
Abstract
Voltage-gated sodium (NaV) channels are a family of transmembrane ion channel proteins. They function by forming a gated, water-filled pore to help establish and control cell membrane potential via control of the flow of ions between the intracellular and the extracellular environments. Blockade of NaVs has been successfully accomplished in the clinic to enable control of pathological firing patterns that occur in a diverse range of conditions such as chronic pain, epilepsy, and cardiac arrhythmias. First generation sodium channel modulator drugs, despite low inherent subtype selectivity, preferentially act on over-excited cells which reduces undesirable side effects in the clinic. However, the limited therapeutic indices observed with the first generation demanded a new generation of sodium channel inhibitors. The structure, function and the state of the art in sodium channel modulator drug discovery are discussed in this chapter.
Abbreviations
| NaV | = | Voltage-gated sodium channel |
| TTX | = | Tetrodotoxin |
| TTX-S | = | Tetrodotoxin sensitive |
| TTX-R | = | Tetrodotoxin resistant |
| PEPD | = | Paroxysmal extreme pain disorder |
| CIP | = | Congenital insensitivity to pain |
| FDA | = | Food and Drug Administration. |
Voltage-gated sodium (NaV) channels are crucial in the initiation and propagation of electrical signals (action potentials) in excitable neuronal cells, muscles, and heart tissues. This has led to the successful clinical exploitation of NaV channel inhibitors as anticonvulsants, antiarrhythmics and local anesthetics e.g. lamotrigine, flecainide and lidocaine.Citation1-3 The NaV channel family has nine members (NaV1.1 to NaV1.9) with a high degree of sequence homology between the nine subtypes. A number of natural toxins such as tetrodotoxin (TTX) block the sodium channel extracellular pore, and the sensitivity of the sodium channel subtypes to blockade by TTX has been used to divide the family into two classes: (i) TTX-S (sensitive), TTX IC50<30 nM against NaV1.1, 1.2, 1.3, 1.4, 1.6, 1.7 and (ii) TTX-R (resistant), TTX IC50 >30 nM against NaV1.5, 1.8 and Nav1.9. The selectivity difference is believed to be explained by the presence of a key cysteine residue in the TTX binding site.Citation4 The NaX channel has also been classified as a subtype of voltage-gated sodium channels although the primary structure of NaX is markedly different from the other NaV channels. NaX is thought to have a physiological role in maintaining body fluid homeostasis through the regulation of sodium concentration ().
Table 1. Sodium channel distribution and classification based on sensitivity to tetrodotoxin (TTX) block
Structure and Function
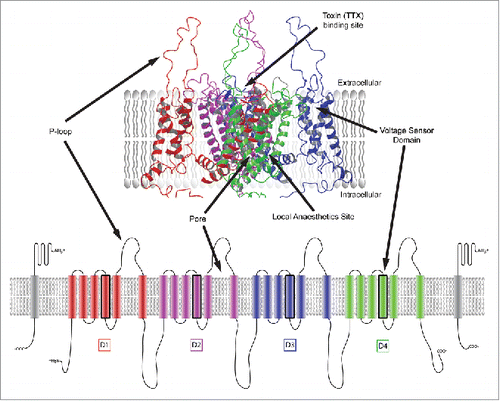
NaV channels are heteromeric complexes comprised of an α subunit, having an approximate mass of 260 kDa, and one or more β subunits of lower molecular weight (). Each α subunit has four homologous domains (D1–D4), with each domain containing six transmembrane segments (S1–S6). The S4 segment of each domain contains positively charged arginine and lysine residues which allow them to act as voltage sensors of cell membrane depolarisation and repolarisation. In combination, the S5 and S6 transmembrane helices from each domain form the sodium channel pore and the p-loop creates the Na+ ion selectivity filter. shows a 3-dimensional representation of a voltage-gated sodium channel and highlights common regions that have been reported to support ligand binding.Citation4 A significant proportion of this evidence has been gathered via site directed mutagenesis. However, there are now several published apo crystal structures of bacterial NaV channels that highlight key structural features.Citation4-8 Moreover, the first co-crystal structures of ligands bound in a NaV channel have been recently generated.Citation9
Figure 1. NaV channel structural topology, highlighting common ligand binding sites and significant structural features. Domains D1-D4 are represented in different colors while β subunits are shown in gray.
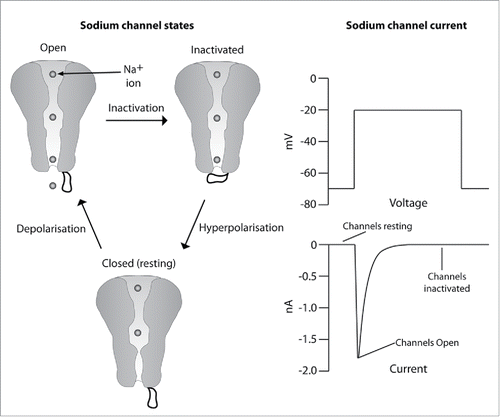
NaV channels exist in essentially three states: open, closed (resting) and inactivated (). Under resting membrane potential the channels are in their non-conducting closed state. Upon depolarization (decrease in membrane voltage) it is believed that the S4 voltage sensors move outward, allowing the pore to open briefly (<1 millisecond), before several processes termed fast and slow inactivation occur that move the channel into a non-conducting inactivated state. The inactivation gate between D3 and D4 is thought to be responsible for fast inactivation. Upon cell membrane hyperpolarisation (increase in membrane voltage) the sodium channel is reprimed back into the resting state ahead of the next depolarization. Many NaV channel blockers have different affinities for each state, often with a preference for the open and/or inactivated state. Since the proportion of channels populating different states is controlled by cell membrane voltage, this state dependence is also termed voltage dependence. Many compounds additionally demonstrate a phenomenon called use dependence which occurs when the compound potency increases upon higher NaV channel firing frequency stimulation. As damaged nerve NaV channels move more rapidly through the three states than a sodium channel in the heart, this phenomenon has been exploited to preferentially target diseased cells and derive an improved therapeutic index over cardiovascular side effects. The improved therapeutic index is accomplished because use dependence imparts functional selectivity over sodium channels in heart.
Figure 2. Sodium channel states: NaV channels cycle between 3 states: open, closed (resting) and inactivated. They cycle from the closed (resting) state to the open state upon membrane depolarisation. The channel is open for less than a millisecond prior to inactivation. The inactivated state reprimes to the resting state when the cell membrane potential has returned to a hyperpolarised resting potential. Sodium channel current: The current associated with the cycling of sodium channels through the resting, open and inactivated state.
Genetic Link to Disease State
A number of channelopathies linked to sodium channels have been identified which has helped to identify their physiological role. This data has been used to classify sodium channel subtypes as potential drug targets and anti-targets. Expression of the mutant sodium channels in transfected cells and biophysical characterization indicate a diverse range of gain- and loss-of-function properties. In addition to measuring mRNA and protein expression levels from ex vivo studies, mice with knock-in, knock-out or conditional expression of sodium channel genes have also been used to better understand the role of the sodium channel subtypes.Citation2,4,10 From the various studies, NaV1.1 and NaV1.2 mutations have a genetic link to epilepsy and related CNS disorders whilst NaV1.5 mutations have significant relation to cardiac arrhythmias. Periodic paralyses are caused by mutations in NaV1.4, while the NaV1.6 channel has been associated with cerebellar atrophy, behavioural deficits and ataxia.Citation11 Due to their differential expression and presence in sensory neurons in humans and animals NaV1.3, 1.7, 1.8 and 1.9 are of interest for their potential role in pain transmission.Citation12,13 There is compelling genetic evidence for the role of NaV1.7 as a key contributor to pain perception in humans. Gain-of-function mutations in NaV1.7 can lead to paroxysmal extreme pain disorder (PEPD) an inherited disease that is characterized by severe rectal, ocular, or sub-mandibular pain and primary erythromelalgia,Citation14 typified by burning pain in the extremities accompanied with hyperemia and inflammation.Citation15 Conversely, congenital insensitivity to pain (CIP) can result from several loss-of-function mutations in NaV1.7 characterized by a complete inability to sense pain.Citation16 While no human loss-of–function mutation of SCN10A, the gene which codes Nav1.8, have been reported gain-of-function mutations which increase the likelihood of depolarization and which are associated with painful neuropathies are known.Citation17 Finally, different gain-of–function mutations of SCN11A, the gene coding for Nav1.9, have recently demonstrated a genetic link to pain.Citation18,19
Toxins Which Act Through Sodium Channels
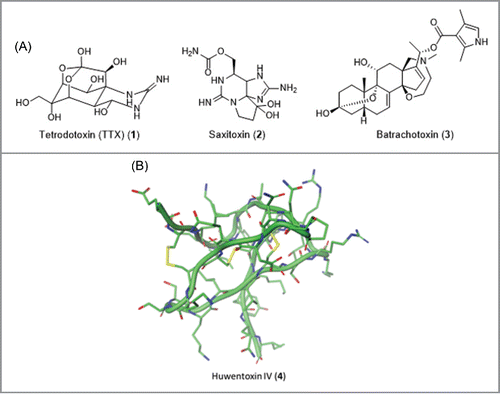
A number of natural toxins are known to exert their effect through sodium channels. Some neurotoxins such as TTX (1), saxitoxin (2) are known to be blockers of NaV channels whereas other toxins such as batrachotoxin (3) and natural pyrethroid insectisides are conversely known to activate NaV channels (). TTX (1) is considered to be a true ion channel current blocker as it has been established to physically occlude the extracellular channel pore.Citation20 In addition to these polar small molecule toxins, a variety of peptide-based venom toxins have been isolated from spider, snail, scorpion and centipede venoms and have been shown to modulate sodium channel function.Citation20 These include protoxins (ProTx), huwentoxins (HwnTx) e.g. HwnTx IV (4) and a variety of other cysteine knot peptides, many of which have been reported to display NaV subtype selective modulation via binding to the extracellular portion of the channel.Citation21,22 NaV toxins have also been taken into clinical trials for use as therapeutic treatments; Wex Pharmaceuticals is currently progressing TTX in Phase III trials for the treatment of cancer pain.
Figure 3. Selected toxin modulators.
Small Molecule Blockade of Sodium Channels
Sodium channels have been implicated as biological targets for some antiarrhythmic, anticonvulsant and local anesthetic medications, but many of these classical clinical agents were discovered prior to appreciating their full pharmacology profiles. Whilst many of these drugs are known to be weak and subtype unselective sodium channel blockers, they also modulate other ion channels. Over the past two decades, based on a more detailed understanding of biology and genetics, NaV channels have been confirmed to be therapeutically desirable targets, leading to a resurgence of medicinal chemistry work in this area. Much of this work has focussed on delivering safer variants of subtype unselective blockers. However, there have also been some recent examples of subtype selective modulators.
First Generation Sodium Channel Modulators
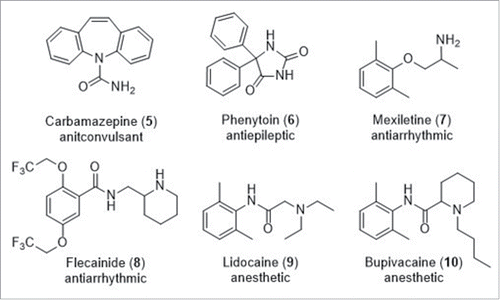
A variety of sodium channel modulating drugs have been applied to the treatment of clinical conditions caused by abnormal cell excitability.Citation2 In particular they have been applied to CNS conditions such as anti-convulsants e.g. carbamazepine (5), and epilepsy therapy e.g. phenytoin (6) via modulation of sodium channels expressed in the brain (). Antiarrhythmics such as mexiletine (7) and flecainide (8) rectify cardiac rhythm by acting on NaV channels in the heart. Finally, local anesthetics e.g. lidocaine (9) and bupivacaine (10) are established injectable or topical agents for the treatment of pain via the blockade of NaV channels in peripheral nerves. These compounds are largely subtype unselective within the sodium channel family leading to the potential for undesirable side effects which limit their application for certain chronic indications. Physicochemically, all of these compounds are either weakly basic or neutral and structurally it has been suggested that they to bind to an intracellular site within the channel pore that is commonly referred to as the local anesthetic binding site ().Citation23 Due to a high degree of amino acid sequence conservation in the channel pore across the NaV subtypes, it is not surprising that imparting subtype selectivity via binding to this site has proven challenging.
Figure 4. Selected first generation sodium channel modulator drugs.
Second Generation Sodium Channel Modulators
Recent research efforts have focused on the purposeful identification of molecules with known sodium channel pharmacology (as opposed to characterisation subsequent to their use in the clinic) with minimal off-target related activity. The main focus of interest has been directed towards identifying molecules that block NaV1.3, NaV1.7, NaV1.8 and NaV1.9. These subtypes are predominately expressed in sensory neurons with a link to nociception and therefore provide strong rationale as targets for the development of novel pain therapeutics.Citation2,10 Whilst subtypes NaV1.1 and NaV1.2 have been associated with the treatment of a variety of disorders they are also implicated in CNS mediated side effects, resulting in a narrow therapeutic index for many of the modulators. Furthermore, pro-arrhythmic effects resulting from block of NaV1.5 channels presents a potential cardiac liability. This increased understanding for the functional roles of sodium channel subtypes, coupled with dramatic advances in automated screening technologies, provided the necessary impetus for the pharmaceutical industry to undertake high-throughput screening campaigns in an effort to identify second generation inhibitors with improved selectivity and side effect profile compared to first generation molecules. However, whilst this has led to the discovery of a diverse array of chemotypes, relatively few have been disclosed with data to support them being subtype selective, implying that delivering subtype selectivity still presents a significant challenge.
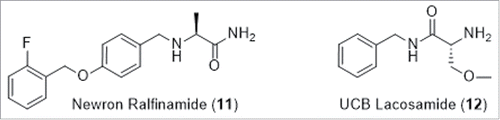
Molecules with sodium channel activity that bridge the gap between first and second generation modulators have been taken into clinical development (). Ralfinamide (11), a state and use dependent NaV modulator from Newron, is also reported to act through N-type calcium channels and N-methyl-D-aspartic acid (NMDA) receptors.Citation24 Lacosamide (12) which also possesses a benzylamino headgroup, is suggested to bind to a novel site that enhances slow inactivation with some selectivity towards TTX-S channels. Lacosamide (12) was approved in 2008 as an anticonvulsant and for the treatment of diabetic neuropathic pain.Citation25
Figure 5. Early examples of second generation sodium channel modulators.
Recently, the number of sodium channel modulators disclosed in the scientific literature has increased substantially with many examples spanning a broad range of chemotypes and physicochemistry. Selected examples described below.Citation2,3,26-28
Patents from Vertex based on TTX-S channel modulators containing a weakly acidic N-heterocylic sulfonamide suggest NaV1.1 and NaV1.3 to be the preferred targets with compound 13 exhibiting IC50 <2 µM at both NaV1.1 and NaV1.3 (). Icagen and a collaboration between Pfizer and Icagen have also reported compounds containing acidic N-heterocyclic sulfonamides which have been reported to modulate TTX-S channels with compound 14 having NaV1.3 IC50 30 nM and examples reported to possess >1000-fold selectivity over cardiac channel NaV1.5.
Figure 6. Examples of NaV subtype selective targeting modulators.
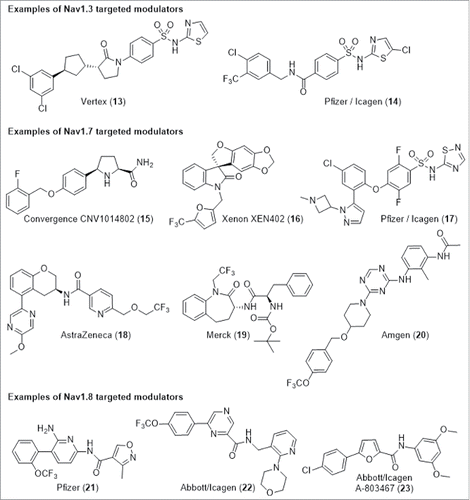
The high confidence in target rationale for NaV1.7 provided by genetic data has sparked an industry-wide search for NaV1.7 preferring modulators for the treatment of pain resulting in the publication of a diverse array of chemotypes with several lead compounds moving forward into clinical trials.Citation2,3 Convergence and Xenon have both entered clinic trials with chemotypes represented by a weakly basic proline 15,Citation29 and a neutral spiro-oxindole 16,Citation30 respectively. AstraZeneca have also published several patents which cover a diversity of chemical series including chromane 18, reporting NaV1.7 IC50 66 nM, NaV1.5 IC50 13 µM and NaV1.2 IC50 >33 µM. Benzazepinone 19 has been reported by Merck to be a state dependent inhibitor of NaV1.7 (IC50 90 nM) and NaV1.8 (IC50 680 nM) whilst displaying ˜10-fold use dependent selectivity over NaV1.5.Citation31 Pfizer, in collaboration with Icagen, have published several patents based on acidic and zwitterionic series (represented here by compound 17) with numerous examples reported to have NaV1.7 potency in the single digit nanomolar range and a molecule currently in clinical trials. Recently, Icagen and Pfizer have also described a unique binding site on the voltage sensor domain away from the pore region for related chemotypes.Citation32 Amgen have described a novel series of triazine derivatives as NaV1.7 modulators and have also determined that 20 binds to a distinct site from that of local anesthetics.Citation33 Modulation of this subtype remains a competitive area and multiple pharmaceutical companies have recently published patents for chemical series with activity against NaV1.7 that cover diverse structures and physicochemistry including basic, neutral and acidic series.
Pfizer has described a series of 6,6-biaryl derivatives optimised for NaV1.8 activity whilst retaining selectivity over the related TTX-R NaV1.5 cardiac channel.Citation34 For example, compound 21 has a whole cell electro-physiology NaV1.8 IC50 of 260 nM and selectivity of >20-fold over NaV1.1, NaV1.5 and NaV1.7. A collaboration between Abbott and Icagen has also discovered 6,6 and 6,5 biaryl lead matter with an alternative amide geometry. Substituted pyrazine 22 showed NaV1.8 IC50 of 30 nM and good selectivity over TTX-S channel NaV1.2 and TTX-R channel NaV1.5. Alternative cores were also tolerated and had similar profiles such as the furan derivative A-803467 (23).Citation35
Despite the recent compelling genetic evidence for Nav1.9 as an analgesic target, development of modulators for this subtype has been hampered by the difficulty of expressing this subtype in recombinant systems and to date no modulators of Nav1.9 have been described.
Compounds in Clinical Development and Outlook
Despite significant interest in sodium channel blockers as pain therapies, only a few second generation compounds have been reported to have entered clinical trials. AstraZeneca has reported the effects of intradermal administration of AZD-3161 in a Phase I UVIH burn study. Xenon Pharmaceuticals reported a positive Phase II readout in acute pain with a compound targeting NaV1.7 where topical application of XEN402 (16) reduced pain in postherpetic neuralgia (PHN). Results from an oral Phase II study with XEN402 (16) suggested positive effects in patients suffering from primary erythromelalgia.Citation30 Xenon recently partnered XEN402 (TV-45070, 16) with Teva and have been granted orphan drug designation by the FDA for the treatment of erythromelalgia. Pfizer have advanced a NaV1.7 compound PF-05089771 into Phase II clinical trials of third molar extraction and primary inherited erythromelalgia.Citation36 Pfizer has also reported advancing selective NaV1.8 blockers into the clinic. Convergence have conducted Phase II trials for lumbosacral radiculopathy and trigeminal neuralgia with their most advanced sodium channel modulator CNV1014802 (15), which is reported to be a highly state dependent subtype preferring NaV1.7 blocker. Convergence also announced that CNV1014802 (15) has been granted orphan drug status by the FDA for the treatment of trigeminal neuralgia. Dainippon Sumitomo has advanced a NaV1.7/NaV1.8 compound DSP-2230 into Phase II for neuropathic pain. Although the second generation sodium channel modulators are making their way through efficacy based clinical trials, the isoform selectivity and biophysical characteristics of many of these clinical compounds have not been disclosed. The hope is that new agents with greater subtype selectivity will achieve therapeutic utility with decreased cardiovascular and CNS side effects.
In conclusion, there have been major advances in sodium channel drug discovery over the last decade. Sodium channels have been implicated in a variety of disease states through both genetic data and a greater understanding of the pharmacology of marketed drugs. Moreover, knowledge around the role of specific sodium channel subtypes has led to an increase in drug discovery efforts for the identification of subtype or functionally selective agents. Advances in screening technology, coupled with the use of sodium channel co-crystal structures in the various sodium channel states for structure based drug design, should further enable future efforts.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, et al. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J Med Chem 2013; 56:593-624; PMID:23121096
- Nardi A, Damann N, Hertrampf T, Kless A. Advances in Targeting Voltage-Gated Sodium Channels with Small Molecules. Chem Med Chem 2012; 7:1712-40.
- Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, Swain NA. Recent progress in sodium channel modulators for pain. Bioorg Med Chem Letters 2014; 24:3690-9.
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature 2011; 475:353-8.
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012; 486:135-9; PMID:22678296
- Tsai C-J, Tani K, Irie K, Hiroaki Y, Shimomura T, McMillan DG, Cook GM, Schertler GFX, Fujiyoshi Y, Li X-D. Two Alternative Conformations of a Voltage-Gated Sodium Channel. J Mol Biol 2013; 425:4074-88; PMID:23831224
- Bagnéris C, DeCaen PG, Hall BA, Naylor CE, Clapham DE, Kay CWM, Wallace BA. Role of the C-terminal domain in the structure and function of tetrameric sodium channels. Nat Commun 2013; 4; 2465; PMID:24051986
- Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, Minor DL, Jr. Structure of a Prokaryotic Sodium Channel Pore Reveals Essential Gating Elements and an Outer Ion Binding Site Common to Eukaryotic Channels. J Mol Biol 2014; 426:467-83; PMID:24120938; http://dx.doi.org/10.1016/j.jmb.2013.10.010
- Bagnéris C, DeCaen PG, Naylor CE, Pryde DC, Nobeli I, Clapham DE, Wallace BA. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc Natl Acad Sci 2014; 111:8428-33; http://dx.doi.org/10.1073/pnas.1406855111
- England S, de Groot MJ. Subtype-selective targeting of voltage-gated sodium channels. British J Pharmacol 2009; 158:1413-25; http://dx.doi.org/10.1111/j.1476-5381.2009.00437.x
- England S. Voltage-gated sodium channels: the search for subtype-selective analgesics. Expert Opin Investig Drugs 2008; 17:1849-64; http://dx.doi.org/10.1517/13543780802514559
- Bhattacharya A, Wickenden AD, Chaplan SR. Sodium channel blockers for the treatment of neuropathic pain. Neurotherapeutics 2009; 6:663-78; PMID:19789071; http://dx.doi.org/10.1016/j.nurt.2009.08.001
- Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neuroscience Letters 2008; 433:65-70; PMID:18242854; http://dx.doi.org/10.1016/j.neulet.2007.12.064
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, et al. SCN9A mutations in paroxysmal clinical study extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 2006; 52:767-74; PMID:17145499; http://dx.doi.org/10.1016/j.neuron.2006.10.006
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, et al. Mutations in SCN9A, encoding a sodium channel α subunit, in patients with primary erythermalgia. J Med Genet 2004; 41:171-4; http://dx.doi.org/10.1136/jmg.2003.012153
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006; 444:894-8; PMID:17167479; http://dx.doi.org/10.1038/nature05413
- Faber CG, Lauria G, Merkies ISJ, Cheng X, Han C, Ahn HS, Persson AK, Hoeijmakers JGJ, Gerrits MM, Pierro T, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA 2012; 109:19444-9, S/1-S/3; http://dx.doi.org/10.1073/pnas.1216080109
- Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stoedberg T, Hennings JC, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nature Genet 2013; 45:1399-404; PMID:24036948; http://dx.doi.org/10.1038/ng.2767
- Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, et al. Gain-of-Function Mutations in SCN11A Cause Familial Episodic Pain. Am J Hum Genet 2013; 93:957-66; http://dx.doi.org/10.1016/j.ajhg.2013.09.016
- Lipkind GM, Fozzard HA. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophysical J 1994; 66:1-13; http://dx.doi.org/10.1016/S0006-3495(94)80746-5
- Xiao Y, Blumenthal K, Jackson JO, II, Liang S, Cummins TR. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol 2010; 78:1124-34; http://dx.doi.org/10.1124/mol.110.066332
- Peng K, Shu Q, Liu Z, Liang S. Function and Solution Structure of Huwentoxin-IV, a Potent Neuronal Tetrodotoxin (TTX)-sensitive Sodium Channel Antagonist from Chinese Bird Spider Selenocosmia huwena. J Biol Chem 2002; 277:47564-71; PMID:12228241; http://dx.doi.org/10.1074/jbc.M204063200
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci 1996; 93:9270-5; PMID:8799190; http://dx.doi.org/10.1073/pnas.93.17.9270
- Cattabeni F. Ralfinamide Newron Pharmaceuticals. IDrugs 2004; 7:935-9; PMID:15478019
- Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther 2008; 326:89-99; http://dx.doi.org/10.1124/jpet.107.133413
- Kemp MI. Structural trends among second-generation voltage-gated sodium channel blockers. Prog Med Chem 2010; 49:81-111; http://dx.doi.org/10.1016/S0079-6468(10)49003-7
- Matulenko MA, Scanio MJC, Kort ME. Voltage-gated sodium channel blockers for the treatment of chronic pain. Curr Top Med Chem 2009; 9:362-76; PMID:19442207; http://dx.doi.org/10.2174/156802609788317883
- England S, Rawson D. Isoform-selective voltage-gated Na+ channel modulators as next-generation analgesics. Future Med Chem 2010; 2:775-90; http://dx.doi.org/10.4155/fmc.10.26
- Zajac MA. Process for preparing α-carboxamide pyrrolidine derivatives. Application: WOWO: (Convergence Pharmaceuticals Limited, UK). 2011:34pp
- Goldberg YP, Price N, Namdari R, Cohen CJ, Lamers MH, Winters C, Price J, Young CE, Verschoof H, Sherrington R, et al. Treatment of Nav1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain 2012; 153:80-5; PMID:22035805; http://dx.doi.org/10.1016/j.pain.2011.09.008
- Williams BS, Felix JP, Priest BT, Brochu RM, Dai K, Hoyt SB, London C, Tang YS, Duffy JL, Parsons WH, et al. Characterization of a New Class of Potent Inhibitors of the Voltage-Gated Sodium Channel Nav1.7. Biochemistry 2007; 46:14693-703; PMID:18027973; http://dx.doi.org/10.1021/bi7018207
- McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci U S A 2013; 110:E2724-E32; PMID:23818614; http://dx.doi.org/10.1073/pnas.1220844110
- Bregman H, Berry L, Buchanan JL, Chen A, Du B, Feric E, Hierl M, Huang L, Immke D, Janosky B, et al. Identification of a Potent, State-Dependent Inhibitor of Nav1.7 with Oral Efficacy in the Formalin Model of Persistent Pain. J Medicinal Chem 2011; 54:4427-45; http://dx.doi.org/10.1021/jm200018k
- Bagal SK, Bungay PJ, Denton SM, Gibson KR, Glossop MS, Hay TL, Kemp MI, Lane CAL, Lewis ML, Maw GN, et al. Discovery and Optimization of Selective Nav1.8 Modulator Series That Demonstrate Efficacy in Preclinical Models of Pain. ACS Med Chem Letters 2015; 6:650-4; PMID:26101568; http://dx.doi.org/10.1021/acsmedchemlett.5b00059
- Triggle DJ. Calcium channel antagonists: Clinical uses-Past, present and future. Biochemical Pharmacol 2007; 74:1-9; http://dx.doi.org/10.1016/j.bcp.2007.01.016
- Nantermet PG, Henze DA. Recent advances toward pain therapeutics. Annual Rep Med Chem 2011; 46:19-32; http://dx.doi.org/10.1016/B978-0-12-386009-5.00025-4