
ABSTRACT
The renal thiazide-sensitive sodium-chloride cotransporter (NCC), located in the distal convoluted tubule (DCT) of the kidney, plays an important role in blood pressure regulation by fine-tuning sodium excretion. The human SLC12A3 gene, encoding NCC, gives rise to three isoforms, of which only the third isoform (NCC3) has been extensively investigated so far. However, recent studies unraveled the importance of the isoforms 1 and 2, collectively referred to as NCC splice variant (NCCSV), in several (patho)physiological conditions. In the human kidney, NCCSV localizes to the apical membrane of the DCT and could constitute a functional route for renal sodium-chloride reabsorption. Analysis of urinary extracellular vesicles (uEVs), a non-invasive method for measuring renal responses, demonstrated that NCCSV abundance changes in response to acute water loading and correlates with patients’ thiazide responsiveness. Furthermore, a novel phosphorylation site at serine 811 (S811), exclusively present in NCCSV, was shown to play an instrumental role in NCCSV as well as NCC3 function. This review aims to summarize these new insights of NCCSV function in humans that broadens the understanding on NCC regulation in blood pressure control.
Introduction
Hypertension (high blood pressure) is a major risk factor for stroke, myocardial infarction, heart failure, chronic kidney disease and overall increased risk of mortality [Citation1–Citation4]. It is the most critical and expensive public health problem with a global prevalence of 1.4 billion people, representing one of every four adults worldwide. This is predicted to increase to 1.6 billion people by 2025 [Citation1,Citation2,Citation5–Citation7]. About 90% of hypertensive patients have primary or essential hypertension, which means that the origin of their disorder is unknown and secondary causes such as monogenic disease, renal failure, aldosteronism and renovascular disease are not present [Citation8,Citation9].
It is well known that salt intake plays a significant role in the development of hypertension. The body’s salt homeostasis is maintained by the kidneys through regulated processes of sodium (and chloride) reabsorption [Citation10]. Specifically, the sodium-chloride cotransporter (NCC) is an important player in the maintenance of salt homeostasis as it controls the fine-tuning (~ 7%) of sodium excretion in the distal convoluted tubule (DCT) [Citation11]. NCC is an important pharmacological target in the treatment of hypertension as thiazide-type diuretics, which specifically block NCC, are considered first-line therapy [Citation12,Citation13]. Furthermore, over 180 loss-of-function mutations have been identified in the SLC12A3 gene, encoding NCC, in relation to Gitelman syndrome (OMIM 263,800) [Citation14–Citation16]. This is an autosomal recessive disease that is portrayed by hypocalciuria, hypomagnesemia, hypokalemic metabolic alkalosis, sodium wasting and lower blood pressure levels in comparison to their age-matched unaffected relatives [Citation17,Citation18].
The human SLC12A3 gene yields three separate isoforms as a result of an alternative splicing. NCC isoforms 1 and 2 (NCC1 and NCC2), collectively termed NCCSV, are nine amino acids longer than the third isoform (NCC3) [Citation18–Citation20]. In the present review, we aim to outline the newest findings on the molecular regulation of NCCSV and its role in renal salt handling in various (patho)physiological conditions. A better understanding of all three isoforms is crucial to unravel the molecular events underlying the pathogenesis of essential hypertension, and to develop effective therapeutic approaches to combat the dysregulation of blood pressure.
Characterization of NCCSV
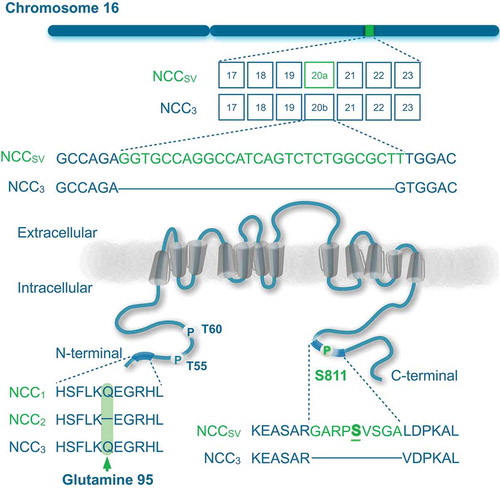
The molecular cloning of human NCC took place in 1996, showing that there exist three different isoforms of the protein as a result of alternative splicing of the SLC12A3 gene [Citation18,Citation21]. Simon et al. reported an mRNA sequence encoding a 1,030 amino acid protein, which was NCC1 [Citation18]. Mastroianni et al. reported later the mRNA sequence corresponding to the NCC3, which encoded for a protein of 1,021 amino acids [Citation21]. The exon 20 of NCC3 is nine amino acids shorter in comparison to NCC1. A possible cryptic splicing at the donor site leads to both long and short forms of exon 20 [Citation19]. In comparison to NCC1, the isoform 2 (NCC2) lacks one amino acid at glutamine residue 95 within amino (N)-terminal domain, making these two isoforms practically indistinguishable (). Hence, both isoforms are collectively referred to as NCC splice variant (NCCSV), which is only present in humans and higher primates.
Figure 1. Splicing of NCC gene.
Schematic representation of the SLC12A3 gene including exon 20a that encodes for the NCCSV (top). The encoded protein NCC contains 12 transmembrane domains and intracellular amino (N)- and carboxy (C)-terminal domains (bottom). Serine (S811) is highlighted as phosphorylation site in the nine additional amino acids in NCCSV. Left bottom panel shows a multiple protein sequence alignment of the three NCC isoforms, demonstrating the lack of glutamine 95 in NCC2.
Recently, the presence of both NCC3 and NCCSV was shown in human urinary extracellular vesicles (uEVs) using mass spectrometry and immunoblot analysis [Citation20]. This study confirmed multiple overlapping peptides of NCCSV and NCC3, which revealed that SCL12A3 leads to the expression of a mixture of NCC3 and at least one but possibly both NCC1 and NCC2 [Citation20]. Total NCC mRNA in the human kidneys consisted on average of ~ 45% of NCCSV and ~ 50% of NCC3 [Citation20]. However, the ratio of NCCSV to NCC3 mRNA expression in human kidney varied between 20 and 60%, which is likely the result of a highly regulated splicing at exon 20 [Citation20]. It has been shown that ~ 95% of human genes are alternatively spliced and the production of alternative mRNA splices is diversely regulated by a variety of external factors. Mechanisms of alternative splicing are highly variable and not well understood yet [Citation22].
Generation of antibodies specific against NCCSV led to confirmation of NCCSV abundance at the apical membrane of the DCT in human kidney, as well in kidney membrane fractions and uEVs [Citation20]. This demonstrated the potential importance of the formerly underrepresented NCCSV in renal salt handling.
Functional role of NCCSV
Regulators of NCC
NCC is a member of the solute carrier 12 (SLC12) family of electroneutral cation-coupled chloride cotransporters. It is a membrane glycoprotein and contains 12 transmembrane segments (S) with large intracellular N- and carboxy (C)-terminal regions, and a large hydrophilic extracellular loop between S7 and S8 [Citation23]. This loop contains glycosylation sites that are essential for the cell surface expression, thiazide sensitivity and activity of NCC [Citation24]. Functional NCC proteins represent as homodimer at the plasma membrane [Citation25,Citation26]. Other posttranslational modifications that play a crucial role in the regulation of NCC activity include ubiquitylation and phosphorylation (reviewed in [Citation27–Citation29]. According to recent large-scale proteomics studies, NCC is ubiquitylated on several conserved lysine residues [Citation30,Citation31]. Generally, NCC ubiquitylation is associated with increased endocytosis from the plasma membrane, as well as protein degradation [Citation32,Citation33]. A recent study pointed out that NCC exhibits site-specific ubiquitylation exerting different roles on NCC function, via modulating NCC phosphorylation or its total abundance at the plasma membrane [Citation33]. Interestingly, several studies indicate that phosphorylation of NCC can interfere with its ubiquitylation, thereby playing a dual role in modulating NCC function [Citation34,Citation35]. Generally, phosphorylation is considered one of the most important posttranslational modification processes in the regulation of NCC activity. The N-terminal domain of NCC contains several key phosphorylation sites including threonines 48, 55 and 60 (T48, T55, T60) and serines 73, 91 and 124 (S73, S90, S124) [Citation36–Citation38]. Phosphorylation at these sites is important for plasma membrane abundance and influence NCC activity [Citation29,Citation39]. Interestingly, several studies have shown that changing T60 to a non-phosphorylated amino acid (T60A) prevented or reduced the phosphorylation at other sites (T48, T55, S73 or S91), suggesting that T60 is a key modulator in the regulation of NCC activity [Citation29,Citation35,Citation37]. This is highlighted by a loss-of-function mutation at T60 in NCC that is linked to Gitelman Syndrome [Citation35,Citation40].
These phosphorylation sites (apart from S124) are targets of the serine-threonine kinases SPAK (STE20/SPS1-related, proline alanine-rich kinase) and OSR1 (oxidative stress responsive protein type 1), which are activated by with no lysine (WNK) kinases [Citation37,Citation41]. Mutations in the human genes encoding WNK1 and WNK4 are causing Gordon’s syndrome [Citation42], which is characterized by hypertension, hyperkalemia, metabolic acidosis, and hypercalciuria. This phenotype is the mirror image of Gitelman syndrome, and it was shown to result from gain-of-function of NCC [Citation43]. While the exact mode of action of the different WNK kinases is still under debate, there are three abundant WNK kinases along the DCT, namely WNK1, WNK3, and WNK4, that affect NCC function by modulating trafficking and/or transport activity via phosphorylation [Citation44]. Regulation of NCC by the WNK-SPAK signaling is an area of intense research and several studies also provided evidence for hormone regulation of the WNK-SPAK-NCC pathway () reviewed in [Citation45] and [Citation29].
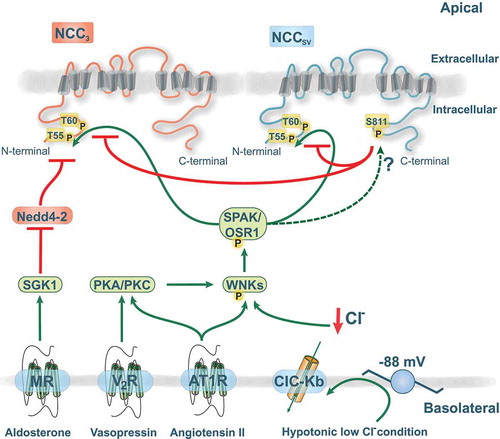
Figure 2. Functional role of the novel phosphorylation site serine 811 (S811).
The amino (N)-terminal domain of NCC contains several key phosphorylation sites (T55/T66) that are essential for NCC activity and plasma membrane abundance. S811 acts as a dominant regulatory site for phosphorylation of T60 and T55 in NCCSV and NCC3. The various interactions of the NCC regulatory pathway are shown as green arrows (stimulatory) or red lines (inhibitory). Phosphorylation is indicated with the symbol P. The with no lysine kinases (WNKs) are capable of activating SPAK (STE20/SPS1-related, proline alanine-rich kinase) and OSR1 (oxidative stress responsive protein type 1). The hormone receptors are located in the basolateral membrane with indicated signaling pathways involving either serum glucocorticoid regulated kinase 1 (SGK1)-Nedd4-2 pathway, protein kinase A/C (PKA/PKC), or WNKs. V2R, vasopressin receptor; AT1R, angiotensin receptor 1; MR, mineralocorticoid receptor.
Novel phosphorylation site in NCCSV
All three human NCC isoforms contain the T55 and T60 residues that are essential mediators of NCC activity. Recently, a new phosphorylation site uniquely located at S811 in NCCSV was identified [Citation46]. Tutakhel et al. further investigated the role of NCCSV S811 phosphorylation on activity of both NCCSV and NCC3 [Citation47]. They showed that a non-phospho-mimetic (S811A) mutant of NCCSV, but not the phospho-mimetic S811D, prevents the phosphorylation at T55 and T60. Importantly, NCCSV S811A was able to inhibit NCC3 phosphorylation at T55 and T60 upon co-expression of NCCSV and NCC3 [Citation47]. In both phosphorylation sites the decrease was more than 50%. This indicates that S811 not only regulates NCCSV activity, but also affects the function of NCC3 in a dominant-negative fashion (). This effect may be due to the heterodimerization of NCCSV with NCC3. The translocation of NCC to the plasma membrane or stability of NCC did not seem to be affected by S811 [Citation47]. This is in agreement with a study demonstrating that NCC3 surface abundance is not affected by the phosphorylation status of T60 [Citation35,Citation36,Citation48]. Based on these insights, NCCSV might have a crucial role in renal salt handling.
Although it is acknowledged that T60 and T55 are phosphorylated via activation of the WNK-SPAK signaling pathway [Citation36,Citation49], the kinase involved in S811 phosphorylation remains to be clarified (). Prediction software (pkaPS) from the IMP-IMBA Bioinformatics group [Citation50] suggested that the recognition motif around S811 of NCCSV shows high similarity to a protein kinase A (PKA) consensus site. Interestingly, several hormonal systems have been shown to activate the cAMP-PKA pathway [Citation51,Citation52]. For example, the sodium-potassium-chloride cotransporter (NKCC2), a SLC family member in the thick ascending limb of Henle’s loop (TAL), can be phosphorylated by PKA signaling downstream of hormones like arginine vasopressin (AVP) or parathyroid hormone (PTH) [Citation53,Citation54]. Additionally, an increasing amount of evidence suggests that the abundance and phosphorylation of NCC is affected by AVP and angiotensin II, which might include PKA or protein kinase C (PKC) [Citation55–Citation58] (). While there is no information yet on direct effects on NCC, it was recently shown that activation of PKC by angiotensin II prevents the binding of WNK4 by kelch-like 3 (KLHL3). KLHL3, together with the ubiquitin ligase cullin 3 (CUL3), normally targets WNK4 for ubiquitylation and degradation [Citation59]. PKC can phosphorylate S433 in KLHL3, a site that was previously postulated to be targeted by PKA as well [Citation60]. In addition, PKC was shown to phosphorylate WNK4 directly, leading to an increase in kinase activity [Citation55]. Hence, PKA and PKC activation can stimulate WNK4 signaling, indirectly leading to NCC activation. The NCC cell surface abundance and phosphorylation are also increased by the mineralocorticoid aldosterone, likely through a mechanism involving the serum glucocorticoid regulated kinase 1 (SGK1)-Nedd4-2 pathway or WNK4 [Citation45,Citation61] (). However, recent evidence suggests that modulation of NCC in response to aldosterone is mediated by secondary changes in plasma potassium concentration that result from aldosterone effects on ENaC [Citation62,Citation63]. Interestingly, while most of the studies on NCC regulation have been performed in animals, this point was recently corroborated in humans [Citation64]. Mineralocorticoid administration in humans resulted in increased NCC and WNK4 abundance, as well as enhanced NCC phosphorylation. This was negatively correlated with plasma potassium levels, supporting the role of potassium effects on NCC, possibly via WNKs [Citation64].
Altogether, NCC regulation in response to the renin-angiotensin-aldosterone (RAAS) system and AVP has been studied extensively and is well documented [Citation45]. It will be interesting to study whether these hormones also signal towards NCCSV and identify which pathway is involved in S811 phosphorylation.
The role of NCCSV in (patho)physiology
Urinary extracellular vesicles, a tool to study NCC
Over the past years, uEVs have been established as a non-invasive tool to study NCC regulation in several physiological and pathophysiological conditions. The uEVs are composed of nucleic acids and proteins that reflect the (patho)physiological state of cells lining the nephron and the urinary tract. They contain membrane and cytosolic proteins, DNA and RNA that are conserved and protected from degradation [Citation65,Citation66]. The cargo of uEVs reflects changes in the expression of different proteins present in the epithelial cells of the nephron, including the total and phosphorylated forms of NCC [Citation20,Citation67,Citation68]. Indeed, it has been demonstrated that patients with Gitelman syndrome display a decreased NCC abundance in uEVs [Citation67,Citation69]. On the other hand, patients with Gordon syndrome show higher levels of NCC in the kidney and this increased abundance was also observed in the uEVs [Citation68]. Moreover, a recent study on kidney transplant recipients treated with calcineurin inhibiters (CNIs) suggested the use of NCC abundance in uEVs as biomarker to predict hypertensive patient’s response to thiazide diuretics [Citation70]. The CNIs cyclosporine A and tacrolimus are commonly used to prevent rejection of transplanted organs, and a common side effect of these drugs is hypertension that can be accompanied by hyperkalemia and metabolic acidosis [Citation71]. Recent studies showed increased levels of total and phosphorylated NCC in uEVs of patients with chronic CNI treatment [Citation70], which suggests that NCC might be involved in the pathogenesis of hypertension in kidney transplant recipients. Hence, the NCC abundance in uEV was analyzed in these patients following treatment with thiazide diuretics, the main anti-hypertensive drug. In line with several studies in animal models [Citation72,Citation73], NCC abundance was increased after thiazide treatment [Citation70]. Furthermore, pre-treatment abundance of NCC in uEVs correlated with blood pressure response to thiazide diuretics, which implies that this method could be used to predict patients’ thiazide sensitivity. Upon dividing the group in responders (blood pressure decrease ≥ 10 mmHg) and non-responders, it became apparent that the non-responder group had a decrease in the ratio between phosphorylated and total NCC (pNCC/tNCC) [Citation70]. Moreover, the serum potassium concentration was lower in responders compared to non-responders, which might be explained by the fact that blood pressure response to thiazides is accompanied by altered renal potassium handling. Blocking of NCC likely results in increased distal delivery of sodium that results in increased potassium secretion via the renal outer medullary potassium channel (ROMK).
NCCSV in physiology
Both NCCSV and NCC3 are present as glycosylated oligomeric structures in uEVs [Citation74,Citation75]. Hence, uEVs were used to assessed the role of NCCSV in vivo. In response to water loading, a strong decrease in expression of NCC3 as well as NCCSV was shown [Citation20]. Water loading results in transient extracellular volume expansion and decrease in serum osmolality, which is known to deactivate RAAS and reduce AVP release [Citation76]. AVP is mainly known for its water-retaining action on collecting duct (CD) by increasing the abundance of the water channel aquaporin 2 (AQP2) [Citation77]. Two recent studies demonstrated that water loading indeed resulted in significantly reduced AQP2 expression in uEVs of the healthy volunteers [Citation20,Citation78]. In line with this, urine osmolality was also decreased, reflecting the diluted urine after water loading [Citation20,Citation78]. Since several studies have described the regulation of NCC by angiotensin, aldosterone, and AVP (reviewed in [Citation45]), the decrease in NCCSV abundance in water-loaded subjects is likely the result of the decline in RAAS activity and AVP release [Citation20]. So far, it indicates that NCCSV, like NCC3, is highly regulated under physiological conditions and suggests a key role in renal salt handling. As abovementioned, future studies should examine whether AVP or the RAAS hormones are directly involved in phosphorylation of NCCSV.
NCCSV in pathophysiology
To understand the role of NCCSV in pathophysiological conditions, the abundance and phosphorylation of all three NCC isoforms were compared in uEVs of essential hypertensive patients before and after hydrochlorothiazide or valsartan treatment [Citation75]. The thiazide-like diuretics work as an anti-hypertensive medication by direct blocking of NCC [Citation79], whereas valsartan is a well-known anti-hypertensive drug that reduces blood pressure by antagonizing the angiotensin II type 1 receptor [Citation80].
While plasma sodium levels remained the same in both treatments, plasma renin levels increased considerably after valsartan treatment. Plasma potassium concentrations were reduced in patients treated with hydrochlorothiazide but not upon valsartan treatment [Citation75]. Interestingly, the abundance of NCCSV and NCC3 was increased upon chronic thiazide treatment, but not by valsartan [Citation75]. However, dividing the hydrochlorothiazide patient group into responders (blood pressure decrease ≥ 5 mmHg) and non-responders revealed that the NCCSV and NCC3 abundance was significantly higher in responders than in patients who did not respond to thiazide treatment. There was no significant difference of the phosphorylated form of NCC (T55/T60 abundance) [Citation75]. These data highlighted that the blood pressure decrease upon thiazide treatment correlates with NCCSV abundance in uEVs and with plasma potassium levels. Together, these observations demonstrate the importance of NCCSV in pathophysiological processes and its involvement in blood pressure control.
Future perspectives
NCCSV is significantly expressed in the human kidney and is not a redundant transcription product, but a fully functional thiazide-sensitive sodium-chloride transporting protein. Besides, the phosphorylation site S811 in NCCSV acts as a dominant-negative regulatory site for phosphorylation of T55 and T60 in all isoforms. While the WNK-SPAK kinases are a well-studied signaling mechanism for NCC regulation, it is yet unclear whether which molecular pathways and kinases are involved in phosphorylation of S811. Future experiments should focus on delineating the regulation of NCCSV. These discoveries reveal a new regulatory mechanism of NCC function, which might be important in renal salt handling, consequently playing a significant role in the pathogenesis of essential hypertension. Future experiments developing S811 phospho-specific antibodies and samples from different human pathological states could provide further understanding of the precise role of NCCSV. Here, uEVs can serve as a non-invasive biomarker source as recent data revealed its potential in guiding anti-hypertensive therapy in patients with essential hypertension. This could ultimately lead to personalized anti-hypertensive treatment. The development and standardization of high-throughput assays for uEV analysis will be necessary for future clinical applications.
Author Contributions
H.W. drafted the manuscript. O.A.Z.T. prepared the figures. O.A.Z.T. and J.W. edited the manuscript.
Acknowledgements
JW is supported by the European Union’s Horizon 2020 Marie Skłodowska-Curie (grant agreement No 748,058) and by The Netherlands Organisation for Health Research and Development (Off Road grant 451,001 004).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mills KT, Bundy JD, Kelly TN, et al. Global disparities of hypertension prevalence and control. Circulation. 2016;134(6):441–450.
- Murray CJ, Lopez AD. Measuring the global burden of disease. New England J Med. 2013;369(5):448–457.
- Narayan KV, Ali MK, Koplan JP. Global noncommunicable diseases—where worlds meet. New England J Med. 2010;363(13):1196–1198.
- Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292.
- Mancia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC task force on the management of arterial hypertension. J Hypertens. 2007;25(9):1751.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). Jama. 2014;311(5):507–520.
- Chockalingam A. Impact of world hypertension day. Can J Cardiol. 2007;23(7):517–519.
- Carretero OA, Oparil S. Essential hypertension: part I: definition and etiology. Circulation. 2000;101(3):329–335.
- Messerli FH, Williams B, Ritz E. Essential hypertension. The Lancet. 2007;370(9587):591–603.
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–556.
- Ellison DH, Velazquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol-Renal Physiol. 1987;253(3):F546–F554.
- Wright JM, Lee C-H, Chambers GK. Systematic review of antihypertensive therapies does the evidence assist in choosing a first-line drug? Can Med Assoc J. 1999;161(1):25–32.
- Cutler JA, Davis BR. Thiazide-type diuretics and β-adrenergic blockers as first-line drug treatments for hypertension. Circulation. 2008;117(20):2691–2705.
- Valdez-Flores MA, Vargas-Poussou R, Verkaart S, et al. Functionomics of NCC mutations in Gitelman syndrome using a novel mammalian cell-based activity assay. Am J Physiol-Renal Physiol. 2016;311(6):F1159–F1167.
- Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22(4):693–703.
- Urbanova, M., Reiterova J, Stekrova J, et al. DNA analysis of renal electrolyte transporter genes among patients suffering from bartter and gitelman syndromes-summary of mutation screening. Folia Biol. 2011;57(2):65.
- Gitelman H, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235.
- Simon DB, Thompson ME, Jetton TL, et al. Gitelman’s variant of Barter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–cl cotransporter. Nat Genet. 1996;12(1):24.
- Mastroianni N, Wang Z, Udar N, et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet. 1996;59(5):1019.
- Tutakhel OA, Jeleń S, Valdez-Flores M, et al. Alternative splice variant of the thiazide-sensitive NaCl cotransporter: a novel player in renal salt handling. Am J Physiol-Renal Physiol. 2015;310(3):F204–F216.
- Mastroianni N, De Fusco M, Zollo M, et al. Molecular cloning, expression pattern, and chromosomal localization of the human Na–cl thiazide-sensitive cotransporter (SLC12A3). Genomics. 1996;35(3):486–493.
- Lu ZX, Jiang P, Xing Y. Genetic variation of pre‐mRNA alternative splicing in human populations. Wiley Interdiscip Rev: RNA. 2012;3(4):581–592.
- Gamba G, Saltzberg SN, Lombardi M, et al. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci. 1993;90(7):2749–2753.
- Hoover RS, Poch E, Monroy A, et al. N-glycosylation at two sites critically alters thiazide binding and activity of the rat thiazide-sensitive Na+: cl− cotransporter. J Am Soc Nephrol. 2003;14(2):271–282.
- de Jong JC, Willems PHGM, Mooren FJM, et al. The structural unit of the thiazide-sensitive NaCl cotransporter is a homodimer. J Biol Chem. 2003;278(27):24302–24307.
- Gamba G. The thiazide-sensitive Na+-Cl− cotransporter: molecular biology, functional properties, and regulation by WNKs. Am J Physiol-Renal Physiol. 2009;297(4):F838–F848.
- Bazúa-Valenti S, Gamba G. Revisiting the NaCl cotransporter regulation by with-no-lysine kinases. Am J Physiol-Cell Physiol. 2015;308(10):C779–C791.
- Moes AD, van der Lubbe N, Zietse R, et al. The sodium chloride cotransporter SLC12A3: new roles in sodium, potassium, and blood pressure regulation. Pflügers Archiv-Eur J Physiol. 2014;466(1):107–118.
- Gamba G. Regulation of the renal Na+-Cl− cotransporter by phosphorylation and ubiquitylation. Am J Physiol-Renal Physiol. 2012;303(12):F1573–F1583.
- Wagner SA, Beli P, Weinert BT, et al. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11(12):1578–1585.
- Huebner AR, Cheng L, Somparn P, et al. Deubiquitylation of protein cargo is not an essential step in exosome formation. Mol Cell Proteomics. 2016;15(5):1556–1571.
- Ko B, Kamsteeg E-J, Cooke LL, et al. RasGRP1 stimulation enhances ubiquitination and endocytosis of the sodium-chloride cotransporter. Am J Physiol-Renal Physiol. 2010;299(2):F300–F309.
- Rosenbaek LL, Rizzo F, Wu Q, et al. The thiazide sensitive sodium chloride co-transporter NCC is modulated by site-specific ubiquitylation. Sci Rep. 2017;7(1):12981.
- Rosenbaek LL, Kortenoeven MLA, Aroankins TS, et al. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J Biol Chem. 2014;289(19):13347–13361.
- Yang S-S, Fang Y-W, Tseng M-H, et al. Phosphorylation regulates NCC stability and transporter activity in vivo. J Am Soc Nephrol. 2013;24(10):1587–1597.
- Pacheco-Alvarez D, Cristóbal PS, Meade P, et al. The Na+: cl–cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281(39):28755–28763.
- Richardson C, Rafiqi FH, Karlsson HKR, et al. Activation of the thiazide-sensitive Na+-Cl–cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121(5):675–684.
- Rosenbaek L, Assentoft M, Pedersen NB, et al. Characterization of a novel phosphorylation site in the sodium–chloride cotransporter, NCC. J Physiol. 2012;590(23):6121–6139.
- Ferdaus MZ, Barber KW, López-Cayuqueo KI, et al. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol. 2016;594(17):4945–4966.
- Lin S-H, Shiang J-C, Huang C-C, et al. Phenotype and genotype analysis in Chinese patients with Gitelman’s syndrome. J Clin Endocrinol Metabolism. 2005;90(5):2500–2507.
- Vitari AC, Deak M, Morrice NA, et al. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391(1):17–24.
- Wilson FH, Disse-Nicodème S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112.
- Wilson FH, Kahle KT, Sabath E, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na–cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci. 2003;100(2):680–684.
- Yang C-L, Angell J, Mitchell R, et al. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111(7):1039–1045.
- Rojas-Vega L, Gamba G. Mini-review: regulation of the renal NaCl cotransporter by hormones. Am J Physiol-Renal Physiol. 2015;310(1):F10–F14.
- Gonzales PA, Pisitkun T, Hoffert JD, et al. Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am Soc Nephrol. 2009;20(2):363–379.
- Tutakhel, O.A., Bianchi F, Smits DA, et al. Dominant functional role of the novel phosphorylation site S811 in the human renal NaCl cotransporter. FASEB J. 2018;32(8):4482–4493.
- Khan MZH, Sohara E, Ohta A, et al. Phosphorylation of Na–cl cotransporter by OSR1 and SPAK kinases regulates its ubiquitination. Biochem Biophys Res Commun. 2012;425(2):456–461.
- Alessi DR, Zhang J, Khanna A, et al. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal. 2014;7(334):re3–re3.
- Neuberger G, Schneider G, Eisenhaber F. pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol Direct. 2007;2(1):1.
- Chabardes D, Gagnan-Brunette M, Imbert-Teboul M, et al. Adenylate cyclase responsiveness to hormones in various portions of the human nephron. J Clin Invest. 1980;65(2):439–448.
- Elalouf J, Roinel N, de Rouffignac C. Stimulation by human calcitonin of electrolyte transport in distal tubules of rat kidney. Pflügers Archiv. 1983;399(2):111–118.
- Gunaratne R, Braucht DWW, Rinschen MM, et al. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci. 2010;107(35):15653–15658.
- Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol-Renal Physiol. 2011;301(6):F1143–F1159.
- Castañeda-Bueno M, Arroyo JP, Zhang J, et al. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc Natl Acad Sci. 2017;114(5):E879–E886.
- Mutig K, Saritas T, Uchida S, et al. Short-term stimulation of the thiazide-sensitive Na+-Cl− cotransporter by vasopressin involves phosphorylation and membrane translocation. Am J Physiol-Renal Physiol. 2009;298(3):F502–F509.
- Pedersen NB, Hofmeister MV, Rosenbaek LL, et al. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 2010;78(2):160–169.
- Saritas, T., Borschewski A, McCormick JA, et al. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol. 2013;24(3):407–418.
- Shibata S, Arroyo JP, Castaneda-Bueno M, et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci. 2014;111(43):15556–15561.
- Yoshizaki Y, Mori Y, Tsuzaki Y, et al. Impaired degradation of WNK by Akt and PKA phosphorylation of KLHL3. Biochem Biophys Res Commun. 2015;467(2):229–234.
- Kim G-H, Masilamani S, Turner R, et al. The thiazide-sensitive Na–cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci. 1998;95(24):14552–14557.
- Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21(1):39–50.
- Terker AS, Yarbrough B, Ferdaus MZ, et al. Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol. 2016;27(8):2436–2445.
- Wolley MJ, Wu A, Xu S, et al. In primary aldosteronism, mineralocorticoids influence exosomal sodium-chloride cotransporter abundance. J Am Soc Nephrol. 2017;28(1):56–63.
- Pisitkun T, Shen R-F, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101(36):13368–13373.
- Salih M, Zietse R, Hoorn EJ. Urinary extracellular vesicles and the kidney: biomarkers and beyond. Am J Physiol-Renal Physiol. 2014;306(11):F1251–F1259.
- Isobe K, Mori T, Asano T, et al. Development of enzyme-linked immunosorbent assays for urinary thiazide-sensitive Na-Cl cotransporter measurement. Am J Physiol-Renal Physiol. 2013;305(9):F1374–F1381.
- van der Lubbe N, Jansen PM, Salih M, et al. The phosphorylated sodium chloride cotransporter in urinary exosomes is superior to prostasin as a marker for aldosteronismnovelty and significance. Hypertension. 2012;60(3):741–748.
- Corbetta S, Raimondo F, Tedeschi S, et al. Urinary exosomes in the diagnosis of Gitelman and Bartter syndromes. Nephrol Dial Transplant. 2014;30(4):621–630.
- Tutakhel OA, Moes AD, Valdez-Flores MA, et al. NaCl cotransporter abundance in urinary vesicles is increased by calcineurin inhibitors and predicts thiazide sensitivity. PLoS One. 2017;12(4):e0176220.
- Hoorn EJ, Walsh SB, McCormick JA, et al. Pathogenesis of calcineurin inhibitor–induced hypertension. J Nephrol. 2012;25(3):269.
- Na KY, Oh YK, Han JS, et al. Upregulation of Na+ transporter abundances in response to chronic thiazide or loop diuretic treatment in rats. Am J Physiol-Renal Physiol. 2003;284(1):F133–F143.
- Nijenhuis T, Vallon V, van der Kemp AWCM, et al. Enhanced passive Ca 2+ reabsorption and reduced Mg 2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115(6):1651–1658.
- Arroyo JP, Ronzaud C, Lagnaz D, et al. Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology. 2011;26(2):115–123.
- Pathare G, Tutakhel OAZ, van der Wel MC, et al. Hydrochlorothiazide treatment increases the abundance of the NaCl cotransporter in urinary extracellular vesicles of essential hypertensive patients. Am J Physiol-Renal Physiol. 2017;312(6):F1063–F1072.
- Schweda F. Salt feedback on the renin-angiotensin-aldosterone system. Pflügers Archiv-Eur J Physiol. 2015;467(3):565–576.
- Fenton RA, Moeller HB. Recent discoveries in vasopressin-regulated aquaporin-2 trafficking. Prog Brain Res. 2008;170:571–579.
- Salih M, Fenton RA, Knipscheer J, et al. An immunoassay for urinary extracellular vesicles. Am J Physiol-Renal Physiol. 2016;310(8):F796–F801.
- Hughes AD. How do thiazide and thiazide-like diuretics lower blood pressure? J Renin-Angiotensin-Aldosterone Syst. 2004;5(4):155–160.
- Wood JM, Schnell CR, Levens NR. Kidney is an important target for the antihypertensive action of an angiotensin II receptor antagonist in spontaneously hypertensive rats. Hypertension. 1993;21(6 Pt 2):1056–1061.