
ABSTRACT
Myotonia congenita (MC) is a rare genetic disease caused by mutations in the skeletal muscle chloride channel gene (CLCN1), encoding the voltage-gated chloride channel ClC-1 in skeletal muscle. Our study reported the clinical and molecular characteristics of six patients with MC and systematically review the literature on Chinese people. We retrospectively analyzed demographics, clinical features, family history, creatine kinase (CK), electromyography (EMG), treatment, and genotype data of our patients and reviewed the clinical data and CLCN1 mutations in literature. The median ages at examination and onset were 26.5 years (range 11–50 years) and 6.5 years (range 1.5–11 years), respectively, in our patients, and 21 years (range 3.5–65 years, n = 45) and 9 years (range 0.5–26 years, n = 50), respectively, in literature. Similar to previous reports, myotonia involved limb, lids, masticatory, and trunk muscles to varying degrees. Warm-up phenomenon (5/6), percussion myotonia (3/5), and grip myotonia (6/6) were common. Menstruation triggered myotonia in females, not observed in Chinese patients before. The proportion of abnormal CK levels (4/5) was higher than data from literature. Electromyography performed in six patients revealed myotonic changes (100%). Five novel CLCN1 mutations, including a splicing mutation (c.853 + 4A>G), a deletion mutation (c.2010_2014del), and three missense mutations (c.2527C>T, c.1727C>T, c.2017 G > C), were identified. The c.892 G > A (p.A298T) mutation was the most frequent mutation in the Chinese population. Our study expanded the clinical and genetic spectrum of patients with MC in the China. The MC phenotype in Chinese people is not different from that found in the West, while the genotype is different.
Introduction
Myotonia congenita (MC) is a genetic disease caused by mutations in CLCN1 on chromosome 7q35 (OMIM # 118,425), coding for the main voltage-dependent chloride channel CLC-1 in skeletal muscle cells [Citation1]. The most characteristic symptom is impaired muscle relaxation after forceful contraction (myotonia). Stiffness worsens after rest and improves with repeated activity (warm-up phenomenon). The most common stiffness sites are the legs, while the face is less commonly affected [Citation2].
The disease can be distinguished into the autosomal dominant-type Thomsen (TMC) (OMIM #160,800) and the autosomal recessive-type Becker (BMC) (OMIM #255,700) by genetic findings and inheritance patterns [Citation3]. There were 348 pathogenic mutations throughout the CLCN1 gene identified in the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php) to date. TMC is usually caused by a heterozygous mutation on one allele, while compound heterozygous mutations inherited from each parent are common in BMC patients. Some mutations may lead to autosomal dominant MC in some families and a recessive form in others [Citation4,Citation5]. Certain clinical features may help differentiate TMC from BMC. TMC may be associated with milder characteristics and age of onset tends to be earlier in this type [Citation5,Citation6]. However, the phenotype can differ significantly between siblings or children and parents [Citation7]. The genotypic and phenotypic heterogeneity of the disease creates challenges in distinguishing between dominant and recessive types distinctly, especially in sporadic cases or small kindreds. A classic Mendelian pattern does not always elucidate myotonia congenita; terms, such as incomplete dominance with variable penetrance or expressivity, better describe the modes of transmission in some families [Citation8].
With the development of gene sequencing, especially the next-generation sequencing (NGS) technology, increasing cases of genetically confirmed MC have been reported in China since 2010. All studies in the Chinese population have been case reports or case series to date. And a recent literature review found that there was no significant difference in main clinical presentation between the two types of MC, however other aspects of the diseases, such as triggers, CK levels and treatment, were not fully discussed [Citation9]. We analyzed the data on genetically confirmed MC diagnosed in our center in detail, and compared our results with the systematic literature review on the clinical features of all patients with MC, including probands and their affected family members, regardless of inheritance pattern. To describe the mutation spectrum of CLCN1 in the Chinese population more accurately, the same mutation with different expressions were identified.
Methods
Patients
Fourteen patients referred to the Neurology Clinic of the Chinese PLA General Hospital were diagnosed with nondystrophic myotonias (NDMs) by clinical and electrophysiological features from October 11th, 2010, to October 21st, 2020. Of these, six had a genetically confirmed diagnosis of MC, and one was diagnosed with paramyotonia congenita. We obtained a complete medical history of the patients; an experienced neurologist performed physical and neurological examinations. Each patient with MC received a telephone follow-up on September 2021. The study was approved by the Ethics Committee of the Chinese PLA General Hospital. All patients provided written informed consent.
We performed a detailed search of all relevant English language literature published between 1960 and 2021 via PubMed (http://www.ncbi.nlm.nih.gov/pubmed), using “myotonia congenita” and “Chinese” as keywords. Chinese literature was also searched via CNKI (www.cnki.net), using “myotonia congenita” as keywords. The diagnosis of all patients was supported by molecular genetic testing. Clinical data and mutations were recorded by investigating original articles. Four MC patients comorbid with other genetic diseases (one patient with hypokalemic periodic paralysis type 1, one patient with paroxysmal kinesigenic dyskinesia, one patient with Hoffmann syndrome) were excluded from clinical analysis. We included every pedigree with CLCN1 mutations to obtain the complete picture of the mutation spectrum. We enrolled 58 MC patients and 47 pedigrees in the literature review ().
Table 1. Mutational spectrum of the CLCN1 gene in the 47 pedigrees reported in Chinese population
Clinical data collection
The following data were collected and analyzed: demographics, age of onset, distribution of myotonia, other concomitant symptoms (weakness, muscle hypertrophy, myalgia, cramps), myotonia characteristics (warm-up, grip myotonia, percussion myotonia), triggers (cold, stress, exercise, menstruation), elevated CK, family history, anti-myotonic medications, and efficacy.
Exon sequencing for ion channelopathy genes
For screening potential pathogenic genes for MC, we performed gene panel sequencing for ion channelopathy containing 12 genes (CACNA1A, CACNA1S, CAV3, CLCN1, KCNA1, KCNE3, KCNE4, KCNH2, KCNJ16, KCNJ2, KCNQ1, SCN4A) (GenCap Enrichment technologies, MyGenostics Inc, Beijing, China), using HiSeq 2000 high-throughput sequencing system (Illumina Inc, USA). The DMPK gene was analyzed in the patient with myopathic changes on EMG.
Bioinformatics analysis
After filtering out the low-quality reads and adaptor sequences using the Solexa QA package and the Cutadapt program (http://code.google.com/p/cutadapt/), respectively, we used the SOAPaligner program to align the clean read sequences to the human reference genome (hg19). After removing the PCR duplicates by the Picard software, we first identified the SNPs using the SOAPsnp program (http://soap.genomics.org.cn/soapsnp.html). Subsequently, we realigned the reads to the reference genome using BWA and identified the insertions or deletions (InDels) using the GATK program (http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page). The identified SNPs and InDels were annotated using the Exome-assistant program (http://122.228.158.106/exomeassistant). MagicViewer was used to view the short read alignment and validate the candidate SNPs and InDels. Nonsynonymous variants were evaluated by four algorithms, PolyPhen2, SIFT, Mutation Taster, and REVEL (as described previously), to determine pathogenicity. Mutations were searched in the 1000 Genomes Project database (www.1000genomes.org), Exome Aggregation Consortium (ExAC) database (www.exac.broadinstitute.org), Genome Aggregation Database (gnomAD), and dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) followed by the Human Genome Mutation Database (http://www.biobase-international.com/product/hgmd HGMD) and the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) to determine whether the variant was a reported pathogenic mutation.
Sanger sequencing validation
PCR amplification was optimized, following the standard PCR protocol using FastStart Taq DNA Polymerase, dNTPack (Roche Applied Science). A sequencing reaction was performed using the BigDye® v.1.1 Terminator cycle sequencing kit and the ABI Prism® 3130xl Genetic Analyzer (Life Technologies).
Statistical analysis
Statistical analysis was performed with IBM SPSS Statistics 26.0. Descriptive statistics of demographic characteristics were made using medians and range for continuous variables and frequency analysis for categorical variables.
Results
Clinical features
The clinical diagnosis of MC was established in all six patients according to the Diagnostic Criteria for Neuromuscular Disorders [Citation24]. The demographics and clinical features are summarized in .
Table 2. Demographics and clinical features of this cohort of patients
There were four males and two females, with median ages 26.5 years (range 11–50 years) at examination and 6.5 years (range 1.5–11 years) at the onset. No family history was reported, except in patient 3. We observed similar symptoms in one of his elder sisters but not in other family members (). Stiffness in the legs was more prominent in four patients at the early stage, involving all four limbs eventually. Lids, masticatory muscles, and trunk muscles were also affected (2/5, 3/4, 1/6, respectively). No patient experienced muscle weakness. Patient 2 had slight muscle hypertrophy in the buttocks and thigh; patient 6, a fitness coach, had a muscular appearance. Only one patient reported myalgia, muscle cramps, and dysphagia. No patient reported dysphonia. Myotonia improved with exercise (warm-up phenomenon) in five of the six patients. Grip myotonia was detectable in all patients and percussion myotonia in half of the patients. Myotonia was most commonly exacerbated by cold and stress and became apparent during menstruation in females. No patient had exercise-induced myotonia. The CK levels were 1.4–2.9 folds (range 275.2–579.6 U/L) higher than the upper limit of the normal reference level in four patients. We prescribed phenytoin to four patients, but only two patients took the medication; one patient showed a response to the treatment. Two patients (patients 5 and 6) had mild symptoms and required no pharmacological intervention. Patient 3 could minimize his symptoms by avoiding situations and stimuli that triggered myotonic episodes. Needle EMG revealed myotonic discharges in all patients, with additional myopathic changes in patient 3, whose muscle biopsy (left biceps brachii) showed scattered muscle fiber atrophy. No expansion of CTG repeats was found in the DMPK gene. At follow-up, the symptoms were stable in patients 1 to 3 and relieved without treatment in patients 5–6. Patient 1 discontinued the medication due to low efficacy. Patient 2 used the medication intermittently on aggravation of symptoms during winter.
Figure 1. The pedigree and genetic analysis of the six patients with myotonia congenita. a. Patient 1, CLCN1 c.871 G > A (p.E291K) and c.1262insC (p.R421PfsX9) mutation. b. Patient 2, CLCN1 c.1262insC (p.R421PfsX9) mutation. c. Patient 3, CLCN1 c.853 + 4A>G (splicing) and c.2527C>T (p.L843F) mutation. d. Patient 4, CLCN1 c.1657A>T (p.I553F) and c.2010_2014del (p.L671Rfs*41) mutation. e. Patient 5, CLCN1 c.1727C>T (p.S576F) mutation. f. Patient 6, CLCN1 c.937 G > A (p.A313T) and c.2017 G > C (p.A673P) mutation.
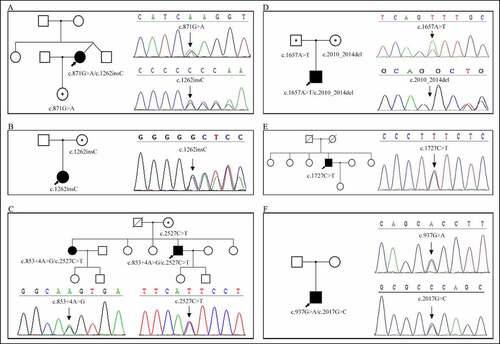
CLCN1 mutations
Gene panel testing showed that the six patients carried 10 mutations in the CLCN1 gene; of these, five mutations were novel and not reported elsewhere (). According to the mutated genes, three cases with compound heterozygous mutations were in the AR inheritance pattern. The inheritance patterns in other cases were uncertain. Patient 1 harbored two compound heterozygous mutations, c.871 G > A (p.E291K) and c.1262insC (p.R421PfsX9). The mutation c.1262insC (p.R421PfsX9) was also detected in patient 2, but the heterozygous state. Patient 3 had two novel heterozygous mutations in the CLCN1 gene, a splicing mutation (c.853 + 4A>G) and a missense mutation (c.2527C>T), leading to abnormal splicing and an L843F amino acid change, respectively. Molecular analysis of patient 4 revealed a novel compound heterozygous c.2010_2014del (p.L671Rfs*41) mutation in exon 17 and a previously noted c.1657A>T (p.I553F) mutation in exon 14 [Citation25]. The new mutation was a 5 base pair deletion of nucleotides, 2010 to 2014, leading to a frame-shifted sequence beginning with codon 670 and ending with a stop at codon 711. We detected a novel heterozygous transition in nucleotide position 1727 (codon 198) of C to T, resulting in an amino acid change of serine to phenylalanine in patient 5. We identified two heterozygous variants, c.937 G > A (p.A313T) in exon 8 and c.2017 G > C (p.A673P) in exon 17 in patient 6.
Table 3. Mutations associated with nondystrophic myotonia identified in the study
Pathogenicity analysis
The pathogenicity of the five novel mutations was predicted using Mutation Taster, PolyPhen-2, SIFT, and REVEL. The results are listed in . According to the bioinformatics analysis results and the American College of Medical Genetics and Genomics (ACMG) guidance for the interpretation of sequence variants, we considered these five mutations as potential pathogenic variants.
Table 4. Bioinformatics analysis of the novel mutations of CLCN1 gene
Pedigree study
Sanger sequencing was performed in the probands and family members of four patients (patients 1, 2, 3, 4) to determine whether their relatives carried the same mutation as the proband. The daughter of patient 1, whose neurological examination and EMG findings were normal, carried a heterozygous mutation c.871 G > A, but not the c.1262insC (p.R421PfsX9) mutation (). The same mutation (c.1262insC) was also detected in the mother of patient 2 (). However, this truncating mutation showed no clinical manifestation in his mother. The healthy mother of patient 3 had the same missense mutation (c.2527C>T) but a normal sequence at the other position. His affected sister had the same heterozygous mutations (c.853 + 4A>G, c.2527C>T) as the proband (). Both parents of patient 4 were heterozygous carriers of the mutations and remained unaffected clinically. His mother carried the c.2010_2014del mutation and his father c.1657A>T the mutation (). Unfortunately, other family members of patients 5 and 6 were unavailable for genetic testing ().
Clinical Findings of the literature
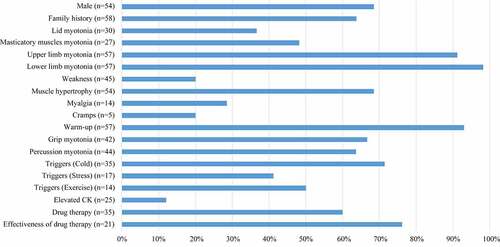
In literature, the median ages at examination and onset were 21 years (range 3.5–65 years, n = 45) and 9 years (range 0.5–26 years, n = 50), respectively. According to the clinical phenotype and genotype, 21 pedigrees (52.5%) were TMC, and 16 (40%) were BMC. It was difficult to determine the inheritance pattern of the three sporadic cases with only one heterozygous mutation in CLCN1. As shown in , 68.5% (37/54) of the patients were male, and 63.8% (37/58) had a family history. The most frequent symptoms were myotonia of the upper and lower limbs (92.3% vs. 98.1%), followed by the warm-up phenomenon. Myotonia at onset most frequently involved lower limbs (76.5%). Other symptoms, including muscle hypertrophy, grip myotonia, and percussion myotonia, were observed in over half of the patients. Cold temperature environment was the most common trigger for myotonia in the reported patients (80%). Other uncommon triggers involved stress and exercise. Elevated CK was reported only in 10% (2/20) of Chinese patients. EMG detected myotonic runs in 96.3% (52/54) of MC patients; myopathic discharges occurred in 13% (7/54) of these patients. Anti-myotonic drugs were effective in 76.2% (16/21) of patients. Patients on mexiletine or carbamazepine (15/30) reported relief of symptoms. However, patients treated with phenytoin (5/30) showed poor response to medication. Only one patient received propafenone, which was effective.
Figure 2. Clinical features of the 58 Chinese patients in literature. (The number in the brackets were the number of patients available for analysis).
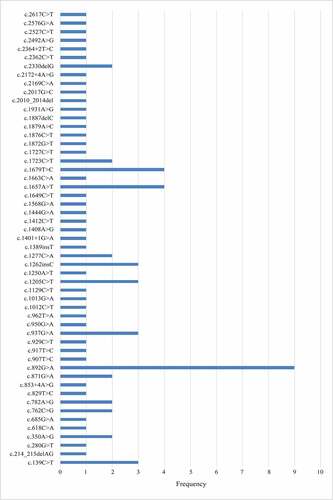
Mutation spectrum of the Chinese population
To date, there are 53 Chinese pedigrees reported, including our study. Fifty-three different variants have been found scattered across the 23 exons of the CLCN1 gene. We identified a region of high mutation frequency that includes exons 8 and 15 of the CLCN1 gene: 34 (41.5%) unrelated probands harbored mutations in this region; other exons with a high rate of mutations, although to a less frequency, included exons 11, 12, and 17. Of the known mutations, three missense mutations were more frequent: c.892 G > A (p.A298T) (n = 9) in exon 8, 1657A>T (p.I553F) (n = 4), and c.1679 T > C (p.M560T) (n = 4) in exon 15 ().
Figure 3. Mutational spectrum of the 53 Chinese pedigrees including our study.
Discussion
Our study illustrated the clinical characteristics of genetically diagnosed Chinese patients with MC from muscular features, inheritance patterns, and auxiliary examination to treatment. Besides, we identified five novel mutations: c.853 + 4A>G (splicing change), c.2010_2014del (p.L671Rfs*41), c.2527C>T (p.L843F), c.2017 G > C (p.A673P) in the compound heterozygous state, and c.1727C>T (p.S576F) in the heterozygous state, which expanded the genetic spectrum of patients with MC in the Chinese population.
Myotonia congenita is a rare inherited disease. Chinese patients with MC with proven mutations in the CLCN1 gene were first reported in 2004 by Taiwan scholars [Citation26]. The diagnosis of NDMs was solely based on clinical and electrophysiological findings until next-generation sequencing was used in the clinical setting in our center in 2014. To our knowledge, there was no large cohort of Chinese MC patients have been described. We reprocessed and analyzed the clinical data on the previously reported 58 MC cases as a cohort, and compared it with our clinical findings in family history, muscular features, myotonia triggers, CK levels, and drug therapy. To depict the mutation spectrum of CLCN1 in the Chinese population, we analyzed pedigrees instead of patients. We checked every mutation carefully to identify the same mutation with different expressions because different transcripts were used. Mutations c.1657A>T (p.I553F), c.1663C>A(p.H555N) and c.937 G > A (p.A313T) were also expressed as c.1744A>T, c.1750C>A [Citation25], and c.1024 G > A [Citation27], respectively. In this case, we renamed the nucleotide position by the commonly used transcript.
Clinical manifestations in our patients were similar to previous reports. Males were slightly more affected than females. We found that myotonia worsened during menstruation in female patients, implying that sex hormones could alter the function of CLC-1 [Citation28]. Menstruation is not reported as a factor worsening symptoms in Chinese people. CK levels increased in about 10% of Chinese patients, while most of our patients (4/5) had abnormal CK. Our data indicated that the elevation of CK levels was common in MC, as reported in a large German cohort [Citation29]. EMG detected myotonic discharges in almost all patients with MC in the literature and all patients in our study, implying that EMG was the most valuable diagnostic tool for identifying patients with MC and indicating the need for genetic testing. Only two patients had no myotonia discharges on EMG at the examination. One patient with normal EMG at 44 years experienced myotonia at the age of 18. The symptoms showed almost complete resolution by 40 years without treatment [Citation25]. The other patient who carried S471F mutations only showed a positive wave and increased recruitment in the EMG [Citation26]. Myopathic motor unit action potentials (MUAPs) occurred in about 13% of patients in previous reports and 17% (1/6) of patients in our study. Noteworthy that all patients with myopathy changes were BMC, which was in agreement with some published reviews of other populations [Citation30]. However, a more systematic quantitative analysis of MUAPs in genetically proven MC found that myopathic MUAPs occurred in more than 20% of muscles in the entire MC group. The incidence of myopathic MUAPs was 21.1% in the BMC subgroup and 30.4% in the TMC subgroup, and they occurred most often in distal limb muscles in BMC and upper limb muscles in the Thomsen MC group [Citation31]. Our patient exhibited low amplitude and short duration MUAPs with interference patterns in proximal and distal muscles of four limbs. The incidence of myopathic MUAPs in Chinese patients with MC, especially in TMC subgroups, was unknown.
From our literature review, 60% of Chinese patients received anti-myotonic drugs. All patients treated with anti-myotonic drugs received monotherapy. Phenytoin and gabapentin were the most effective therapies in recessive CLCN1-myotonia [Citation32]. However, recent studies showed that mexiletine and lamotrigine were more effective [Citation33–35], consistent with our findings. However, randomized controlled trials in the Chinese population are challenging due to the rarity and genetic heterogeneity of the disorder. A large prospective cohort study in Chinese patients is needed to obtain accurate data.
According to the clinical phenotype and genotype, MC pedigree with dominant inheritance accounts for about 50% in the Chinese literature, which is higher than the percentage of Western countries (UK 36%, South-Italian 36.8%, French-Canadian 25%) [Citation32,Citation36,Citation37] but lower than another report from Japan (67%) [Citation38]. A comprehensive prevalence study is needed to determine the prevalence of dominant versus recessive in Asia. In our study, patients 1, 3, and 4 who carried compound heterozygous mutations were BMC. Of the six mutations identified in three BMC patients, c.853 + 4A>G, c.2010_2014del, and c.2527C>T mutations were first reported. The mutation c.1657A>T (p.I553F) is reported in the literature as inherited in a recessive mode [Citation25,Citation39]. One previously reported mutation, c.871 G > A (p.E291K), was recessive in one family [Citation40] but dominant in the other [Citation4]. The c.1262insC (p.R421PfsX9) variant is also reported in the literature as inherited in a recessive [Citation40] or dominant mode [Citation41]. Family history was negative for patients 2 and 5 who carried only one mutated allele; therefore, it was difficult to determine their inheritance pattern. The heterozygous c.1262insC mutation presented in patient 2 and her apparently normal mother who haven’t received EMG test, indicating that this mutation could be recessive or dominant with incomplete penetrance, as suggested previously for G230E [Citation40,Citation42], A313T [Citation8], R317Q [Citation40,Citation42], R338Q [Citation43], and R894X [Citation40,Citation43,Citation44]. Patient 5 had a novel heterozygous mutation S576F, but his parents and children showed no myotonia. One cannot exclude the presence of a second (yet undetected) mutation, and therefore, firm conclusions regarding the inheritance of mutations require functional experiments and the study of other family members carrying the same mutation. Patient symptoms showed complete resolution without treatment, which supported the assumption that this mutation had a dominant-negative effect. This assumption needs confirmation by accumulating more pedigrees and further pathophysiological study. Patient 6 carried two compound heterozygous mutations. As mentioned above, mutation A313T is reported as autosomal recessive or dominant with incomplete penetrance. The other mutation, A673P, is reported in the ExAc database (frequency 0.0016) and the dbSNP database as rs200385034. However, there are no reports on its pathogenicity. PolyPhen-2 software predicted the effects of the A673P mutation as “benign,” SIFT software predicted its effects as “tolerated,” and Mutation Taster indicated “polymorphism.” According to the ACMG standards, this variant could be classified as a variant of uncertain significance. The A673P may be a single nucleotide polymorphism not associated with MC, and therefore, the A313T mutation may be an autosomal-dominant mutation in this patient. Alternatively, the A673P mutation had a recessive effect, and the A313T mutation acted as an autosomal-recessive mutation. The patient was the only affected family member and expressed a mild phenotype, which gradually disappeared with age; his healthy parents refused to undergo genetic evaluation.
Of the known mutations, the most frequent mutation is c.892 G > A (p.A298T) in exon 8 (11%), followed by the c.1657A>T (p.I553F) (4.9%) and c.1679 T > C (p.M560T) (4.9%) in exon 15. A298T was associated with both autosomal dominant and recessive inheritance. This mutation was not frequently reported in other European [Citation45–47] or American [Citation48] cohorts. However, it was most common in Japanese cohorts [Citation38]. Further haplotype studies on these families are necessary to determine whether the founder effect plays a role in the higher frequency of the c.892 G > A mutation.
In addition to the five novel mutations of CLCN1 identified, our study elaborated the clinical manifestations, drug therapy, and molecular characteristics of Chinese patients with MC. However, no systematic research has been conducted in China. Due to the rarity of the disease, multi-center collaboration is an effective strategy to conduct large-scale cohort studies on phenotype–genotype relationships and randomized controlled trials on treatment.
Availability of data
The data that support the findings of this study are openly available in Dryad at https://doi.org/10.5061/dryad.0cfxpnw42.
Acknowledgments
We thank all patients and their family members for cooperation in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Altamura C, Lucchiari S, Sahbani D, et al. The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel. Hum Mutat. 2018;39(9):1273–1283.
- Trivedi JR, Bundy B, Statland J, et al. Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain. 2013;136(Pt 7):2189–2200.
- Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257(5071):797–800.
- Yang X, Jia H, An R, et al. Sequence CLCN1 and SCN4A in patients with Nondystrophic myotonias in Chinese populations: genetic and pedigree analysis of 10 families and review of the literature. Channels (Austin). 2017;11(1):55–65.
- Lossin C, George AL Jr. Myotonia congenita. Adv Genet. 2008;63:25–55.
- Cherian A, Baheti NN, Kuruvilla A. Muscle channelopathies and electrophysiological approach. Ann Indian Acad Neurol. 2008;11(1):20–27.
- Colding-Jorgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve. 2005;32(1):19–34.
- Plassart-Schiess E, Gervais A, Eymard B, et al. Novel muscle chloride channel (CLCN1) mutations in myotonia congenita with various modes of inheritance including incomplete dominance and penetrance. Neurology. 1998;50(4):1176–1179.
- Hu C, Shi Y, Zhao L, et al. Myotonia congenita: clinical characteristic and mutation spectrum of CLCN1 in Chinese patients. Front Pediatr. 2021;9:759505.
- Kuo HC, Hsiao KM, Chang LI, et al. Novel mutations at carboxyl terminus of CIC-1 channel in myotonia congenita. Acta Neurol Scand. 2006;113(5):342–346.
- Gao F, Ma FC, Yuan ZF, et al. Novel chloride channel gene mutations in two unrelated Chinese families with myotonia congenita. Neurol India. 2010;58(5):743–746.
- Ma FC, Gao F, and Yuan ZF. Clinical analysis and gene mutation screening of CLCN1 in two myotonia congenita families. J Clin Pediatr. 2011;29(11):1041–1043. Chinese.
- Kong LN, Wu QY, and Shen YS, et al. Study of CLCN1 gene mutations in a family with myotonia congenital. J Clin Neurol. 2012;25(6):407–409. Chinese.
- Li HF, Chen WJ, Ni W, et al. Paroxysmal kinesigenic dyskinesia and myotonia congenita in the same family: coexistence of a PRRT2 mutation and two CLCN1 mutations. Neurosci Bull. 2014;30(6):1010–1016.
- Liu XL, Huang XJ, Shen JY, et al. Myotonia congenita: novel mutations in CLCN1 gene. Channels (Austin). 2015;9(5):292–298.
- Gu P, Sun ZQ, Wang WT, et al. A case of myotonia congenita. J Chin J Nerv Ment Dis. 2017;43(6):377–378. Chinese.
- Jing F, Li H, Yang D, et al. Analysis of CLCN1 gene mutations in a family affected with myotonia congenita. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35(3):400–402. Chinese.
- Zhang W, Meng HX, Wang XM, et al. Identification of two novel compound heterozygous CLCN1 mutations associated with autosomal recessive myotonia congenita. Neurol Res. 2019;41(12):1069–1074.
- Yang HJ, Zhao HJ, Yang XL. Clinical, electrophysiological, and genetic studies of one family and one sporadic patient with congenital myotonia. J Int Neurol Neurosurg. 2019;46(4):364–367. Chinese.
- Zhao CY, Tang DF, and Huang H, et al. Myotonia congenita and periodic hypokalemia paralysis in a consanguineous marriage pedigree: coexistence of a novel CLCN1 mutation and an SCN4A mutation. PLoS One. 2020;15(5):e0233017.
- Cao XL, Qin QY, Huang W. Clinical and genetic analysis for a family with myotonia congenita of CLCN1 gene mutations. Chin J Pract Nerv Dis. 2020;23(23):2112–2116. Chinese.
- Su MX, Da YW, Yang BF, et al. A case report of young female with lowerlimnbs stiffness for 20 years, fatigue and weakness for 1 month-Myotonia congenita and Hoffmann syndrome. Chin J Nerv Ment Dis. 2020;46(12):759–762R. Chinese.
- Song J, Zhang JW, Li G, et al. Clinical and genetic features of three Chinese patients with myotonia congenita. J Apoplexy Nervous Dis. 2021;38(3):249–252. Chinese.
- Lehmann-Horn F, Rudel R, Ricker K. Non-dystrophic myotonias and periodic paralyses. A European neuromuscular center workshop held 4-6 October 1992, Ulm, Germany. Neuromuscul Disord. 1993;3(2):161–168.
- Burgunder JM, Huifang S, Beguin P, et al. Novel chloride channel mutations leading to mild myotonia among Chinese. Neuromuscul Disord. 2008;18(8):633–640.
- Jou SB, Chang LI, Pan H, et al. Novel CLCN1 mutations in Taiwanese patients with myotonia congenita. J Neurol. 2004;251(6):666–670.
- Chen ZT, He J, and Chen WJ, et al. Analysis of CLCN1 gene mutations in 2 patients with myotonia congenita. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2012;29(6):690–692. Chinese.
- Fialho D, Kullmann DM, Hanna MG, et al. Non-genomic effects of sex hormones on CLC-1 may contribute to gender differences in myotonia congenita. Neuromuscul Disord. 2008;18(11):869–872.
- Vereb N, Montagnese F, Glaser D, et al. Non-dystrophic myotonias: clinical and mutation spectrum of 70 German patients. J Neurol. 2021;268(5):1708–1720.
- Heatwole CR, Statland JM, Logigian EL. The diagnosis and treatment of myotonic disorders. Muscle Nerve. 2013;47(5):632–648.
- Nojszewska M, Lusakowska A, Gawel M, et al. The needle EMG findings in myotonia congenita. J Electromyogr Kinesiol. 2019;49:102362.
- Dupre N, Chrestian N, Bouchard JP, et al. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord. 2009;19(5):330–334.
- Stunnenberg BC, Raaphorst J, Groenewoud HM, et al. Effect of mexiletine on muscle stiffness in patients with nondystrophic myotonia evaluated using aggregated N-of-1 trials. JAMA. 2018;320(22):2344–2353.
- Andersen G, Hedermann G, Witting N, Andersen G, Hedermann G, Witting N, et al. The antimyotonic effect of lamotrigine in non-dystrophic myotonias: a double-blind randomized study. Brain. 2017;140(9):2295–2305.
- Modoni A, D’Amico A, Primiano G, et al. Long-term safety and usefulness of mexiletine in a large cohort of patients affected by non-dystrophic myotonias. Front Neurol. 2020;11:300.
- Orsini C, Petillo R, D’Ambrosio P, et al. CLCN1 molecular characterization in 19 South-Italian patients with dominant and recessive type of myotonia congenita. Front Neurol. 2020;11:63.
- Fialho D, Schorge S, Pucovska U, et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007;130(Pt 12):3265–3274.
- Sasaki R, Nakaza M, Furuta M, et al. Mutation spectrum and health status in skeletal muscle channelopathies in Japan. Neuromuscul Disord. 2020;30(7):546–553.
- Miao J, Wei XJ, Liu XM, et al. A case report: autosomal recessive Myotonia congenita caused by a novel splice mutation (c.1401+1G>A) in CLCN1 gene of a Chinese Han patient. BMC Neurol. 2018;18(1):154.
- Meyer-Kleine C, Steinmeyer K, Ricker K, et al. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet. 1995;57(6):1325–1334.
- Meng YX, Zhao Z, Shen HR, et al. Identification of novel mutations of the CLCN1 gene for myotonia congenital in China. Neurol Res. 2016;38(1):40–44.
- Esteban J, Neumeyer AM, McKenna-Yasek D, et al. Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Becker’s generalized myotonia. Neurogenetics. 1998;1(3):185–188.
- Zhang J, George AL Jr., Griggs RC, et al. Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology. 1996;47(4):993–998.
- Duno M, Colding-Jorgensen E, Grunnet M, et al. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet. 2004;12(9):738–743.
- Milla CP, De Castro CP, Gomez-Gonzalez C, et al. Myotonia congenita: mutation spectrum of CLCN1 in Spanish patients. J Genet. 2019;98:98.
- Brugnoni R, Kapetis D, Imbrici P, et al. A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet. 2013;58(9):581–587.
- Skalova D, Zidkova J, Vohanka S, et al. CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS One. 2013;8(12):e82549.
- Koty PP, Pegoraro E, Hobson G, et al. Myotonia and the muscle chloride channel: dominant mutations show variable penetrance and founder effect. Neurology. 1996;47(4):963–968.