:901-3; PMID:20935651; http://dx.doi.org/10.1038/nmeth.1515
- Pardo B, Gómez-González B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci 2009; 66:1039-56; PMID:19153654; http://dx.doi.org/10.1007/s00018-009-8740-3
- Holtzman S, Miller D, Eisman RC, Kuwayama H, Niimi T, Kaufman TC. Transgenic tools for members of the genus Drosophila with sequenced genomes. Fly (Austin) 2010; 4:349-62; PMID:20890112; http://dx.doi.org/10.4161/fly.4.4.13304
- Dong S, Lin J, Held NL, Clem RJ, Passarelli AL, Franz AW. Heritable CRISPR/Cas9-Mediated genome editing in the yellow fever mosquito, Aedes aegypti. PLoS One 2015; 10:e0122353; PMID:25815482; http://dx.doi.org/10.1371/journal.pone.0122353
- Griswold CM, Roebuck J, Anderson RO, Stam LF, Spana EP. A toolkit for transformation and mutagenesis in Drosophila using piggyBac. Drosoph Inf Serv 2002; 85:129-32
- Perez C, Guyot V, Cabaniols J-P, Gouble A, Micheaux B, Smith J, Leduc S, Pâques F, Duchateau P. Factors affecting double-strand break-induced homologous recombination in mammalian cells. Biotechniques 2005; 39:109-15; PMID:16060375; http://dx.doi.org/10.2144/05391GT01
- Wittkopp PJ, Vaccaro K, Carroll SB. Evolution of yellow gene regulation and pigmentation in Drosophila. Curr Biol 2002; 12:1547-56; PMID:12372246; http://dx.doi.org/10.1016/S0960-9822(02)01113-2
- Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, Carroll SB. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 2008; 132:783-93; PMID:18329365; http://dx.doi.org/10.1016/j.cell.2008.01.014
- Yassin A, Bastide H, Chung H, Veuille M, David JR, Pool JE. Ancient balancing selection at tan underlies female colour dimorphism in Drosophila erecta. Nat Commun 2016; 7:10400; PMID:26778363; http://dx.doi.org/10.1038/ncomms10400
- Martin A, Serano JM, Jarvis E, Bruce HS, Wang J, Ray S, Barker CA, O'Connell LC, Patel NH. CRISPR/Cas9 mutagenesis reveals versatile roles of hox genes in crustacean limb specification and evolution. Curr Biol 2016; 26:14-26; PMID:26687626; http://dx.doi.org/10.1016/j.cub.2015.11.021
- Kistler KE, Vosshall LB, Matthews BJ. Genome engineering with CRISPR-Cas9 in the mosquito Aedes aegypti. Cell Rep 2015; 11:51-60; PMID:25818303; http://dx.doi.org/10.1016/j.celrep.2015.03.009
- Port F, Muschalik N, Bullock SL. Systematic evaluation of drosophila CRISPR tools reveals safe and robust alternatives to autonomous gene drives in basic research. G3 (Bethesda) 2015; 5:1493-502; PMID:25999583; http://dx.doi.org/full_text
- Ren X, Sun J, Housden BE, Hu Y, Roesel C, Lin S, Liu L-P, Yang Z, Mao D, Sun L, et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc Natl Acad Sci 2013; 110:19012-7; PMID:24191015; http://dx.doi.org/10.1073/pnas.1318481110
- Beumer KJ, Trautman JK, Bozas A, Liu J-L, Rutter J, Gall JG, Carroll D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc Natl Acad Sci U S A 2008; 105:19821-6; PMID:19064913; http://dx.doi.org/10.1073/pnas.0810475105
- Wong N, Liu W, Wang X. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol 2015; 16:218; PMID:26521937; http://dx.doi.org/10.1186/s13059-015-0784-0
- Ren X, Yang Z, Xu J, Sun J, Mao D, Hu Y, Yang S-J, Qiao H-H, Wang X, Hu Q, et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep 2014; 9:1151-62; PMID:25437567; http://dx.doi.org/10.1016/j.celrep.2014.09.044
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat Biotechnol 2014; 32:1262-7; PMID:25184501; http://dx.doi.org/10.1038/nbt.3026
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 2007; 104:3312-7; PMID:17360644; http://dx.doi.org/10.1073/pnas.0611511104
- Werner T, Koshikawa S, Williams TM, Carroll SB. Generation of a novel wing colour pattern by the Wingless morphogen. Nature 2010; 464:1143-8; PMID:20376004; http://dx.doi.org/10.1038/nature08896
- Gloor G, Engels W. Single-fly DNA preps for PCR. Drosoph Inf Serv 1992; 71:148-9