
ABSTRACT
CACNA1A-associated epilepsy and ataxia frequently accompany cognitive impairments as devastating co-morbidities. However, it is unclear whether the cognitive deficits are consequences secondary to the neurological symptoms elicited by CACNA1A mutations. To address this issue, Cacna1a mutant mice tottering (tg), and in particular tg/+ heterozygotes, serve as a suitable model system, given that tg/+ heterozygotes fail to display spontaneous absence epilepsy and ataxia typically observed in tg/tg homozygotes. Here, we examined hippocampus-dependent behaviors and hippocampal learning-related synaptic plasticity in tg mice. In behavioral analyses of tg/+ and tg/tg, acquisition and retention of spatial reference memory were characteristically impaired in the Morris water maze task, while working memory was intact in the eight-arm radial maze and T-maze tasks. tg/+ heterozygotes showed normal motor function in contrast to tg/tg homozygotes. In electrophysiological analyses, Schaffer collateral–CA1 synapses showed a deficit in the maintenance of long-term potentiation in tg/+ and tg/tg mice and an increased paired-pulse facilitation induced by paired pulses with 100 ms in tg/tg mice. Our results indicate that the tg mutation causes a dominant disorder of the hippocampus-related memory and synaptic plasticity, raising the possibility that in CACNA1A-associated human diseases, functionally aberrant CaV2.1 Ca2+ channels actively induce the observed cognitive deficits independently of the neurological symptoms.
Introduction
CACNA1A encodes the P/Q-type voltage-gated Ca2+ channel α1 subunit CaV2.1, which is responsible for the Ca2+ entry that triggers neurotransmitter release and synaptic plasticity [Citation1,Citation2]. Human CACNA1A mutations are associated with various neurological diseases, such as absence epilepsy [Citation3,Citation4], episodic ataxia type 2 [Citation5], familial hemiplegic migraine [Citation5], and spinocerebellar ataxia type 6 [Citation6]. These CACNA1A-associated disorders are frequently accompanied with cognitive impairments [Citation7–10], which are particularly recognized as devastating co-morbidities of epilepsy and other neuronal disorders [Citation11]. In some patients, this co-morbidity may be of greater consequence than the neuronal disorders themselves because cognitive impairments are linked to social and educational problems and diminish quality of life [Citation12,Citation13]. Recently, the basic mechanisms of cognitive impairment in epilepsy have come under increased scrutiny [Citation14,Citation15]. However, an understanding of the mechanisms underlying the cognitive co-morbidities in CACNA1A-associated disorders is yet to be attained.
Different mouse mutant alleles, such as tottering (tg), have been reported for the Cacna1a gene [Citation16–19]. tg was the first described Cacna1a gene mutation and provides a well-established mouse model of spontaneous absence epilepsy [Citation16,Citation20]. Previously, we revealed that the tg mutation causes loss of function of the P/Q-type Ca2+ channel [Citation21]. Consistently, human studies have shown that loss of function mutations in CACNA1A lead to absence epilepsy [Citation3,Citation4,Citation10], a generalized epilepsy frequently associated with cognitive impairments [Citation12]. We also found that tg mice showed developmental abnormalities in Cl− transporter expression and GABAA receptor compositions in hippocampal neurons and that compromised maturation of GABAergic inhibition contributes to the abnormal synchrony in the hippocampus, presumably resulting in cognitive impairment [Citation15]. Thus, tg mice are a useful animal model for studying the cognitive co-morbidities of CACNA1A-associated disorders, and epilepsy in particular.
Previous studies using functional brain imaging in humans and neuropsychological analyses in humans and animals with hippocampal damage have suggested elemental cognitive processes mediated by hippocampal networks [Citation22]. Also, hippocampal synaptic plasticity, which exists in both short- and long-term forms, is generally believed to contribute toward learning and memory storage [Citation23]. Therefore, it is plausible to hypothesize that hippocampal dysfunction may play important roles in the cognitive co-morbidities of CACNA1A-associated disorders. Prompted by this idea, in this study, we investigated tg mice in terms of hippocampus-dependent behaviors and learning-related synaptic plasticity in the hippocampus. Importantly, heterozygous tg (tg/+) mice serve as a suitable model for evaluating cognitive function independent of ataxia and dyskinesia, since no clear evidence has been provided for motor dysfunction in tg/+ mice [Citation24]. Our results revealed that in tg/+ and homozygous tg (tg/tg) mutants, impaired behaviors were linked to the spatial reference memory in the Morris water maze task but not to the spatial working memory in the eight-arm radial maze and T-maze tasks. In electrophysiological analyses, Schaffer collateral–CA1 synapses showed a deficit in the maintenance of long-term potentiation (LTP) in tg/+ and tg/tg mice and increased paired-pulse facilitation (PPF) induced by paired pulses of 100 ms in tg/tg mice. These results indicated that the Cacna1a mutation tg contributes toward dominant disorders of hippocampus-dependent memory and learning-related hippocampal synaptic plasticity in mice.
Materials and methods
Animals
The C57BL/6J-tg strain of tg mice was obtained from the Jackson Laboratory. The tg mice were provided with a commercial diet (CE-2, Nihon Clea) and water ad libitum under conventional conditions with controlled temperature (22 ± 2°C), humidity (55 ± 5%), and lighting (12 h light/dark cycle). Genotyping of tg mice was performed using PCR-restriction fragment length polymorphism. A PCR fragment was obtained using a pair of primers, 5′-GGAAACCAGAAGCTGAACCA-3′ (sense) and 5′-GAAATGGAGGAATTCAGGG-3′ (antisense), and genomic DNA as a template. Digestion of the fragment with AciI yielded the following fragments: 295 bp in tg/tg; 127 and 168 bp in wild-type control (+/+); and 127, 168, and 295 bp in tg/+ [Citation21]. All animal studies described herein were reviewed and approved by the ethical committee of National Institute for Physiological Sciences and were performed according to the institutional guidelines concerning the care and handling of experimental animals.
Behavioral studies
All of the behavioral studies were undertaken between 10:00 and 16:00 by a well-trained researcher unaware of the genotypes. Seven-to-eight 2-month-old male mice were used for each group. The same mice were used across the different tests. When the behavioral studies commenced, the average body weight of the tg/tg mice (19.9 ± 0.5 g), but not that of the tg/+ mice (24.5 ± 0.5 g), was lower than that of the +/+ mice (23.3 ± 0.6 g).
Morris water maze test
Spatial learning was assessed through three variants of the Morris water maze task [Citation25] adapted for mice. The maze consisted of a 150 cm plastic pool filled to a depth of 31 cm with 23–25°C water. Hidden-platform task: A circular transparent acrylic platform (diameter 12 cm) was submerged 1 cm below the surface of the water in the southeast (SE) quadrant throughout the hidden-platform task. Each mouse was subjected to four trials per day over 7 days. There were four starting points located at the center of each quadrant, and the mouse was dropped at a different starting point location for each of the four daily trials. A mouse was placed in the water facing the wall of the pool but not touching it. The time taken to reach the platform (escape latency) was recorded. When the mouse found the platform within 60s, it was allowed to stay there for 30s. Mice that failed to find the platform within 60s were placed onto the platform by hand and remained on it for 30s. Probe trial: A single probe trial was carried out after the series of hidden-platform tasks had been completed. In this trial, the platform was removed, and the movement of each mouse in the pool was monitored using a computer-based video tracking system (BTA-2, Muromachi Kikai). Each mouse was placed in the pool at the northwest (NW) position and was allowed to swim for 60s. The time spent in each quadrant, the number of times the platform site was crossed, and swimming path length were calculated. Cue-platform task: A circular platform (diameter 12 cm) was made visible by attaching a black board (9 × 19 cm) to the platform, and the mouse had to locate the visible platform. This task consisted of four trials per day for three consecutive days. The placement of the platform varied among four possible locations for each of the four trials daily. Each mouse was always initially placed at the eastern position and was given 60s to locate the platform. Other procedures were the same as those for the hidden-platform task.
Eight-arm radial maze test
The body weights of the mice were maintained at within 80% to 90% of their initial values by mild food restriction for this task. An elevated eight-arm radial maze [Citation26] adapted for mice was employed. Briefly, each arm was 30 cm long and 60 cm wide, and the center arena was 15 cm in diameter. Food pellets were placed into a well at the end of each maze arm, and a mouse was introduced into the central arena of the maze. The animal was left in the maze until all eight pellets were obtained or 5 min had elapsed. Each mouse underwent two trials per session each day for 7 days. A correct choice was recorded when the mouse entered an unvisited arm during the trial, while reentering an arm that the mouse had already visited was recorded as an error.
Delayed non-matching-to-position task in the T-maze
Mice were raised individually and their body weights were reduced and maintained to within 80% to 90% of their initial values. Water was offered ad libitum. Before testing, the mice were fed food pellets (20 mg, dustless precise pellets, Bio-Serv) in their home cage. A T-maze made of gray Plexiglas was used [Citation27]. The stem and arms were 35 cm long, 10 cm wide, and 15 cm high. The start box (10 × 10 × 15 cm high) was separated from the stem by a horizontal sliding door. Sliding doors were also placed at the entrance of each arm. Each goal arm had a small well (diameter 2 cm) at the distal end to hold a food pellet. Following a 10 min period of exploration, mice were trained to run down from the start box to one of the goal boxes to get a pellet (six trials a day for 6 days). Mice were first forced to enter one of the goal arms (i.e. sample run) and, immediately after consuming a pellet, were returned to the start box. The start door was opened 5 s later and mice were allowed to make a choice (i.e. choice run). During this run, the opposite arm was baited. If the animal entered the incorrect unbaited arm, it was not reinforced. Each animal underwent six trials per day for 5 days. The mice were then subjected to the delay trials. In these trials, three kinds of delays (0, 1, and 2 min) intervened between the end of the sample run and the start of the choice run. A total of 18 trials, which consisted of six trials for each delay, were carried out for 4 days for each mouse. The testing order of delays was pseudo-randomized. The proportion of correct responses for every delay was calculated.
Spontaneous motor activity test
Locomotor activity was measured by placing individual animals in a clear Plexiglas box (30 × 20 × 13 cm) that was positioned in a frame mounted with infrared beams (Scanet SV-10, Toyo Industry). Beam interruptions were summed over a period of 60 min.
Rotating rod test
Motor coordination was assessed with a rotating rod apparatus (KN-75, Natsume Seisakusho), which consisted of a plastic rod (3 cm diameter, 8 cm long) with a gritted surface flanked by two large discs (40 cm diameter) [Citation28]. A mouse was placed on the rod, which was then rotated at a speed of 0 (stationary), 5, 10, and 20 rpm. Latency until a fall occurred was recorded for four trials for each speed.
Footprint test
To obtain footprints, black ink was applied to the hindpaws of each mouse and the mouse was allowed to walk forward in a narrow alley (9 × 25 × 10 cm) on white paper.
Hanging test
The apparatus was made of a stainless-steel bar (50 cm, 2 mm diameter) placed between two vertical supports and elevated 37 cm from a flat surface. A mouse was placed on the bar at a point midway between the supports and observed four times for 30s. The amount of time spent hanging was recorded.
Traction test
The grip strength of a mouse was measured with a traction apparatus (FU-1, Muromachi Kikai, Tokyo, Japan) to which a horizontal stainless-steel bar (2 mm diameter) was attached for holding. A mouse was lifted by the tail and made to grasp the holding bar with its forepaws. The researcher then slowly pulled the mouse back by the tail, and the maximum tension in the cable was recorded.
Elevated plus maze test
The elevated plus maze constituted a cross of gray plastic consisting of two arms that were open to the environment (30.5 × 5.5 cm, open arms) and two arms that were enclosed by side and end walls (30.5 × 5.5 × 15 cm, closed arms) [Citation29]. The arms were connected by a central area (5.5 × 5.5 cm). The maze was elevated from the floor (46.5 cm). Behavioral testing began by placing an animal in the central area of the maze facing an open arm. Explorative behavior was recorded via a video camera and a remote monitor located in an adjacent room. The number of open- and closed-arm entries and the time spent in both types of arms were determined. An entry was registered when all four paws crossed into one arm.
Electrophysiological experiments with multi-electrode array
Four-to-five-week-old tg mice were euthanized by decapitation after anesthesia with isoflurane, in accordance with the Kyoto University guidelines for animal experiments. The brain was immediately soaked in an ice-cold oxygenated preparation of artificial cerebrospinal fluid (aCSF) of the following composition (in mM): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 KH2PO4, 2 CaCl2, and 1 MgSO4, for approximately 2 min. Appropriate portions of the brain were trimmed and placed on the ice-cold stage of a vibrating tissue slicer (LinearSlicer PRO7, Dosaka EM Co., Ltd., Kyoto, Japan). The stage was immediately filled with oxygenated and frozen aCSF. The thickness of each tissue slice was 300 μm. Sections were soaked in the oxygenated preparation buffer for 1 h at 27.5°C. Procedures for the electrophysiological experiments with the Multi-Electrode Dish (MED probe, Alpha MED Scientific, Inc., Ibaraki, Osaka, Japan) have been described in previous studies [Citation30]. The device has an array of 64 planar microelectrodes (50 × 50 μm) arranged in an 8 × 8 pattern with interelectrode spacing of 150 μm (MED-P515A, Alpha MED Scientific, Inc.). The slices, positioned to cover the 8 × 8 array on the MED probe, were placed in a small incubator at 32°C, and responses were collected in aCSF. Oxygenated, fresh recording aCSF was infused at 1.5 mL/min. Field potentials at all 64 sites were simultaneously recorded with the multichannel recording system (MED system, Alpha MED Scientific, Inc.) at a 20 kHz sampling rate. One of the electrodes in the Schaffer collateral fibers projecting toward the CA1 region was selected as a stimulating electrode, while another one in the stratum radiatum was selected as a recording electrode. Bipolar constant current pulses were delivered at 30% intensity of the current that produced the maximum field excitatory postsynaptic potential (fEPSP). For the LTP experiment, baseline fEPSPs were recorded for 15 min before the conditioning stimulation. LTP was induced by theta burst conditioning stimulation [Citation31], which consisted of 10 bursts every 200 ms and each burst consisted of four pulse (0.2 ms width) every 10 ms. The fEPSP slopes were expressed as a proportion of the average values measured during the 15 min baseline recording period. For the PPF experiment, paired stimuli at 20, 30, 40, 50, 100, 200, 500, and 800 ms intervals were applied. Three traces for each interval were recorded. The paired-pulse ratio was calculated by dividing the second fEPSP slope by the first fEPSP slope.
Statistical analyses
Statistical analyses were performed as described previously [Citation32]. Data were analyzed using one-way ANOVA or two-way repeated measures ANOVA followed by post-hoc analysis with Scheffe or Tukey HSD tests. Behavioral scores were subjected to ANOVAs with repeated measures for time factors (sessions for Morris water maze, T-maze, and eight-arm radial maze tests; trials for rotating rod test).
Results
Acquisition and retention of spatial reference memory are impaired in tg/+ and tg/tg mice
To understand the hippocampal-dependent memory in tg mice, we first performed the Morris water maze task, a standard measure of spatial learning and memory in rodents [Citation33,Citation34]. Both tg/tg and tg/+ mice showed a significantly longer escape latency to locate the hidden platform compared with that of +/+ mice through sessions [genotype effect, F(2, 20) = 7.914, p = 0.0029; Scheffe post-hoc test: tg/tg vs. +/+: p = 0.0042; tg/+ vs. +/+: p = 0.0246] ()). A significant genotype × session interaction was observed in the escape latency [genotype × session effect, F(12, 120) = 2.596, p = 0.0042], statistically validating our comparison of escape latency among three genotypes at each session. In the 1, 2, 5, 6, and 7 sessions, tg/tg mice showed a significantly longer latency to locate the hidden platform than +/+ mice (session 1, tg/tg vs. +/+, p = 0.0209; session 2, tg/tg vs. +/+, p = 0.0083; session 5, tg/tg vs. +/+, p = 0.0231; session 6, tg/tg vs. +/+, p = 0.0023; session 7, tg/tg vs. +/+, p = 0.0035). In the last three sessions, tg/+ mice showed a significant increase in escape latency to reach the hidden platform compared with +/+ mice (session 5, tg/+ vs. +/+, p = 0.0155; session 6, tg/+ vs. +/+, p = 0.0013; session 7, tg/+ vs. +/+, p = 0.0025). These results suggest that acquisition of spatial reference memory is impaired in tg/+ and tg/tg mice.
Figure 1. Acquisition and retention of spatial reference memory assessed by the Morris water maze task in tg/+ and tg/tg mice. (a) Mean escape latencies in the hidden-platform task averaged over four trials per session (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). *p < 0.05 (tg/tg vs +/+), **p < 0.01 (tg/tg vs +/+), ***p < 0.005 (tg/tg vs +/+), †p < 0.05 (tg/+ vs +/+), and †††p < 0.005 (tg/+ vs +/+). (b) Mean time spent in the four quadrants during the probe trial (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). NE, SE, SW, and NW indicate northeast, southeast, southwest, and northwest positions of the quadrants, respectively. The platform was placed in the SE quadrant (filled columns) during acquisition. N.S., p > 0.05, *p < 0.05, and ***p < 0.005. (c) The number of platform crossings during the probe trial (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). *p < 0.05. (d) The total distance during the probe trial (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). *p < 0.05 (genotype effect). (e) Mean escape latencies in the cue-platform task averaged over four trials per session (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). *p < 0.05 (tg/tg vs +/+), ***p < 0.005 (tg/tg vs +/+), and †p < 0.05 (tg/+ vs +/+). Data represent the mean ± SEM or mean + SEM.
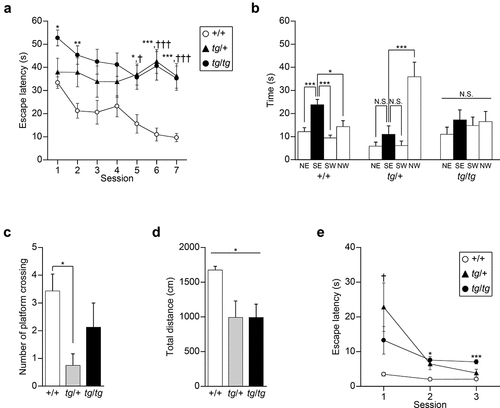
In the probe test, the platform was removed from the pool after the hidden platform task, and the trained mice were allowed to swim freely for 60s. Tracking analysis of swimming revealed that +/+ mice spent a significantly greater proportion of the trial swimming time in the SE quadrant where the platform had been placed in the previous task [F(3, 24) = 9.590, p = 0.0002; Scheffe post-hoc test: NE vs. SE, p = 0.0048; SW vs. SE, p = 0.0005; NW vs. SE, p = 0.0262], whereas tg/tg mice did not focus on the training SE quadrant [F(3, 28) = 0.500, p = 0.6853] and tg/+ mice swam in the starting NW quadrant for a longer time [F(3, 28) = 13.518, p < 0.0001; Scheffe post-hoc test: NE vs. SE, p = 0.8339; SW vs. SE, p = 0.8486; NW vs. SE, p = 0.0014] ()). The number of platform crossings was significantly different among the genotypes [F(2, 20) = 3.946, p = 0.0359], and the post-hoc test showed that the crossing number of tg/+ mice was lower than that of +/+ mice (tg/+ vs. +/+: p = 0.0362; tg/tg vs. +/+: p = 0.4103) ()). Although the swimming distance was significantly different among the three genotypes [F(2, 20) = 4.298, p = 0.0280], its post-hoc analysis failed to show a statistical difference (tg/+ vs. +/+: p = 0.0580; tg/tg vs. +/+: p = 0.0579) ()). These results showed that tg/+ and tg/tg mice failed to develop an effective strategy to locate a hidden platform through training, suggesting the impaired retention of spatial reference memory in tg/+ and tg/tg mice.
In the cue task, mice of the three genotypes as a whole showed an improvement in their ability to find the visible platform across sessions [session effect, F(2, 40) = 10.641, p = 0.0002] ()). A significant genotype × session interaction was observed in the escape latency [genotype × session effect, F(4, 40) = 3.606, p = 0.0133], statistically validating our comparison of escape latency among three genotypes at each session. Compared to +/+ mice, escape latency to reach the visible platform was significantly increased in tg/tg mice for the last two sessions (session 1, p = 0.3914; session 2, p = 0.0154; session 3, p = 0.0012) and in tg/+ mice for the first session (session 1, p = 0.0393; session 2, p = 0.0562; session 3, p = 0.3305). Thus, it is possible that additional alterations, such as motor dysfunction (see results below), other than cognitive deficits, might affect the performance of the tg mutant mice in the Morris water maze task.
Spatial working memory is intact in tg/+ and tg/tg mice
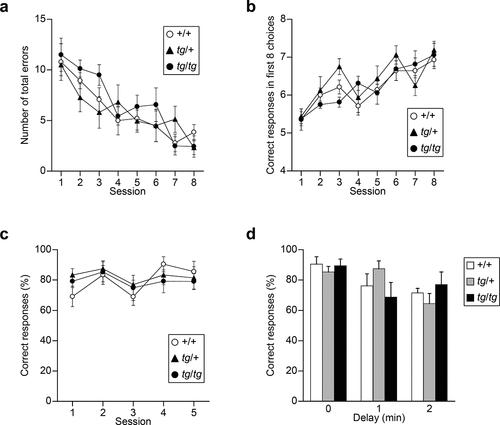
To evaluate spatial working memory, tg mutants were subjected to an eight-arm radial maze and T-maze tasks. In the eight-arm radial maze task, mice were required to remember and avoid previously visited arms to get food rewards. No significant effect of genotype and genotype × session interaction was observed for the number of total errors [genotype effect, F(2, 20) = 0.682, p = 0.5171; genotype × session effect, F(14, 140) = 1.020, p = 0.4371] ()). Also, for the analysis of correct responses in the first eight choices, as seen in total errors, no significant effect of genotype and genotype × session interaction was observed [genotype effect, F(2,20) = 0.727, p = 0.4958; genotype × session effect, F(14, 140) = 1.093, p = 0.3690] ()). Next, mice were subjected to a delayed non-matching-to-position T-maze task using food reward after food restriction. In this task, mice were required to remember the previous direction of the sample run to respond correctly in the choice run of this task and were subjected to five consecutive sessions. No significant effect of genotype and genotype × session interaction was observed for the correct proportion [genotype effect, F(2, 20) = 0.440, p = 0.6501; genotype × session effect, F(8, 80) = 0.661, p = 0.7243] ()). With 1 and 2 min delays between the forced and free choices, all groups showed reduction in correct % according to the increase in delay time [delay effect, F(2, 40) = 5.884, p = 0.0058], and no significant genotype effect and genotype × delay interaction were found for the correct proportion [genotype effect, F(2, 20) = 0.016, p = 0.9842; genotype × delay effect, F(4, 40) = 1.799, p = 0.1481] ()). Thus, tg/+ and tg/tg mice showed intact spatial working memory in the T-maze and eight-arm radial maze tasks.
Figure 2. Spatial working memory assessed by the eight-arm radial maze and T-maze tasks in tg/+ and tg/tg mice. (a), (b) Acquisition of the eight-arm radial maze task of tg mutant mice (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). Total errors in a trial (a) and correct responses in the first eight choices (b) in the eight-arm radial maze averaged over two trials per session. (c), (d) Delayed non-matching-to-position task in the T-maze (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). Proportion of correct responses during training (c) and in delay trials (d). Data represent the mean ± SEM or mean + SEM.
Motor function is intact in tg/+ mice but impaired in tg/tg mice
Considering the possibility that water maze performance can be influenced by other deficits, such as motor dysfunction (see above), we assessed motor function in tg/+ and tg/tg mice. Homozygous tg/tg mice all showed ataxic gait, whereas heterozygous tg/+ mice failed to show any signs of abnormal gait, being indistinguishable from +/+ mice at the whole-body level. Spontaneous locomotor activity in a new environment was indexed by infrared beam breaks, for which a significant effect of the genotype was seen [F(2, 20) = 4.405, p = 0.0260]. tg/tg mice were significantly hypoactive compared to +/+ mice (p = 0.0328), whereas the locomotor activity of tg/+ mice was comparable to that of +/+ mice (p = 0.1126) ()).
Figure 3. Motor activity and function assessed in tg/+ and tg/tg mice. (a) Spontaneous motor activity in a novel environment (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). Note that tg/+ mice, but not tg/tg mice, showed normal motor function. *p < 0.05. (b) Retention time for remaining on the rotating rod (n = 7 for +/+, n = 7 for tg/+, and n = 8 for tg/tg). Note that tg/tg mice showed a significant decrease in locomotor activity and motor discoordination, while the performance of tg/+ mice was comparable to that of +/+ mice. ***p < 0.005 (tg/tg vs +/+) and §§§p < 0.005 (tg/+ vs tg/tg). (c) Step width in the footprint test (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). ***p < 0.005. (d) Step length in the footprint test (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). *p < 0.05 (genotype effect). (e) Retention time in the hanging test (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). tg/tg mice, but not tg/+ mice, showed motor dysfunction. *p < 0.05 and ***p < 0.005. (f) Grip strength in the traction test (n = 7 for +/+, n = 8 for tg/+, and n = 8 for tg/tg). N.S., p > 0.05 (genotype effect). Data represent the mean ± SEM or mean + SEM.
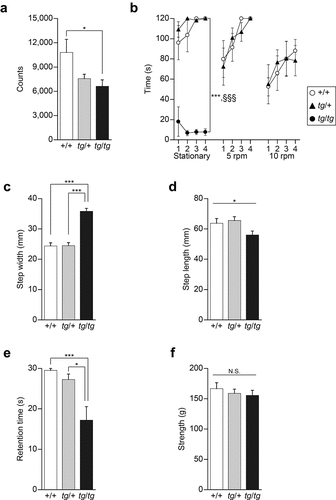
In the rotating rod test, a sensitive test for detecting motor dysfunctions, tg/tg mice failed to remain even on the stationary rod [genotype effect, F(2, 19) = 121.045, p < 0.0001; genotype × time effect, F(6, 57) = 1.467, p = 0.2058. Scheffe post-hoc test: tg/tg vs. +/+, p < 0.0001; tg/tg vs. tg/+, p < 0.0001], whereas no significant difference was found between tg/+ and +/+ mice in terms of retention time on the stationary rod [Scheffe post-hoc test: tg/+ vs. +/+, p = 0.6962] ()). When the rods were rotated at 5 and 10 rpm, the retention time of tg/+ mice was comparable to that of +/+ mice [5 rpm: genotype effect, F(1,12) = 0.066, p = 0.8022; genotype × trial effect, F(3, 36) = 0.516, p = 0.6736. 10 rpm: genotype effect, F(1, 12) = 0.002, p = 0.9613; genotype × trial effect, F(3, 36) = 0.386, p = 0.7635] ()).
The footprint test revealed that the step width of tg/tg mice was significantly wider than that of +/+ or tg/+ mice [F(2, 20) = 44.013, p < 0.0001. Scheffe post-hoc test: tg/tg vs. +/+: p < 0.0001; tg/+ vs. +/+: p = 0.9981; tg/tg vs. tg/+: p < 0.0001] ()). A significant genotype effect was observed in step length [F(2, 20) = 3.533, p = 0.0485], but a post-hoc test showed no significant difference among genotypes (tg/tg vs. +/+: p = 0.1675; tg/+ vs. +/+: p = 0.9051; tg/tg vs. tg/+: p = 0.0646) ()).
The hanging test showed that the retention time of tg/tg mice, but not that of tg/+ mice, was shorter compared to that of +/+ mice [F(2, 20) = 8.926, p = 0.0017. Scheffe post-hoc test: tg/tg vs. +/+: p = 0.0034; tg/+ vs. +/+: p = 0.7708; tg/tg vs. tg/+: p = 0.0131] ()). On the other hand, the traction test, in which grip strength was tested, showed no significant genotype effect on strength to tract [F(2, 20) = 0.430, p = 0.6565], excluding the possibility that the observed motor dysfunction was not due to muscular weakness in tg/tg mice ()).
Thus, our results indicated that the motor function of heterozygous tg/+ mice is intact and comparable to that of +/+ mice, while motor dysfunction, which is unlikely to be due to muscular weakness, is displayed by homozygous tg/tg mice.
Synaptic plasticity is impaired in the hippocampi of tg/tg and tg/+ mice
To understand the synaptic basis of abnormal hippocampus-related behaviors exhibited by tg/tg and tg/+ mice, we investigated synaptic plasticity in the Schaffer collateral–CAl synapses of the hippocampus by recording the response of fEPSP to theta burst conditioning stimulation that induces LTP [Citation31]. In our electrophysiological recordings, we used a 64-electrode array with the multichannel recording system (MED system), which allowed simultaneous recording of field potentials in mouse hippocampal slices [Citation15]. Upon post-tetanic potentiation (PTP), 1 min after theta burst stimulation, the slopes of the fEPSP were comparable among tg/tg, tg/+ and +/+ mice [F(2, 16) = 1.314, p = 0.2962] (), indicating that tg/tg and tg/+ mice exhibited intact LTP induction. The fEPSP slopes of tg/tg and tg/+ mice returned to the baseline level 50–60 min after theta burst stimulation, while the fEPSP slope from slices of +/+ mice was continually augmented ()). Quantitative analysis revealed that averaged values of the fEPSP slope of tg/tg and tg/+ mice significantly decreased 50–60 min after theta burst stimulation compared with those of +/+ mice [F(2, 16) = 11.294, p = 0.0009. Tukey HSD post-hoc test: tg/tg vs. +/+: p = 0.0110; tg/+ vs. +/+: p = 0.0008; tg/tg vs. tg/+: p = 0.4928] ()). The results suggested that the maintenance of the LTP at the Schaffer collateral–CA1 synapses is defective in the hippocampal slices from tg/tg and tg/+ mice.
Figure 4. Hippocampal synaptic plasticity recorded by the MED system at the Schaffer collateral–CAl synapses in tg/+ and tg/tg mice. (a) Field potentials were recorded from the CA1 region of hippocampal slices of +/+, tg/+, and tg/tg mice. LTP was induced by theta burst conditioning stimulation. Averaged time courses of normalized fEPSP slopes are plotted (n = 6 slices from five mice for +/+, n = 7 slices from five mice for tg/+, and n = 6 slices from four mice for tg/tg). The inset traces show fEPSP at baseline (black) and LTP (red) in +/+ (upper), tg/+ (middle), and tg/tg (lower) mice. (b) Comparison of normalized fEPSP slopes at 1 min (PTP) and the last 10 min (LTP) after theta burst conditioning stimulation recorded from the CA1 regions of hippocampal slices in tg/tg, tg/+, and +/+ mice. *p < 0.05 and ***p < 0.005. (c) PPF induced by paired stimuli with 20, 30, 40, 50, 100, 200, 500, and 800 ms pulse intervals from the CA1 regions of hippocampal slices (n = 10 slices from three mice for +/+, n = 8 slices from three mice for tg/+, and n = 9 slices from three mice for tg/tg). The paired-pulse ratio was expressed by the ratio of the second fEPSP slope to the first fEPSP slope. *p < 0.05. Data represent the mean ± SEM or mean + SEM.
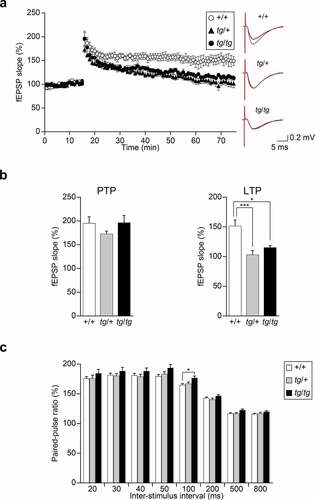
We also examined the PPF of CA1 responses to paired stimuli with 20, 30, 40, 50, 100, 200, 500, and 800 ms pulse intervals ()). We noted that PPF by a paired stimulus with 100 ms pulse interval was significantly augmented in tg/tg compared with +/+ mice [F(2, 24) = 4.576, p = 0.0207. Tukey HSD post-hoc test: tg/tg vs. +/+: p = 0.0217; tg/+ vs. +/+: p = 0.8729; tg/tg vs. tg/+: p = 0.0852], suggesting that neurotransmitter release at the Schaffer collateral–CA1 synapse was impaired in tg/tg, presumably due to reduced Ca2+ influx [Citation21]. These results indicated impaired hippocampal synaptic plasticity of tg/tg and tg/+ mice.
Discussion
The present study revealed that hippocampus-dependent behaviors and learning-related synaptic plasticity in the hippocampus are impaired in the tg point-mutant Cacna1a mice, the well-established model of spontaneous absence epilepsy. The results suggest that the tg mutation in the P/Q-type Ca2+ channel leads to a dominant disorder of hippocampal synaptic plasticity, causing abnormal hippocampus-related behaviors independently of neurological phenotypes, such as epilepsy and ataxia. Thus, hippocampus-related deficits may be an important underlying hallmark of cognitive impairments accompanied with CACNA1A-associated disorders.
Cognitive impairments are critical co-morbidities of epilepsy and other neuronal disorders, given their impacts on social and educational issues [Citation11]. CACNA1A-associated disorders, such as epilepsy and ataxia, are frequently accompanied by cognitive impairments [Citation7–10]. In this study, we showed that the acquisition and retention of spatial reference memory in the Morris water maze task were impaired by both homozygous and heterozygous tg mutations in the Cacna1a gene. Our observation is consistent with those of previous studies on Cacna1a mutants showing spatial learning and memory impairment in the Morris water maze for 4- to 6-month-old homozygous tg mice [Citation35] and for 6-month-old heterozygous leaner mice [Citation36]. Since water maze performance can also be influenced by other deficits, such as motor dysfunction and emotional peculiarities [Citation37], it was important to examine the impact of tg mutation on cognitive function under the condition that maintains normal motor functions in mice. Indeed, our present results indicated that heterozygous tg/+ mice are intact in the series of used motor function tests in contrast to homozygous tg/tg mice that display obvious motor dysfunction (). Therefore, motor dysfunction is unlikely to be the primary cause of the learning deficit induced by the tg mutation in the Morris water maze task. Interestingly, evaluation of the emotional state using an elevated plus maze, which is frequently used to pharmacologically assess anxiolytics [Citation29,Citation38], showed alterations in the emotional state of tg/tg mice, such as reduced fearfulness or increased impulsivity; tg/+ mice were again normal (data not shown). Also, in tg/+ mice, spike-wave discharges, the hallmark of absence seizures, are not observed [Citation20,Citation39]. Thus, in spatial reference memory in the Morris water maze task, the observed abnormality was predominantly explained by cognitive impairments in the tg/+ mice, in contrast to the tg/tg mice, in which motor and emotional dysfunction contributes toward the behavioral scores. Moreover, considering that tg/+ mice failed to show clear epileptic and ataxic phenotypes, in CACNA1A-associated disorders, cognitive impairments are more likely to be the direct consequence of dysfunctions of CaV2.1-mediated synaptic transmission and its plasticity than a secondary consequence of neurological symptoms.
The P/Q-type Ca2+ channel is the major source of Ca2+ that controls depolarization-evoked neurotransmitter release at mammalian central synapses [Citation40]. This is not an exception to Schaffer collateral–CA1 synapses, in which the P/Q-type Ca2+ channel currents is essential for neurotransmitter release [Citation41]. At the Schaffer collateral–CA1 synapses, in the paired pulse protocol with 100 ms pulse intervals, PPF, which reflects the amount of residual Ca2+ in the presynaptic terminal [Citation42], was significantly augmented in tg/tg compared with +/+ mice ()). Given that the most striking effect of the tg mutation on the P/Q-type Ca2+ channel is a marked reduction in current density in cerebellar Purkinje neurons [Citation21], our findings may suggest that the amount of Ca2+ influx into the tg/tg nerve terminal evoked by a single activation is too low for reliable synaptic transmission in the recording condition. A similar result was obtained at the Schaffer collateral–CA1 synapses by Qian and Noebels [Citation43]. In addition, the suppressive effect of the tg mutation on synaptic transmission has been reported in the cerebral cortex [Citation44], thalamus [Citation45], and cerebellum [Citation46] in tg/tg mice.
Postsynaptic intracellular concentration of Ca2+ increase and AMPA receptor trafficking are critical for hippocampal LTP [Citation47]. In the postsynaptic membrane, specific association of CaV2.1 with AMPA receptors and functional coupling of CaV2.1 and AMPA receptors have been reported [Citation48]. Also, the direct interaction and functional coupling between metabotropic glutamate receptor subtype 1 and CaV2.1 have been reported [Citation49,Citation50]. In our electrophysiological recordings using the MED system, tg/+ and tg/tg mice showed impairments in LTP at the Schaffer collateral–CA1 synapses, suggesting that P/Q-type Ca2+ channels play an important role at the postsynapse. Interestingly, a Cacna1a mutant rocker showed a decrease in the number and density of postsynaptic AMPA receptors in parallel fiber–Purkinje cell synapses [Citation51]. Therefore, impairment of interaction and/or functional coupling between CaV2.1 and glutamate receptors in the postsynaptic membrane may contribute to the reduction of LTP in tg/+ and tg/tg mice.
Our results suggest that the hippocampus is a critical onset site for cognitive co-morbidities of absence epilepsy. In terms of the generation of spike-wave discharges, the hallmark of absence seizures, abnormal hyper-synchronized oscillatory activities in the thalamocortical network have been classically proposed as an underlying mechanism [Citation52]. Interestingly, frequency and temporal correlation analysis of local field potentials have revealed that spike-wave discharges in a pharmacological rat model increased synchronization between the hippocampus and thalamocortical network, suggesting that the hippocampus participates in absence seizures in addition to the thalamocortical network [Citation53]. Moreover, electroencephalography and functional magnetic resonance imaging showed an increased functional connectivity between the hippocampus and thalamus during the appearance of spike-wave discharges in a pharmacological rat model [Citation54]. Therefore, in tg mice, the apparent independence of cognitive co-morbidities from absence epilepsy cannot be simply attributed to the difference of the onset site within the central nervous system. In this context, it is worth noting that the hippocampus is essential for mesial temporal lobe epilepsy, a different form of epilepsy, in contrast to absence epilepsy [Citation55].
The present study provides a mechanistic insight into the cognitive co-morbidities of absence epilepsy, which will aid in establishing their diagnostic criteria and potential therapeutic strategies. A recent comprehensive clinical and radiological study suggested that CACNA1A-associated phenotypes are neurodevelopmental disorders [Citation8]. In support of this concept, in tg/tg mice, our group previously demonstrated developmental abnormalities in Cl− transporter expression and GABAA receptor compositions in hippocampal neurons, proposing an idea of “compromised maturation” of GABAergic inhibition, which contributes toward the abnormal synchrony in the hippocampus [Citation15]. Similar to tg/tg mice, a mouse model of Down syndrome, which is one of the most common neurodevelopmental disorders, showed the compromised maturation of GABAergic inhibition and the deficits in hippocampal LTP and hippocampus-dependent memory [Citation56]. Therefore, CACNA1A-associated disorders and neurodevelopmental disorders, including Down syndrome, may share a common mechanism of compromised maturation of the hippocampus. Thus, cognitive impairments in CACNA1A-associated human disorders can be generalized as hippocampus-related functional deficits. Interestingly, tg/tg mice have been reported to show decreased hippocampal volume, increased cell densities in the hippocampal CA1 region and the dentate gyrus hilus, and increased cell proliferation in the hippocampal CA2 region and dentate gyrus hilus [Citation35]. An analysis of neuronal arborization by Sholl analysis revealed alterations of dendritic complexity in hippocampal CA1 pyramidal neurons in tg/tg mice. Further electrophysiological characterization of different hippocampal regions of wild-type and Cacna1a mutant mice using the MED system would provide new insights into cognitive impairments in CACNA1A-associated disorders.
Author contributions
A.N., Y.M., and K.I. designed the project. A.N., K.H., and H.O. performed experiments and analyses. K.I. supervised the project. A.N., Y.M., and K.I. generated the first draft of the manuscript. All authors discussed the results and contributed to the final manuscript.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Mori Y, Friedrich T, Kim MS, et al. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350(6317):398–402.
- Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. 2018;98(3):466–481.
- Imbrici P, Jaffe SL, Eunson LH, et al. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain. 2004;127(12):2682–2692.
- Jouvenceau A, Eunson LH, Spauschus A, et al. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358(9284):801–807.
- Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543–552.
- Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62–69.
- Damaj L, Lupien-Meilleur A, Lortie A, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015;23(11):1505–1512.
- Humbertclaude V, Riant F, Krams B, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol. 2020;62(3):330–337.
- Lupien-Meilleur A, Jiang X, Lachance M, et al. Reversing frontal disinhibition rescues behavioural deficits in models of CACNA1A-associated neurodevelopment disorders. Mol Psychiatry. 2021;26(12):7225–7246.
- Rajakulendran S, Graves TD, Labrum RW, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. 2010;588(11):1905–1913.
- Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. 2016;15(1):106–115.
- Caplan R, Siddarth P, Stahl L, et al. Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities. Epilepsia. 2008;49(11):1838–1846.
- Sillanpää M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338(24):1715–1722.
- Kleen JK, Scott RC, Lenck-Santini PP, et al. Cognitive and behavioral co-morbidities of epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al. editors. Jasper’s basic mechanisms of the epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012. https://www.ncbi.nlm.nih.gov/books/NBK98139/
- Nakao A, Miki T, Shimono K, et al. Compromised maturation of GABAergic inhibition underlies abnormal network activity in the hippocampus of epileptic Ca2+ channel mutant mice, tottering. Pflügers Arch - Eur J Physiol. 2015;467(4):737–752.
- Fletcher CF, Lutz CM, O’Sullivan TN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87(4):607–617.
- Miki T, Zwingman TA, Wakamori M, et al. Two novel alleles of tottering with distinct CaV2.1 calcium channel neuropathologies. Neuroscience. 2008;155(1):31–44.
- Mori Y, Wakamori M, Oda S, et al. Reduced voltage sensitivity of activation of P/Q-type Ca2+ channels is associated with the ataxic mouse mutation rolling Nagoya (tgrol). J Neurosci. 2000;20(15):5654–5662.
- Zwingman TA, Neumann PE, Noebels JL, et al. Rocker is a new variant of the voltage-dependent calcium channel gene Cacna1a. J Neurosci. 2001;21(4):1169–1178.
- Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. 1979;204(4399):1334–1336.
- Wakamori M, Yamazaki K, Matsunodaira H, et al. single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273(52):34857–34867.
- Eichenbaum H. Hippocampus: cognitive processes and neural representations that underlie declarative memory. Neuron. 2004;44(1):109–120.
- Humeau Y, Choquet D. The next generation of approaches to investigate the link between synaptic plasticity and learning. Nat Neurosci. 2019;22(10):1536–1543.
- Syapin PJ. Effects of the tottering mutation in the mouse: multiple neurologic changes. Exp Neurol. 1982;76(3):566–573.
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11(1):47–60.
- Olton DS, Samuelson RJ. Remembrance of places passed: spatial memory in rats. J Exp Psychol Anim Behav Process. 1976;2(2):97–116.
- Béracochéa DJ, Jaffard R. The effects of mammillary body lesions on delayed matching and delayed non-matching to place tasks in the mice. Behav Brain Res. 1995;68(1):45–52.
- Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46(3):208–209.
- Lister RG. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology (Berl). 1987;92(2):180–185.
- Hoang TH, Yoshihara T, Nakao A, et al. Deficiency of the RIβ subunit of protein kinase A causes body tremor and impaired fear conditioning memory in rats. Sci Rep. 2021;11(1):2039.
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368(2):347–350.
- Nakao A, Miki T, Shoji H, et al. Comprehensive behavioral analysis of voltage-gated calcium channel beta-anchoring and -regulatory protein knockout mice. Front Behav Neurosci. 2015;9:141.
- Morris RGM, Garrud P, Rawlins JNP, et al. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297(5868):681–683.
- Silva AJ, Paylor R, Wehner JM, et al. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257(5067):206–211.
- Nigussie F, Huang PS, Lukauskis K, et al. Neural cell proliferation and survival in the hippocampus of adult CaV2.1 calcium ion channel mutant mice. Brain Res. 2016;1650:162–171.
- Alonso I, Marques JM, Sousa N, et al. Motor and cognitive deficits in the heterozygous leaner mouse, a CaV2.1 voltage-gated Ca2+ channel mutant. Neurobiol Aging. 2008;29(11):1733–1743.
- Wolfer DP, Müller U, Stagliar M, et al. Assessing the effects of the 129/Sv genetic background on swimming navigation learning in transgenic mutants: a study using mice with a modified β-amyloid precursor protein gene. Brain Res. 1997;771(1):1–13.
- Rodgers RJ, Lee C, Shepherd JK. Effects of diazepam on behavioural and antinociceptive responses to the elevated plus-maze in male mice depend upon treatment regimen and prior maze experience. Psychopharmacology (Berl). 1992;106(1):102–110.
- Noebels JL. Isolating single genes of the inherited epilepsies. Ann Neurol. 1984;16(S1):S18–S21.
- Iwasaki S, Momiyama A, Uchitel OD, et al. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20(1):59–65.
- Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366(6451):156–158.
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64(1):355–405.
- Qian J, Noebels JL. Presynaptic Ca2+ influx at a mouse central synapse with Ca2+ channel subunit mutations. J Neurosci. 2000;20(1):163–170.
- Ayata C, Shimizu-Sasamata M, Lo EH, et al. Impaired neurotransmitter release and elevated threshold for cortical spreading depression in mice with mutations in the α1A subunit of P/Q type calcium channels. Neuroscience. 2000;95(3):639–645.
- Caddick SJ, Wang C, Fletcher CF, et al. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4lh) and tottering (Cacna1atg) mouse thalami. J Neurophysiol. 1999;81(5):2066–2074.
- Matsushita K, Wakamori M, Rhyu IJ, et al. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J Neurosci. 2002;22(11):4388–4398.
- Nicoll RA. A brief history of long-term potentiation. Neuron. 2017;93(2):281–290.
- Kang MG, Chen CC, Wakamori M, et al. A functional AMPA receptor–calcium channel complex in the postsynaptic membrane. Proc Natl Acad Sci U S A. 2006;103(14):5561–5566.
- Beqollari D, Kammermeier PJ. The interaction between mGluR1 and the calcium channel CaV2.1 preserves coupling in the presence of long Homer proteins. Neuropharmacology. 2013;66:302–310.
- Kitano J, Nishida M, Itsukaichi Y, et al. Direct interaction and functional coupling between metabotropic glutamate receptor subtype 1 and voltage-sensitive CaV2.1 Ca2+ channel. J Biol Chem. 2003;278(27):25101–25108.
- Kodama T, Itsukaichi‐Nishida Y, Fukazawa Y, et al. A CaV2.1 calcium channel mutation rocker reduces the number of postsynaptic AMPA receptors in parallel fiber–Purkinje cell synapses. Eur J Neurosci. 2006;24(11):2993–3007.
- Cheong E, Shin HS. T-type Ca2+ channels in absence epilepsy. Pflügers Arch - Eur J Physiol. 2014;466(4):719–734.
- Arcaro J, Ma J, Chu L, et al. The hippocampus participates in a pharmacological rat model of absence seizures. Epilepsy Res. 2016;120:79–90.
- Mousavi SR, Arcaro JA, Leung LS, et al. Functional connectivity of the hippocampus to the thalamocortical circuitry in an animal model of absence seizures. Epilepsy Res. 2017;137:19–24.
- Engel J. Mesial temporal lobe epilepsy: what have we learned? Neuroscientist. 2001;7(4):340–352.
- Deidda G, Parrini M, Naskar S, et al. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat Med. 2015;21(4):318–326.