
ABSTRACT
Degenerin/Epithelial Sodium Channels (DEG/ENaCs) are a large family of animal-specific non-voltage gated ion channels, with enriched expression in neuronal and epithelial tissues. While neuronal DEG/ENaCs were originally characterized as sensory receptor channels, recent studies indicate that several DEG/ENaC family members are also expressed throughout the central nervous system. Human genome-wide association studies have linked DEG/ENaC-coding genes with several neurologic and psychiatric disorders, including epilepsy and panic disorder. In addition, studies in rodent models further indicate that DEG/ENaC activity in the brain contributes to many behaviors, including those related to anxiety and long-term memory. Although the exact neurophysiological functions of DEG/ENaCs remain mostly unknown, several key studies now suggest that multiple family members might exert their neuronal function via the direct modulation of synaptic processes. Here, we review and discuss recent findings on the synaptic functions of DEG/ENaCs in both vertebrate and invertebrate species, and propose models for their possible roles in synaptic physiology.
Introduction
Members of the DEG/ENaC superfamily of non-voltage-gated, animal-specific cation channels, are expressed in diverse cell types, including neurons [Citation1]. Structure-function studies have revealed that the mature channel is comprised of three independent subunits, which can be either homomeric or heteromeric. Each subunit consists of two transmembrane domains that are connected by a large, extracellular loop that includes several structurally conserved domains [Citation2–Citation5]. Several studies suggest that the extracellular loops play important roles in channel gating by a broad spectrum of extracellular stimuli, including protease activation, mechanical forces, and extracellular chemical ligands [Citation6–Citation8]. The first family members to be identified molecularly were named Degenerin (Deg) because their mutant alleles were associated with a neuronal degeneration phenotype in the roundworm Caenorhabditis elegans [Citation9,Citation10]. Later, mammalian orthologs of DEG proteins were molecularly identified as the elusive amiloride-sensitive epithelial sodium channels (ENaCs), which play an essential role in regulating mammalian blood pressure via salt reabsorption in kidneys [Citation11–Citation13]. Several additional vertebrate family members, namely the Acid Sensing Ion Channels (ASICs; also referred to as Amiloride-sensitive Cation Channels Neuronal; ACCNs), were shown to be activated by low extracellular pH [Citation14–Citation17]. To date, genes that encode putative members of the DEG/ENaC superfamily have been identified in all animal genomes that have been sequenced thus far. Yet, for reasons that are not well understood, the number of independent DEG/ENaC-encoding genes varies dramatically across animal phylogeny, which suggests that the family has expanded and contracted several independent times throughout animal evolution [Citation1,Citation6,Citation18,Citation19].
While the physiological function of DEG/ENaC channels in epithelial tissues is relatively well-understood, the neurophysiological functions of most family members remain unknown. In mammals, neuronally-enriched DEG/ENaC genes primarily belong to the ASIC family branch. To date, five independent ASIC-coding genes have been identified in mammalian genomes (ASIC1-5), which are expressed in both central and peripheral neurons, as well as glia [Citation20–Citation25]. In the peripheral nervous system, some ASICs are localized to sensory terminals, where they may play a role in modulating mechanosensation and nociception [Citation26–Citation28]. In the central nervous system, ASICs seem to act as pH sensors, responding to fluctuations in extracellular pH that can be a product of normal neuronal metabolism, acidification from neuronal pathologies, or cell death associated with ischemia and epilepsy [Citation20,Citation29]. In humans, mutations in ASIC genes have been associated with various neurologic and psychiatric disorders [Citation20], including epilepsy [Citation30,Citation31], multiple sclerosis [Citation32,Citation33], and panic disorder [Citation34]. Together, these data suggest that DEG/ENaC channels play an important role in modulating neural functions in humans.
In invertebrate species, DEG/ENaCs have been studied most extensively in the worm C. elegans and the fruit fly Drosophila melanogaster, which encode 30 and 31 independent DEG/ENaC genes respectively [Citation18,Citation35]. Studies of various family members in these two species indicate that individual DEG/ENaC genes are expressed in diverse cell types, including neurons in both the central and peripheral nervous systems, glia and muscle [Citation36–Citation42]. Some invertebrate DEG/ENaCs that are expressed in the peripheral nervous system have been shown to be gated by mechanosensation or chemical ligands [Citation39,Citation42–Citation45], yet the gating mechanisms and physiological functions for the majority of DEG/ENaCs found in C. elegans and Drosophila remain mostly unknown.
More recently, several studies suggested that DEG/ENaCs also play an important modulatory role at the synapse in both vertebrate and invertebrate species [Citation41,Citation46–Citation53]. However, the exact cellular and physiological mechanisms that mediate the effects of DEG/ENaCs on synaptic physiology remain poorly understood. Here we review the recent literature on synaptic functions of DEG/ENaC genes, and propose mechanistic models that may explain the observed phenotypes.
Roles of DEG/ENaCs in behavior
Studies of DEG/ENaC mutant alleles in genetically tractable species such as the mouse, the fly, and the worm revealed the importance of the neuronal functions of DEG/ENaCs to various organismal behavioral phenotypes. For example, studies of ASIC1a knockout mice showed that these animals exhibit reduced depression and anxiety related behavior [Citation54], reduced fear behavior [Citation55,Citation56], and increased addiction related behavior [Citation49]. Additionally, ASIC1a knockout mice display impaired performance in several learning paradigms, including hippocampal-dependent spatial learning and cerebellar-dependent eyeblink conditioning [Citation57], amygdala-dependent impaired cue and contextual fear conditioning paradigms [Citation51,Citation58–Citation61], and impaired extinction of conditioned taste avoidance [Citation53]. Similarly, mutations in the C. elegans ASIC-1 gene cause abnormal performance in an associative learning paradigm [Citation62]. Although the precise neurophysiological processes affected by DEG/ENaC signaling remain poorly understood, these data suggest a conserved role for DEG/ENaC signaling in behaviors that are associated with changes in neuronal plasticity, which could be mediated at least in part by DEG/ENaC function at the synapse.
Roles of DEG/ENaC channels in synaptic physiology and neuronal plasticity
Several studies of the neuronal functions of DEG/ENaCs have suggested that these channels might play a role in mediating synaptic plasticity, which was first highlighted by studies of the function of ASIC1a in the mouse hippocampus. Specifically, electrophysiological recordings in hippocampal slices have shown that the loss of ASIC1a has no effect on baseline synaptic transmission, or long-term depression (LTD), but impairs long-term potentiation (LTP) [Citation48,Citation57]. ASIC1a knockout mice also display a decreased paired-pulse ratio [Citation63], and impaired EPSP facilitation in response to high frequency stimulation in central synapses [Citation57]. Together, these data suggest that ASIC1a plays a role in mediating hippocampal synaptic plasticity. However, a follow-up study that used a different transgenic strategy to knock out ASIC1a in the nervous system, found no impact of the mutation on hippocampal LTP [Citation64]. Therefore, whether DEG/ENaC-dependent signaling plays a role in long-term neuronal plasticity in the mammalian hippocampus, or under which conditions, remains uncertain. Nevertheless, more recently, several studies have explored the neuronal functions of ASICs in additional brain regions. These studies showed that ASIC1a knockout mice also display impaired LTP in the amygdala [Citation50,Citation51] and stronger short-term depression at the calyx of Held synapse [Citation65]. Although knocking out ASIC genes in these specific brain regions leads to decreased synaptic activity, studies in other brain regions have found a contrary effect. For example, studies of the mouse insular cortex have found that genetic knockout or pharmacological inhibition of ASIC1a channels decreased the probability of LTD, with no effect on LTP [Citation53], and studies at the rodent neuromuscular junction (NMJ) have found that ASIC1a knockout enhances synaptic facilitation [Citation52]. Together, these studies suggest that the effects of DEG/ENaCs on synaptic plasticity vary across neuronal types and brain regions, and can serve to either enhance or dampen synaptic activity.
The effects of mutations in DEG/ENaCs on LTP and LTD suggest that DEG/ENaCs might play a direct role in synaptic physiology. Additional support for a synaptic role for DEG/ENaCs comes from studies demonstrating that some ASIC channels are specifically localized to synaptic sites in mammalian neurons [Citation58], including presynaptic terminals [Citation52] and dendritic spines [Citation57,Citation66,Citation67]. However, it should be noted that other studies have reported a broader subcellular distribution of ASICs in individual central neurons, in contrast to synaptic enrichment [Citation68]. Invertebrate DEG/ENaCs have also been identified at synaptic sites in C. elegans and Drosophila [Citation47,Citation62], although the majority of localization data in these species has only been assessed by using the overexpression of tagged transgenes, and therefore, the subcellular distribution of native DEG/ENaC channels has yet to be determined in the majority of invertebrate species. In addition to the immunohistochemistry data, immunoprecipitation studies in heterologous expression systems have found that specific ASIC subunits interact with both pre- and postsynaptic proteins, including clathrin and PSD95 [Citation67,Citation69]. Although these molecular interactions are yet to be replicated with natively-expressed proteins in vivo, these studies suggest that at least some DEG/ENaCs are localized to either the pre- or the postsynaptic neuronal subcellular compartment.
The precise physiological functions of DEG/ENaC channels at the synapse remain mostly unknown. Electrophysiological studies in cultured neurons have shown that homomeric ASIC1a and heteromeric ASIC1a/ASIC2a channels can mediate an influx of cations, primarily sodium ions, in response to decreased extracellular pH [Citation50,Citation57,Citation58,Citation70]. The sensitivity of these channels to increases in the extracellular concentration of H+ suggests the provocative hypothesis that synaptic ASICs might be playing the role of a synaptic pH sensor, which directly responds to synaptic acidification associated with neurotransmitter release. This hypothesis is supported by electrophysiological studies in mouse brain slices, which have shown that synaptic transmission is sufficient to acidify extracellular fluid [Citation71,Citation72], likely due to the highly acidic lumen of synaptic vesicles [Citation73]. However, as we discuss below, several recent studies in vertebrate and invertebrate models suggest that the synaptic roles of DEG/ENaCs might be more complex than as simple synaptic pH sensors.
DEG/ENaC modulation of presynaptic physiology
In invertebrate and vertebrate model systems, DEG/ENaC channels have been shown to play presynaptic roles, modulating both evoked and spontaneous neurotransmission. Direct evidence for a presynaptic role for DEG/ENaCs was first described in C. elegans, where ASIC1 is localized to dopaminergic presynaptic terminals, and asic-1 mutant worms have been shown to exhibit decreased dopaminergic release [Citation62]. In Drosophila, several individual DEG/ENaC subunits have been shown to play presynaptic roles in modulating synaptic plasticity at the larval NMJ, a well-studied excitatory glutamatergic synapse that has many similarities to mammalian central glutamatergic synapses [Citation74,Citation75]. The Drosophila NMJ displays robust synaptic homeostatic plasticity, whereby partial blockage of postsynaptic glutamate receptors with either pharmacological or genetic manipulations leads to a homeostatic increase in evoked neurotransmitter release, which subsequently produces “normal” postsynaptic currents in response to stimulation [Citation76]. Recently, a Drosophila genetic screen identified three DEG/ENaC-encoding genes, ppk1, ppk11 and ppk16, as presynaptic proteins that are required in motor neurons for this form of homeostatic synaptic plasticity [Citation46,Citation47].
Although currently available genetic tools in vertebrate models do not allow precise anatomical localization of synaptically-enriched DEG/ENaC channels to either pre- or postsynaptic sites, physiological and behavioral data suggest that some synaptic DEG/ENaC channels are likely to exert their function at the presynaptic site. For example, ASIC1a knockout in mice has been shown to reduce the paired-pulse ratio [Citation63], a phenotype that typically indicates a presynaptic mechanism [Citation77]. Studies in vertebrates have also revealed that, in addition to their presynaptic impact on evoked release, DEG/ENaCs also play a role in regulating the frequency of spontaneous neurotransmission [Citation52,Citation63,Citation78,Citation79], which further supports a presynaptic role in processes that regulate the rate of synaptic vesicle release [Citation80]. Increased frequency of spontaneous release has been observed in ASIC1a knockout mice at peripheral synapses [Citation52] and central synapses in cultured systems [Citation63]. Similarly, pharmacological treatments of hippocampal slices with an ASIC1a antagonist lead to an increase in the frequency of spontaneous neurotransmitter release [Citation79]. Conversely, in brain slice preparations of the amygdala, application of an ASIC1a antagonist decreases the frequency of spontaneous release [Citation78]. How the activity of DEG/ENaC channels might actually regulate spontaneous neurotransmitter release is unclear. Spontaneous neurotransmission has long been appreciated for its role in the development of synaptic connections, yet more recently, has been shown to affect neurotransmission in fully developed animals, including aspects of synaptic plasticity [Citation81]. Therefore, the impact of DEG/ENaCs on spontaneous neurotransmission may be expected to affect both developmental processes as well as synaptic plasticity in adult animals. Together, evidence from invertebrates and vertebrates show that DEG/ENaCs have presynaptic effects on both evoked and spontaneous synaptic transmission.
DEG/ENaC modulation of postsynaptic physiology
In addition to their function at the presynaptic site, observations that mutations in DEG/ENaC channels also affect processes associated with postsynaptic plasticity, such as LTP and LTD, suggested at least some family members act via postsynaptic processes [Citation48,Citation50,Citation51,Citation53,Citation57]. Particularly, studies have demonstrated that mutations in several ASIC-encoding genes affect the function of postsynaptic glutamate receptors and spine density, further supporting a postsynaptic role for DEG/ENaC channels in modulating synaptic plasticity. Specific examples include the effect of ASIC1a mutations on decreased NMDA receptor function in the hippocampus [Citation48], and their association with lower overall dendritic spine density [Citation66]. Conversely, in the nucleus accumbens, ASIC1A knockout mice display increased density of dendritic stubby spines, altered glutamate receptor function in the form of increased inward rectification of AMPA currents following evoked release, and an increase in the ratio of AMPA to NMDA receptor currents [Citation49]. Consequently, some ASICs appear to modulate synaptic plasticity via postsynaptic mechanisms, contributing to either facilitation or dampening of synaptic activity.
In addition to data from vertebrate models, physiological and behavioral data from studies with Drosophila suggest that DEG/ENaCs contribute to postsynaptic functions in invertebrates as well. Specifically, we recently showed that mutations in the DEG/ENaC-encoding gene ppk29 lead to decreased amplitude of spontaneous neurotransmission at the larval neuromuscular junction (NMJ), which could be rescued by the expression of ppk29 specifically in the postsynaptic muscle cells, but not in the presynaptic motor neurons [Citation41]. Conversely, mutations in ppk1 lead to increased amplitude of spontaneous excitatory neurotransmission at the larval NMJ [Citation47]. Together, these data suggest that in both Drosophila and vertebrates, independent postsynaptic DEG/ENaC subunits play opposing roles to either increase or decrease activity at a single synapse.
The exact biophysical and physiological processes associated with the postsynaptic action of ppk29, and how it might affect postsynaptic excitatory glutamatergic current influx in response to spontaneous neurotransmission, are still unknown. However, molecular and genetic analyses suggest that the effects of the ppk29 mutation might be mediated, at least in part, via changes in the relative transcriptional ratio between the major two postsynaptic glutamate receptor subtypes [Citation41]. These data are in agreement with previous findings about the impact of mutations in mammalian ASICs on AMPA and NMDA receptor ratios, as discussed above, and thereby suggests a possible conserved mechanism for the postsynaptic effects of mammalian and insect DEG/ENaCs on synaptic activity through altered glutamate receptor function.
Models for the synaptic functions of DEG/ENaC channels
Based on our general understanding of the physiological functions of ENaCs in epithelial tissues [Citation1,Citation19,Citation82], it is likely that DEG/ENaCs act as cation channels in the plasma membrane, where they mediate the influx of sodium ions, either as a constitutively open channel, or via extracellular ligand-dependent activation. Indeed, previous reports have shown that ASICs can generate robust currents in response to rapid increases in extracellular acidity [Citation50,Citation57,Citation58,Citation70]. Consistent with this model, DEG/ENaCs appear to have larger effects on neuronal activity following bouts of high firing [Citation48,Citation50,Citation51,Citation57], likely due to the extracellular release of free protons associated with the release of many synaptic vesicles. Thus, although DEG/ENaC channels are not voltage-gated, the activity of some family members is likely to be modulated by neuronal firing rates, and therefore, points to a possible physiological explanation for how they might be modulating neuronal plasticity.
In the mammalian brain, extracellular proton-dependent activation of ASIC channels has been shown to play a role in increasing intracellular calcium levels [Citation66,Citation67], which likely mediates some of the impact of DEG/ENaCs on synaptic plasticity. Although heterologously expressed ASICs can directly mediate calcium influx [Citation14,Citation83], the permeability of ASICs to calcium is low [Citation84,Citation85]. Therefore, the actual mechanism by which DEG/ENaC channel activity modulates calcium currents is not clear. One possible model that may explain the relationship between synaptic DEG/ENaC channels and intracellular changes in neuronal calcium levels might be via the indirect impact of DEG/ENaC-dependent sodium influx, which subsequently leads to increased calcium influx through voltage gated calcium channels [Citation86]. In support of this model, inhibition of voltage gated calcium channels prevents acid induced increases in intracellular calcium [Citation87]. Activation of signaling pathways that trigger calcium release from intracellular stores may also contribute to DEG/ENaC mediated increases in intracellular calcium, although this has not been tested experimentally.
Model for presynaptic DEG/ENaC functions
The primary model for presynaptic DEG/ENaC function posits that during high frequency synaptic vesicle release, acidification of the extracellular space, and the subsequent induction of DEG/ENaC-dependent influx of sodium ions, modulates the opening of presynaptic voltage-gated calcium channels (Figure 1(a)). Calcium influx through these channels would then be expected to increase calcium dependent synaptic vesicle release [Citation88–Citation90]. This model is consistent with several phenotypes associated with DEG/ENaC knockout animals. For example, studies of dopaminergic neurons in C. elegans suggest that mutations in some DEG/ENaC channels lead to decreased calcium influx, which is associated with decreased synaptic vesicle release [Citation62]. Similarly, mutations in some synaptic DEG/ENaC channels have been shown to be associated with decreased glutamate release at the Drosophila NMJ during synaptic homeostatic plasticity [Citation46,Citation47], and decreased paired-pulse ratio and frequency of spontaneous release at some mammalian glutamatergic central synapses [Citation63,Citation78].
However, this model does not explain the increased frequency of spontaneous release observed in some brain regions of ASIC1a knockout mice [Citation52,Citation63,Citation79]. Although no current physiological and biophysical models may explain this phenotype, it has been suggested that this phenotype may be mediated by non-traditional roles for ASICs, such as direct protein-protein interactions between ASICs and other synaptic components [Citation52,Citation63], which are discussed below.
Model for postsynaptic DEG/ENaC functions
Empirical studies suggest that the impact of postsynaptic DEG/ENaCs on synaptic physiology can be broadly categorized into two independent pathways, which can lead to either facilitation or depression of synaptic transmission. The primary and most simple model that has been proposed to explain postsynaptic DEG/ENaC-mediated facilitation of synaptic activity suggests that cation influx via DEG/ENaCs leads to either direct or indirect activation of voltage-gated calcium channels, depolarization of the postsynaptic compartment, and the subsequent release of the NMDA receptor magnesium block, which increases NMDA receptor activity [Citation48,Citation57] (). In support of this model, ASICs in the rodent hippocampus have been shown to modulate LTP in part by increasing NMDA receptor activity [Citation48,Citation57]. Additionally, ASICs have been shown to modulate intracellular calcium levels, and to increase the levels of phosphorylated calcium/calmodulin-dependent protein kinase II (CaMKII), which has been shown to mediate the effect of ASIC activation on increasing spine density [Citation66]. Whether the impact of ASICs on NMDA receptors and spine density are dependent on each other is unknown.
Figure 1. Models for the putative pre- and postsynaptic functions of DEG/ENaC channels. (a) Model for presynaptic DEG/ENaC function. The lumen of synaptic vesicles is acidic. Therefore, high frequency release of synaptic vesicles leads to an increase in proton concentration in the synaptic cleft. The lower pH leads to opening of presynaptic DEG/ENaCs, followed by a presynaptic sodium influx. Subsequently, local depolarization drives the opening of presynaptic voltage-gated calcium channels, and calcium-dependent synaptic vesicle release. (b) Model for postsynaptic DEG/ENaC mediated facilitation of synaptic activity. Upon the presynaptic release of vesicles, the synaptic cleft acidifies, which leads to an influx of cations directly through DEG/ENaC channels or indirectly via voltage-gated calcium channels, and the removal of the extracellular magnesium block from NMDA receptors. Subsequently, the DEG/ENaC-dependent calcium influx also induces the phosphorylation of CaMKII, which increases spine density. (c) Model for postsynaptic DEG/ENaC mediated depression of synaptic activity. As in (b), DEG/ENaC-mediated depolarization due to the acidification of the synaptic cleft leads to a calcium influx, which modulates the dephosphorylation and activation of GSK3β, which promotes internalization of postsynaptic AMPA receptors, and subsequently leads to long-term synaptic depression.
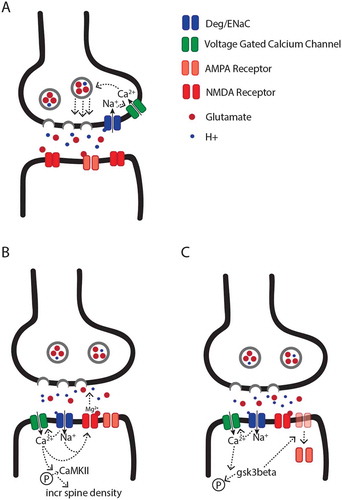
Postsynaptic DEG/ENaCs have also been shown to play a role in synaptic activity depression. For example, ASIC1a knockout mice display a reduced probability of LTD in the insular cortex [Citation53]. Although the exact mechanism for ASIC1a-depedent LTD is not known, it has been hypothesized that direct or indirect DEG/ENaC-dependent modulation of postsynaptic calcium influx leads to the dephosphorylation of glycogen synthase kinase-3β (GSK3β) via phosphatase 1/2A, which drives the internalization of AMPA receptors, and subsequently, depressed synaptic activity () [Citation53]. In support of this model, recent studies suggest that ASIC1a is required for the dephosphorylation of GSK3β in the mammalian brain, and the expression of a constitutively active form of GSK3β is sufficient to rescue some of the behavioral and physiological phenotypes associated with ASIC1a knockout [Citation53].
DEG/ENaC channels may also directly regulate postsynaptic excitability. For example, a recent intriguing study showed that the postsynaptic activity of ASIC1a is capable of mediating action potentials at the calyx of Held synapse even when postsynaptic glutamate receptors are blocked pharmacologically [Citation65]. However, the authors note that ASIC1a does not appear to have a significant contribution to action potentials under physiological conditions, because neither pharmacological block nor genetic knockout of ASIC1a impact action potential generation in the presence of functional glutamate receptors. Nevertheless, these findings suggest that under certain conditions, postsynaptic ASICs may impact synaptic physiology independently of glutamate receptors. Additionally, this study showed that postsynaptic ASIC1a channel activity could modulate short term depression in response to high frequency presynaptic stimulation. Although the mechanism for this effect is unknown, the proposed model suggests that postsynaptic DEG/ENaC channels play a role in a putative retrograde signaling pathway that could modulate presynaptic release probability in response to physiological changes in the postsynaptic cell [Citation65].
The specific mechanistic differences that mediate DEG/ENaC-dependent LTP at some synapses and LTD at others remain mostly unknown. Several non-mutually exclusive explanations include synaptic differences in DEG/ENaC subunit composition, interactions with different classes of voltage gated calcium channels, and differences in intrinsic neuronal activity patterns, which may modulate the time course of acidification of the synaptic cleft. In addition, molecular differences associated with downstream signaling pathways, and the availability of postsynaptic second messengers such as GSK3β, might also contribute to the observed impact of DEG/ENaC channels on glutamatergic neurotransmission.
Here we have presented independent models for synaptic DEG/ENaC function in pre- and postsynaptic compartments, contributing to facilitation or dampening of synaptic activity. However, we suspect these pathways most likely occur in parallel, at least at some synapses. Indeed, at the Drosophila NMJ, DEG/ENaCs are present at both pre- and postsynaptic compartments, mediating both increases and decreases in synaptic activity [Citation41,Citation46,Citation47]. Thus, we suspect that in vertebrate synapses as well, DEG/ENaCs may simultaneously modulate several aspects of synaptic physiology.
Possible contributions of glial DEG/ENaCs to synaptic physiology
Over the past two decades, glial cells have been shown to impact many aspects of neuronal function and synaptic activity [Citation91,Citation92]. Recently, several studies have shown that some DEG/ENaC genes are also expressed in glial cells, including sheath and socket glial cells in C. elegans [Citation39,Citation40,Citation93], as well as astrocytes [Citation23,Citation94], microglia [Citation24], NG2 glial cells [Citation95] and oligodendrocytes [Citation22] in rodents. Although the specific cellular and physiological functions of DEG/ENaCs in glia remain mostly unknown, studies suggest that glial ASICs contribute to inward calcium currents in response to decreases in extracellular pH, possibly by acting as receptors for the protons that are co-released with neurotransmitters [Citation22,Citation24,Citation94,Citation95]. In addition, DEG/ENaCs could regulate glial calcium levels, which have been shown to modulate various aspects of neuronal synaptic function, possibly through the release of gliotransmitters, such as glutamate and ATP [Citation96]. Nonetheless, it should be noted that the majority of studies implicating glial DEG/ENaCs in the regulation of synaptic activity in knockout mouse models are confounded by the gene being missing from both neurons and glia. Therefore, the specific division of labor in terms of the specific contributions of neuronal versus glial DEG/ENaCs to synaptic processes is not yet clear.
Channel activity-independent models for the role of DEG/ENaC proteins in synaptic structure and function
In addition to their possible roles as synaptic ion channels, we would like to consider other possible non-canonical, channel-independent synaptic functions for DEG/ENaC-like proteins. Similar non-canonical neuronal functions have been previously assigned to other classes of ion channels [Citation97–Citation99]. Specifically, as we discuss below, some of the structural features of members of the DEG/ENaC family suggest that these proteins might also contribute to processes associated with synaptic structural integrity and stability.
One of the defining features of all members of the DEG/ENaC protein family is the large extracellular loop [Citation2–Citation5]. Specific structural features of the extracellular loop of several DEG/ENaCs have been hypothesized to resemble peptide neurotoxins that modulate neuronal physiology by directly modifying the functions of voltage-gated sodium and potassium channels [Citation100,Citation101]. In support of this hypothesis, it has been shown that the extracellular loop of ASIC1a can inhibit calcium dependent potassium channels in the BK family, as well as voltage-gated Kv1.3 potassium channels [Citation100,Citation102]. In addition, the inhibitory association between ASIC1a and BK channels has been shown to depend on extracellular pH levels, whereby low pH levels that are sufficient to activate ASICs, are also sufficient to release the inhibition of BK channel activity [Citation100]. In cultured cortical cells, triple knockout of ASIC1a, ASIC2 and ASIC3 was shown to increase action potential firing rate and increase the time of the after hyperpolarization in a manner that was blocked by a BK channel inhibitor [Citation102]. Subsequently, it has been hypothesized that some presynaptic effects of ASICs on the frequency of spontaneous neurotransmitter release could be due to interactions between ASICs and other presynaptic ion channels, or direct interactions with the synaptic release machinery [Citation52,Citation63]. However, to date these hypotheses remain theoretical.
In addition to the studies that have implicated protein-protein interactions between DEG/ENaC channels and other neuronal ion channels [Citation100,Citation102], studies in other tissues such as the kidney suggest that ENaC channels physically interact with sodium chloride cotransporters, leading to specific modulations of the activities of both proteins [Citation103]. Together, these studies suggest that physical interactions between DEG/ENaCs and other membrane bound proteins are probably more common than currently appreciated, and likely represent an important facet of DEG/ENaC-dependent physiological processes in general, and modulation of synaptic activity in particular.
Another non-mutually exclusive hypothesis for the possible synaptic functions of DEG/ENaCs relates to the recent discovery that the intracellular domains of DEG/ENaCs may play enzymatic roles to modulate intracellular signaling cascades. Specifically, it was recently shown that independent of its ion channel function, extracellular acidification drives the intracellular C terminus of ASIC1a to bind to and phosphorylate the serine/threonine kinase receptor interaction protein 1 (RIP1), a key mediator of necroptosis [Citation104]. Additionally, bioinformatic analysis of 28 DEG/ENaC protein sequences, including invertebrate and mammalian sequences, identified similarities between the intracellular N terminus and part of a protease domain [Citation105], suggesting a possible enzymatic function for DEG/ENaCs. Studies of other ion channel families have similarly reported interactions with key enzymes in canonical metabotropic signaling pathways. For example, potassium, calcium and transient receptor potential (TRP) channels have been shown to bind to and modulate protein kinases, including CamKII and serine/threonine kinases [Citation97,Citation98]. Although not yet supported by empirical data, these possible interactions between synaptic ion channels, including DEG/ENaCs, and phosphorylation-dependent signaling cascades, could mediate synaptic plasticity via a myriad of downstream pathways, including short term effects on ion channel function and long-term effects on gene expression [Citation106–Citation108].
Another unexplored aspect of DEG/ENaC channels relates to reports that some family members are localized to intracellular membranes, suggesting that instead of responding to extracellular ligands, these channels play a role in intracellular signaling pathways. For example, in addition to its cell membrane localization, ASIC1a has also been observed in mitochondrial membranes of mouse cortical neurons, where it was shown to physically interact with the inner mitochondrial membrane protein adenine nucleotide translocase (ANT) [Citation109]. While ANTs primary function is in ADP/ATP exchange, ANT is also a key mediator of the mitochondria-driven apoptosis pathway in response to oxidative stress [Citation110]. Based on the observed physical interaction between ASIC1a and ANT, ASIC1a is also likely to modulate mitochondrial ADP/ATP exchange in non-oxidative stress conditions. Therefore, because both the pre- and postsynaptic compartments are packed with mitochondria, which are required for proper neuronal development and function via their regulation of ATP supply and calcium homeostasis [Citation111,Citation112], we hypothesize that some DEG/ENaC channels might also affect synaptic physiology via their role in mitochondria.
Conclusions
In recent years, DEG/ENaCs have emerged as important modulators of neuronal and behavioral plasticity in health and disease. Therefore, understanding their specific action at the synapse is important because they could serve as central targets for drugs that affect neuronal plasticity as a possible solution for diverse cognitive and psychiatric disorders, and for deciphering the basic biological principles that govern synaptic and behavioral plasticity. Although the actual physiological functions of synaptic DEG/ENaCs is still mostly unknown, here we propose several non-mutually exclusive models that might explain such functions, and provide an overview of the empirical data that support at least some of these models. We anticipate that future studies will indicate that individual DEG/ENaC-encoding genes contribute to synaptic physiology and plasticity across neuronal cell types and species via diverse mechanisms and cellular compartments. These studies could be enhanced by a better understanding of the subcellular localization of neuronal DEG/ENaC proteins, especially in the pre- and postsynaptic domains. Because generating antibodies that target membrane bound proteins is notoriously difficult, we expect that the use of CRISPR/Cas9-depedent genome editing approaches in genetically tractable organisms such as the fly, the worm, and the mouse would accelerate this current gap by tagging endogenous DEG/ENaC-encoding genes. Additionally, the use of genome editing for the generation of carefully designed mutant alleles could lead to novel mechanistic insights into the synaptic functions of DEG/ENaCs by directly testing predictions derived from the models we propose here.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002 Jul;82(3):735–767. PubMed PMID: 12087134.
- Jasti J, Furukawa H, Gonzales EB, et al. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007 Sep 20;449(7160):316–323. PubMed PMID: 17882215.
- Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009 Jul 30;460(7255):599–604. PubMed PMID: 19641589; PubMed Central PMCID: PMCPMC2845979.
- Canessa CM, Schild L, Buell G, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994 Feb 03;367(6462):463–467. PubMed PMID: 8107805.
- Lai CC, Hong K, Kinnell M, et al. Sequence and transmembrane topology of MEC-4, an ion channel subunit required for mechanotransduction in Caenorhabditis elegans. J Cell Biol. 1996 Jun;133(5):1071–1081. PubMed PMID: 8655580; PubMed Central PMCID: PMCPMC2120861.
- Ben-Shahar Y. Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Adv Genet. 2011;76:1–26. PubMed PMID: 22099690; PubMed Central PMCID: PMC3298668.
- Eastwood AL, Goodman MB. Insight into DEG/ENaC channel gating from genetics and structure. Physiology (Bethesda). 2012 Oct;27(5):282–290. PubMed PMID: 23026751; PubMed Central PMCID: PMCPMC4012085.
- Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem. 2009 Jul 31;284(31):20447–20451. PubMed PMID: 19401469; PubMed Central PMCID: PMCPMC2742807.
- Chalfie M, Wolinsky E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990 May 31;345(6274):410–416. PubMed PMID: 2342572.
- Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991 Feb 14;349(6310):588–593. PubMed PMID: 1672038.
- Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993 Feb 04;361(6411):467–470. PubMed PMID: 8381523.
- Lingueglia E, Voilley N, Waldmann R, et al. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett. 1993 Feb 22;318(1):95–99. PubMed PMID: 8382172.
- Chalfie M, Driscoll M, Huang M. Degenerin similarities. Nature. 1993 Feb 11;361(6412):504. PubMed PMID: 8429902.
- Waldmann R, Champigny G, Bassilana F, et al. A proton-gated cation channel involved in acid-sensing. Nature. 1997 Mar 13;386(6621):173–177. PubMed PMID: 9062189.
- Waldmann R, Champigny G, Voilley N, et al. The mammalian degenerin MDEG, an amiloride-sensitive cation channel activated by mutations causing neurodegeneration in Caenorhabditis elegans. J Biol Chem. 1996 May 03;271(18):10433–10436. PubMed PMID: 8631835.
- Price MP, Snyder PM, Welsh MJ. Cloning and expression of a novel human brain Na+ channel. J Biol Chem. 1996 Apr 05;271(14):7879–7882. PubMed PMID: 8626462.
- Waldmann R, Bassilana F, de Weille J, et al. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997 Aug 22;272(34):20975–20978. PubMed PMID: 9261094.
- Zelle KM, Lu B, Pyfrom SC, et al. The genetic architecture of Degenerin/Epithelial sodium channels in Drosophila. G3 (Bethesda). 2013 Mar;3(3):441–450. PubMed PMID: 23449991; PubMed Central PMCID: PMC3583452.
- Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016 Apr 1;579(2):95–132. PubMed PMID: 26772908; PubMed Central PMCID: PMCPMC4756657.
- Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013 Jul;14(7):461–471. PubMed PMID: 23783197; PubMed Central PMCID: PMCPMC4307015.
- Bartoi T, Augustinowski K, Polleichtner G, et al. Acid-sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci U S A. 2014 Jun 03;111(22):8281–8286. PubMed PMID: 24847067; PubMed Central PMCID: PMCPMC4050600.
- Feldman DH, Horiuchi M, Keachie K, et al. Characterization of acid-sensing ion channel expression in oligodendrocyte-lineage cells. Glia. 2008 Aug 15;56(11):1238–1249. PubMed PMID: 18452213; PubMed Central PMCID: PMCPMC2895986.
- Huang C, Hu ZL, Wu WN, et al. Existence and distinction of acid-evoked currents in rat astrocytes. Glia. 2010 Sep;58(12):1415–1424. PubMed PMID: 20549751.
- Yu XW, Hu ZL, Ni M, et al. Acid-sensing ion channels promote the inflammation and migration of cultured rat microglia. Glia. 2015 Mar;63(3):483–496. PubMed PMID: 25377529.
- Brockway LM, Zhou ZH, Bubien JK, et al. Rabbit retinal neurons and glia express a variety of ENaC/DEG subunits. Am J Physiol Cell Physiol. 2002 Jul;283(1):C126–C134. PubMed PMID: 12055080.
- Chen CC, Zimmer A, Sun WH, et al. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc Natl Acad Sci U S A. 2002 Jun 25;99(13):8992–8997. PubMed PMID: 12060708; PubMed Central PMCID: PMCPMC124411.
- Chen CC, Wong CW. Neurosensory mechanotransduction through acid-sensing ion channels. J Cell Mol Med. 2013 Mar;17(3):337–349. PubMed PMID: 23490035; PubMed Central PMCID: PMCPMC3823015.
- Omerbasic D, Schuhmacher LN, Bernal Sierra YA, et al. ASICs and mammalian mechanoreceptor function. Neuropharmacology. 2015 Jul;94:80–86. PubMed PMID: 25528740.
- Chu XP, Papasian CJ, Wang JQ, et al. Modulation of acid-sensing ion channels: molecular mechanisms and therapeutic potential. Int J Physiol Pathophysiol Pharmacol. 2011;3(4):288–309. PubMed PMID: 22162785; PubMed Central PMCID: PMCPMC3230262
- Lv RJ, He JS, Fu YH, et al. ASIC1a polymorphism is associated with temporal lobe epilepsy. Epilepsy Res. 2011 Sep;96(1–2):74–80. PubMed PMID: 21664108.
- Ziemann AE, Schnizler MK, Albert GW, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008 Jul;11(7):816–822. PubMed PMID: 18536711; PubMed Central PMCID: PMC2553357.
- Friese MA, Craner MJ, Etzensperger R, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007 Dec;13(12):1483–1489. PubMed PMID: 17994101.
- Bernardinelli L, Murgia SB, Bitti PP, et al. Association between the ACCN1 gene and multiple sclerosis in Central East Sardinia. PLoS One. 2007 May 30;2(5):e480. PubMed PMID: 17534430; PubMed Central PMCID: PMCPMC1868958.
- Smoller JW, Gallagher PJ, Duncan LE, et al. The human ortholog of acid-sensing ion channel gene ASIC1a is associated with panic disorder and amygdala structure and function. Biol Psychiatry. 2014 Dec 01;76(11):902–910. PubMed PMID: 24529281; PubMed Central PMCID: PMCPMC4103972.
- Syntichaki P, Tavernarakis N. Genetic models of mechanotransduction: the nematode Caenorhabditis elegans. Physiol Rev. 2004 Oct;84(4):1097–1153. PubMed PMID: 15383649.
- Liu L, Johnson WA, Welsh MJ. Drosophila DEG/ENaC pickpocket genes are expressed in the tracheal system, where they may be involved in liquid clearance. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):2128–2133. PubMed PMID: 12571352; PubMed Central PMCID: PMCPMC149970.
- Tavernarakis N, Shreffler W, Wang S, et al. unc-8, a DEG/ENaC family member, encodes a subunit of a candidate mechanically gated channel that modulates C. elegans locomotion. Neuron. 1997 Jan;18(1):107–119. PubMed PMID: 9010209.
- Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994 Feb 03;367(6462):467–470. PubMed PMID: 7509039.
- Wang Y, Apicella A Jr., Lee SK, et al. A glial DEG/ENaC channel functions with neuronal channel DEG-1 to mediate specific sensory functions in C. elegans. EMBO J. 2008 Sep 17;27(18):2388–2399. PubMed PMID: 18701922; PubMed Central PMCID: PMCPMC2543049.
- Han L, Wang Y, Sangaletti R, et al. Two novel DEG/ENaC channel subunits expressed in glia are needed for nose-touch sensitivity in Caenorhabditis elegans. J Neurosci. 2013 Jan 16;33(3):936–949. PubMed PMID: 23325233; PubMed Central PMCID: PMCPMC3711640.
- Hill A, Zheng X, Li X, et al. The drosophila postsynaptic DEG/ENaC channel ppk29 contributes to excitatory neurotransmission. J Neurosci. 2017 Mar 22;37(12):3171–3180. PubMed PMID: 28213447; PubMed Central PMCID: PMCPMC5373112.
- Jospin M, Allard B. An amiloride-sensitive H+-gated Na+ channel in caenorhabditis elegans body wall muscle cells. J Physiol. 2004 Sep 15;559(Pt 3):715–720. PubMed PMID: 15254157; PubMed Central PMCID: PMCPMC1665179.
- O’Hagan R, Chalfie M, Goodman MB. The MEC-4 DEG/ENaC channel of caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat Neurosci. 2005 Jan;8(1):43–50. PubMed PMID: 15580270.
- Suzuki H, Kerr R, Bianchi L, et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 2003 Sep 11;39(6):1005–1017. PubMed PMID: 12971899.
- Geffeney SL, Cueva JG, Glauser DA, et al. DEG/ENaC but not TRP channels are the major mechanoelectrical transduction channels in a C. elegans nociceptor. Neuron. 2011 Sep 8;71(5):845–857. PubMed PMID: 21903078; PubMed Central PMCID: PMC3170654.
- Younger MA, Muller M, Tong A, et al. A presynaptic ENaC channel drives homeostatic plasticity. Neuron. 2013 Sep 18;79(6):1183–1196. PubMed PMID: 23973209; PubMed Central PMCID: PMCPMC3784986.
- Orr BO, Gorczyca D, Younger MA, et al. Composition and control of a Deg/ENaC channel during presynaptic homeostatic plasticity. Cell Rep. 2017 Aug 22;20(8):1855–1866. PubMed PMID: 28834749.
- Liu MG, Li HS, Li WG, et al. Acid-sensing ion channel 1a contributes to hippocampal LTP inducibility through multiple mechanisms. Sci Rep. 2016 Mar 21;6:23350. PubMed PMID: 26996240; PubMed Central PMCID: PMCPMC4800407.
- Kreple CJ, Lu Y, Taugher RJ, et al. Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat Neurosci. 2014 Aug;17(8):1083–1091. PubMed PMID: 24952644; PubMed Central PMCID: PMCPMC4115047.
- Du J, Reznikov LR, Price MP, et al. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A. 2014 Jun 17;111(24):8961–8966. PubMed PMID: 24889629; PubMed Central PMCID: PMCPMC4066526.
- Chiang PH, Chien TC, Chen CC, et al. ASIC-dependent LTP at multiple glutamatergic synapses in amygdala network is required for fear memory. Sci Rep. 2015 May 19;5:10143. PubMed PMID: 25988357; PubMed Central PMCID: PMCPMC4437300.
- Urbano FJ, Lino NG, Gonzalez-Inchauspe CM, et al. Acid-sensing ion channels 1a (ASIC1a) inhibit neuromuscular transmission in female mice. Am J Physiol Cell Physiol. 2014 Feb 15;306(4):C396–406. PubMed PMID: 24336653; PubMed Central PMCID: PMCPMC3919981.
- Li WG, Liu MG, Deng S, et al. ASIC1a regulates insular long-term depression and is required for the extinction of conditioned taste aversion. Nat Commun. 2016 Dec 07;7:13770. PubMed PMID: 27924869; PubMed Central PMCID: PMCPMC5150990.
- Coryell MW, Wunsch AM, Haenfler JM, et al. Acid-sensing ion channel-1a in the amygdala, a novel therapeutic target in depression-related behavior. J Neurosci. 2009 Apr 29;29(17):5381–5388. PubMed PMID: 19403806; PubMed Central PMCID: PMC2710967. DOI:10.1523/JNEUROSCI.0360-09.2009
- Taugher RJ, Ghobbeh A, Sowers LP, et al. ASIC1A in the bed nucleus of the stria terminalis mediates TMT-evoked freezing. Front Neurosci. 2015;9:239. PubMed PMID: 26257596; PubMed Central PMCID: PMCPMC4508508.
- Coryell MW, Ziemann AE, Westmoreland PJ, et al. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry. 2007 Nov 15;62(10):1140–1148. PubMed PMID: 17662962.
- Wemmie JA, Chen J, Askwith CC, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002 Apr 25;34(3):463–477. PubMed PMID: 11988176.
- Wemmie JA, Askwith CC, Lamani E, et al. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003 Jul 02;23(13):5496–5502. PubMed PMID: 12843249.
- Coryell MW, Wunsch AM, Haenfler JM, et al. Restoring Acid-sensing ion channel-1a in the amygdala of knock-out mice rescues fear memory but not unconditioned fear responses. J Neurosci. 2008 Dec 17;28(51):13738–13741. PubMed PMID: 19091964; PubMed Central PMCID: PMCPMC2651157.
- Price MP, Gong H, Parsons MG, et al. Localization and behaviors in null mice suggest that ASIC1 and ASIC2 modulate responses to aversive stimuli. Genes Brain Behav. 2014 Feb;13(2):179–194. PubMed PMID: 24256442; PubMed Central PMCID: PMCPMC3998777.
- Du J, Price MP, Taugher RJ, et al. Transient acidosis while retrieving a fear-related memory enhances its lability. Elife. 2017 Jun 26;6. PubMed PMID: 28650315; PubMed Central PMCID: PMCPMC5484615. DOI:10.7554/eLife.22564
- Voglis G, Tavernarakis N. A synaptic DEG/ENaC ion channel mediates learning in C. elegans by facilitating dopamine signalling. EMBO J. 2008 Dec 17;27(24):3288–3299. PubMed PMID: 19037257; PubMed Central PMCID: PMCPMC2609744.
- Cho JH, Askwith CC. Presynaptic release probability is increased in hippocampal neurons from ASIC1 knockout mice. J Neurophysiol. 2008 Feb;99(2):426–441. PubMed PMID: 18094106.
- Wu PY, Huang YY, Chen CC, et al. Acid-sensing ion channel-1a is not required for normal hippocampal LTP and spatial memory. J Neurosci. 2013 Jan 30;33(5):1828–1832. PubMed PMID: 23365222.
- Gonzalez-Inchauspe C, Urbano FJ, Di Guilmi MN, et al. Acid-sensing ion channels activated by evoked released protons modulate synaptic transmission at the mouse calyx of held synapse. J Neurosci. 2017 Mar 8;37(10):2589–2599. PubMed PMID: 28159907.
- Zha XM, Wemmie JA, Green SH, et al. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci U S A. 2006 Oct 31;103(44):16556–16561. PubMed PMID: 17060608; PubMed Central PMCID: PMCPMC1621052.
- Zha XM, Costa V, Harding AM, et al. ASIC2 subunits target acid-sensing ion channels to the synapse via an association with PSD-95. J Neurosci. 2009 Jul 01;29(26):8438–8446. PubMed PMID: 19571134; PubMed Central PMCID: PMCPMC2734339.
- Alvarez de la Rosa D, Krueger SR, Kolar A, et al. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol. 2003 Jan 01;546(Pt 1):77–87. PubMed PMID: 12509480; PubMed Central PMCID: PMCPMC2342460.
- Donier E, Rugiero F, Jacob C, et al. Regulation of ASIC activity by ASIC4–new insights into ASIC channel function revealed by a yeast two-hybrid assay. Eur J Neurosci. 2008 Jul;28(1):74–86. PubMed PMID: 18662336.
- Askwith CC, Wemmie JA, Price MP, et al. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem. 2004 Apr 30;279(18):18296–18305. PubMed PMID: 14960591.
- Krishtal OA, Osipchuk YV, Shelest TN, et al. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res. 1987 Dec 15;436(2):352–356. PubMed PMID: 2829992.
- Chen JC, Chesler M. Modulation of extracellular pH by glutamate and GABA in rat hippocampal slices. J Neurophysiol. 1992 Jan;67(1):29–36. PubMed PMID: 1348085.
- Vukicevic M, Kellenberger S. Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol. 2004 Sep;287(3):C682–C690. PubMed PMID: 15115705.
- Menon KP, Carrillo RA, Zinn K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip Rev Dev Biol. 2013 Sep-Oct;2(5):647–670. PubMed PMID: 24014452; PubMed Central PMCID: PMCPMC3767937.
- Collins CA, DiAntonio A. Synaptic development: insights from Drosophila. Curr Opin Neurobiol. 2007 Feb;17(1):35–42. PubMed PMID: 17229568.
- Frank CA. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology. 2014 Mar;78:63–74. PubMed PMID: 23806804; PubMed Central PMCID: PMCPMC3830618.
- Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994 Sep;14(9):5325–5337. PubMed PMID: 7916043.
- Pidoplichko VI, Aroniadou-Anderjaska V, Prager EM, et al. ASIC1a activation enhances inhibition in the basolateral amygdala and reduces anxiety. J Neurosci. 2014 Feb 26;34(9):3130–3141. PubMed PMID: 24573273; PubMed Central PMCID: PMCPMC3935079.
- Ievglevskyi O, Isaev D, Netsyk O, et al. Acid-sensing ion channels regulate spontaneous inhibitory activity in the hippocampus: possible implications for epilepsy. Philos Trans R Soc Lond B Biol Sci. 2016 Aug 05;371(1700). PubMed PMID: 27377725; PubMed Central PMCID: PMCPMC4938031. DOI:10.1098/rstb.2015.0431
- Kaeser PS, Regehr WG. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol. 2014;76:333–363. PubMed PMID: 24274737; PubMed Central PMCID: PMCPMC4503208.
- Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci. 2015 Jan;16(1):5–16. PubMed PMID: 25524119.
- Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997 Apr;77(2):359–396. PubMed PMID: 9114818.
- Yermolaieva O, Leonard AS, Schnizler MK, et al. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004 Apr 27;101(17):6752–6757. PubMed PMID: 15082829; PubMed Central PMCID: PMCPMC404117.
- Bassler EL, Ngo-Anh TJ, Geisler HS, et al. Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J Biol Chem. 2001 Sep 07;276(36):33782–33787. PubMed PMID: 11448963.
- Zhang P, Canessa CM. Single channel properties of rat acid-sensitive ion channel-1alpha, −2a, and −3 expressed in Xenopus oocytes. J Gen Physiol. 2002 Oct;120(4):553–566. PubMed PMID: 12356856; PubMed Central PMCID: PMCPMC2229538.
- Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Mol Brain. 2013 Jan 02;6:1. PubMed PMID: 23281934; PubMed Central PMCID: PMCPMC3562204.
- Herrera Y, Katnik C, Rodriguez JD, et al. sigma-1 receptor modulation of acid-sensing ion channel a (ASIC1a) and ASIC1a-induced Ca2+ influx in rat cortical neurons. J Pharmacol Exp Ther. 2008 Nov;327(2):491–502. PubMed PMID: 18723775.
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008 Sep 25;59(6):861–872. PubMed PMID: 18817727.
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999 Jun;9(3):305–313. PubMed PMID: 10395573.
- Sudhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol. 2012 Jan 1;4(1):a011353. PubMed PMID: 22068972; PubMed Central PMCID: PMCPMC3249630.
- Wu Y, Dissing-Olesen L, MacVicar BA, et al. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 2015 Oct;36(10):605–613. PubMed PMID: 26431938; PubMed Central PMCID: PMCPMC4841266.
- Chung WS, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. 2015 Feb 6;7(9):a020370. PubMed PMID: 25663667; PubMed Central PMCID: PMCPMC4527946.
- Wang Y, D’Urso G, Bianchi L. Knockout of glial channel ACD-1 exacerbates sensory deficits in a C. elegans mutant by regulating calcium levels of sensory neurons. J Neurophysiol. 2012 Jan;107(1):148–158. PubMed PMID: 21994266; PubMed Central PMCID: PMCPMC3349695.
- Yang F, Sun X, Ding Y, et al. Astrocytic acid-sensing ion channel 1a contributes to the development of chronic epileptogenesis. Sci Rep. 2016 Aug 16;6:31581. PubMed PMID: 27526777; PubMed Central PMCID: PMCPMC4985693.
- Lin YC, Liu YC, Huang YY, et al. High-density expression of Ca2+-permeable ASIC1a channels in NG2 glia of rat hippocampus. PLoS One. 2010 Sep 10;5(9). PubMed PMID: 20844750; PubMed Central PMCID: PMCPMC2937019. DOI:10.1371/journal.pone.0012665
- Haydon PG, Nedergaard M. How do astrocytes participate in neural plasticity? Cold Spring Harb Perspect Biol. 2014 Dec 11;7(3):a020438. PubMed PMID: 25502516; PubMed Central PMCID: PMCPMC4355266.
- Kaczmarek LK. Non-conducting functions of voltage-gated ion channels. Nat Rev Neurosci. 2006 Oct;7(10):761–771. PubMed PMID: 16988652.
- Lee A, Fakler B, Kaczmarek LK, et al. More than a pore: ion channel signaling complexes. J Neurosci. 2014 Nov 12;34(46):15159–15169. PubMed PMID: 25392484; PubMed Central PMCID: PMCPMC4228125.
- Nishimune H. Transsynaptic channelosomes: non-conducting roles of ion channels in synapse formation. Channels (Austin). 2011 Sep-Oct;5(5):432–439. PubMed PMID: 21654201; PubMed Central PMCID: PMCPMC3265764.
- Petroff EY, Price MP, Snitsarev V, et al. Acid-sensing ion channels interact with and inhibit BK K+ channels. Proc Natl Acad Sci U S A. 2008 Feb 26;105(8):3140–3144. PubMed PMID: 18287010; PubMed Central PMCID: PMCPMC2268598.
- Tavernarakis N, Driscoll M. Caenorhabditis elegans degenerins and vertebrate ENaC ion channels contain an extracellular domain related to venom neurotoxins. J Neurogenet. 2000 Jan;13(4):257–264. PubMed PMID: 10858823.
- Petroff E, Snitsarev V, Gong H, et al. Acid sensing ion channels regulate neuronal excitability by inhibiting BK potassium channels. Biochem Biophys Res Commun. 2012 Oct 05;426(4):511–515. PubMed PMID: 22960074; PubMed Central PMCID: PMCPMC3488431.
- Mistry AC, Wynne BM, Yu L, et al. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem J. 2016 Oct 01;473(19):3237–3252. PubMed PMID: 27422782; PubMed Central PMCID: PMCPMC5283799.
- Wang YZ, Wang JJ, Huang Y, et al. Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. Elife. 2015 Nov 02;4. PubMed PMID: 26523449; PubMed Central PMCID: PMCPMC4629285. DOI:10.7554/eLife.05682
- Tavernarakis N, Everett JK, Kyrpides NC, et al. Structural and functional features of the intracellular amino terminus of DEG/ENaC ion channels. Curr Biol. 2001 Mar 20;11(6):R205–R208. PubMed PMID: 11301263.
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002 Mar;3(3):175–190. PubMed PMID: 11994750.
- Giese KP, Mizuno K. The roles of protein kinases in learning and memory. Learn Mem. 2013 Sep 16;20(10):540–552. PubMed PMID: 24042850.
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004 Mar;5(3):173–183. PubMed PMID: 14976517.
- Wang YZ, Zeng WZ, Xiao X, et al. Intracellular ASIC1a regulates mitochondrial permeability transition-dependent neuronal death. Cell Death Differ. 2013 Oct;20(10):1359–1369. PubMed PMID: 23852371; PubMed Central PMCID: PMCPMC3770907.
- Liu Y, Chen XJ. Adenine nucleotide translocase, mitochondrial stress, and degenerative cell death. Oxid Med Cell Longev. 2013;2013:146860. PubMed PMID: 23970947; PubMed Central PMCID: PMCPMC3732615.
- Hollenbeck PJ. Mitochondria and neurotransmission: evacuating the synapse. Neuron. 2005 Aug 04;47(3):331–333. PubMed PMID: 16055057; PubMed Central PMCID: PMCPMC2538582.
- Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139. PubMed PMID: 21423525; PubMed Central PMCID: PMCPMC3059669.