
ABSTRACT
The congenital long QT syndrome (LQTS), one of the most common cardiac channelopathies, is characterized by delayed ventricular repolarization underlying prolongation of the QT interval of the surface electrocardiogram. LQTS is caused by mutations in genes coding for cardiac ion channels or ion channel-associated proteins. The major therapeutic approach to LQTS management is beta blocker therapy which has been shown to be effective in treatment of LQTS variants caused by mutations in K+ channels. However, this approach has been questioned in the treatment of patients identified as LQTS variant 3(LQT3) patients who carry mutations in SCN5A, the gene coding for the principal cardiac Na+ channel. LQT3 mutations are gain of function mutations that disrupt spontaneous Na+ channel inactivation and promote persistent or late Na+ channel current (INaL) that delays repolarization and underlies QT prolongation. Clinical investigation of patients with the two most common LQT3 mutations, the ΔKPQ and the E1784K mutations, found beta blocker treatment a useful therapeutic approach for managing arrhythmias in this patient population. However, there is little experimental data that reveals the mechanisms underlying these antiarrhythmic actions. Here, we have investigated the effects of the beta blocker propranolol on INaL expressed by ΔKPQ and E1784K channels in induced pluripotent stem cells derived from patients carrying these mutations. Our results indicate that propranolol preferentially inhibits INaL expressed by these channels suggesting that the protective effects of propranolol in treating LQT3 patients is due in part to modulation of INaL.
Introduction
The congenital long QT syndrome (LQTS), first described in 1957 [Citation1,Citation2], is one of the most common cardiac channelopathies [Citation3,Citation4]. Congenital LQTS is a disorder characterized by delayed ventricular repolarization reflected in a prolongation of the QT interval of the surface electrocardiogram (EKG) that in turn is caused by mutation-induced prolongation of ventricular muscle cell action potentials [Citation5,Citation6]. There are now 17 different genetic subtypes of LQTS caused by mutations in genes coding for ion channels or ion channel associated proteins [Citation6]. The variants of LQTS were named after the chronological order in which the key genes were identified. The first two genes identified coded for two key cardiac potassium channels: mutations in KCNQ1 the α subunit of the IKS channel are LQT1 mutations [Citation7,Citation8] and mutations in KCNH2 the gene coding for hERG, the α subunit of IKr channels are LQT2 mutations [Citation9,Citation10]. All LQT1 and LQT2 mutations give rise to loss of functional activity of the coded potassium channels that in turn delay repolarization in ventricular muscle cells expressing these genes [Citation6]. Importantly, the IKr channel has also been identified as an off-target effector site of a large number of drugs. This effect also underlies drug-induced LQTS causing pathophysiology very similar to congenital LQT2 [Citation11,Citation12].
Mutations in SCN5A, the gene coding for NaV1.5, the α subunit of the principal cardiac sodium channel were first reported by Keating and colleagues in 1995 [Citation13]. A transient, or peak NaV1.5 channel current (INaP) underlies cardiac excitation, the channels conducting INaP normally close, or inactivate, during the plateau phase of the ventricular action potential that is responsible for the QT interval of the EKG [Citation14]. In contrast to LQT1 and LQT2 mutations, LQT3 mutations are gain of function mutations in that they disrupt channel inactivation causing an increase in Na+ channel activity. The hallmark functional effect of LQT3 mutations is an increase in persistent or late Na+ channel current (INaL) that can produce a prolonged action potential plateau, and prolong the QT interval of the EKG [Citation6]. Enhancement of INaL is often arrhythmogenic and is a drug target for LQT3 therapeutics [Citation14,Citation15].
LQT1 and LQT2 account for almost 85% of all genotyped LQTS patients, and LQT3 accounts for from 5% to 10% of LQTS patients [Citation5]. Triggers for cardiac events in LQTS patients differ according to the underlying LQTS gene, and there are marked differences in events that may cause higher risk of rhythm disturbances [Citation16]. The major therapeutic approach to management of LQTS is beta blocker therapy [Citation3,Citation17], and this rationale is based largely on the arrhythmia risk of LQT1 patients to stimulation of the sympathetic nervous system during exercise [Citation16,Citation18]. β-adrenergic actions enhanced during exercise include an increase in heart rate, an increase in L-type calcium channel current and modulation of intracellular calcium dynamics [Citation19]. The wide use of β-blockers is due in part to this risk being modified by the positive response of LQT1 patients to β-blocker therapy which blunts this adrenergic activity [Citation20–22] and to the fact that LQT1 is the predominant LQTS variant [Citation5].
Because LQT3 patients are at greater risks of serious cardiac events in the setting of slow heart rates [Citation16] it had been proposed that the use of β-blocker therapy would not be useful for treating LQT3, a concept that has been addressed in multiple studies [Citation4,Citation23,Citation24]. Two of the most common LQT3 mutations are the mutation that causes the deletion of three amino acids, KPQ, in the inactivation gate of the NaV1.5 channel, referred to as the ΔKPQ mutation [Citation13]; and the E1784K mutation of the NaV1.5 carboxy terminus [Citation25]. Both mutations cause enhanced persistent or late Nav1.5 channel current, INaL, when expressed in heterologous systems [Citation26–29]. Schwartz and colleagues tested the antiarrhythmic activity of the beta blocker propranolol in transgenic mice expressing ΔKPQ mutant channels and found propranolol to be effective in preventing arrhythmias in this animal model [Citation30]. Fabritz et al. suggested that these antiarrhythmic effects were likely due in part to Na+ channel modulation in addition to a classical anti-adrenergic effect [Citation31]. This work was followed by a large international clinical study in which the effects of β-blocker therapy were investigated in LQT3 genotyped patients [Citation32]. Importantly, this study included 70 patients carrying the E1784K mutation and 64 patients carrying the ΔKPQ mutation. The results of this study supported the findings of the animal studies in that β-blocker therapy was effective in preventing arrhythmia risk, particularly in females, a result that has been supported by meta-analysis [Citation33].
The purpose of this study was to investigate the effects of the β-blocker propranolol on mutant Na+ channels expressed in induced pluripotent stem cell (iPSC) myocytes derived from patients carrying either the ΔKPQ or E1784K mutation or from patients who were mutation-free. The use of patient-derived iPSC myocytes allowed us to investigate the effects of propranolol not only on mutant and wild-type NaV1.5 channels in human cells, but to also test for possible off target effects of propranolol on IKr channels that are also expressed in these cells [Citation34]. Our results indicate that propranolol preferentially inhibits INaL suggest that the protective effects of propranolol in treating LQT3 patients, is due to modulation of NaV1.5 channels along with its anti-adrenergic actions.
Methods
Human fibroblast reprogramming, characterization, differentiation, and culturing of iPSCs
Human fibroblasts were reprogrammed and characterized, and IPSC culture and differentiation were performed as previously described [Citation34,Citation35].
IPSC-CM dissociation
Preparations containing IPSC-CMs were dissociated 25 to 60 days after differentiation. Cells were first washed with a Ca2+ free buffer containing 120 mM NaCl, 5.4 mM KCl, 5 mM MgSO4, 5 mM Na-pyruvate, 20 mM Glucose, 20 mM Taurine, and 10 mM HEPES. They were then dissociated at 37°C using 0.25% trypsin for ~8 to 28 minutes. This reaction was then quenched with 10% FBS in the before mentioned Ca2+ free buffer. Cells were then resuspended in DMEM supplemented with 10% FBS, 1 mM Na-pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM l-glutamine. The cells were plated on 35-mm Petri dishes that had been coated with 0.1% gelatin. Single IPSC-CMs were identified based on beating status and/or morphology using a Nikon object marker and patch clamped 3 to 7 days after dissociation.
Single-cell electrophysiology
INaL was recorded as 1 μM tetrodotoxin (TTX)-sensitive current in previously described internal and external solutions [Citation34]. For recording INaL the external solution also contained 1 μM isradipine to block L-type Ca channels. INaP recordings used in and all steady state inactivation (SSI) recordings were recorded in a reduced Na+ external solution, containing 30 mM NaCl, 10 mM HEPES, 5 mM Glucose, 105 mM TEA-Cl, 2 mM CaCl2, and 1.2 mM MgCl2, pH was adjusted to 7.4 with TEA-OH. When studying INaP and SSI, the internal solution contained 10 mM NaCl, 2.5 mM Na2-ATP, 125 CsCl, 2 mM MgCl2, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, and 10 mM HEPES. pH was adjusted to pH 7.2 with CsOH. INaL and INa,P were measured as the average of the TTX-sensitive currents. INaL measurements were based on the last ~5 ms of a 100 ms pulse to −10 mV from a holding potential of −90 mV. The external solution for IKr measurement contained 132 mM NaCl, 4.8 mM KCl, 2 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, and 5 mM Glucose with 1 μM isradipine and 30 μM chromanol 293B added prior to recording. Pipette internal solution for IKr measurements contained 110 mM KCl, 5 mM ATP-K2, 11 mM EGTA, 10 mM HEPES, 1 mM CaCl2, and 1 mM MgCl2. All solutions were prepared using double distilled H2O.
Figure 1. Characterization of INaL in WT and patient derived ΔKPQ and E1784K IPSC-CMs. A. Representative TTX sensitive traces of total INa and INaL (inset) from WT, ΔKPQ, and E1784K IPSC-CMs. B. INaL of each cell line quantified as percentage of INaP. When two tailed unequal variance t-tests are performed on cells types (WT, ΔKPQ, and E1784K) as a whole, E1784K and ΔKPQ, had significantly more INaL than WT (p = 6.5638E-06 and 6.15E-05 respectively). E1784K also had significantly more INaL than ΔKPQ (p = 0.000235918). Currents were elicited by pulsing from a holding potential of −90 mV to −10 mV for 100 ms.
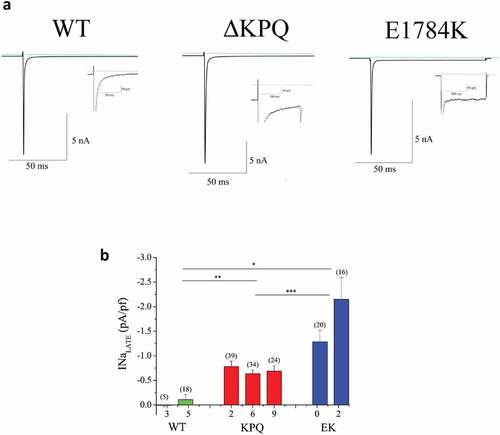
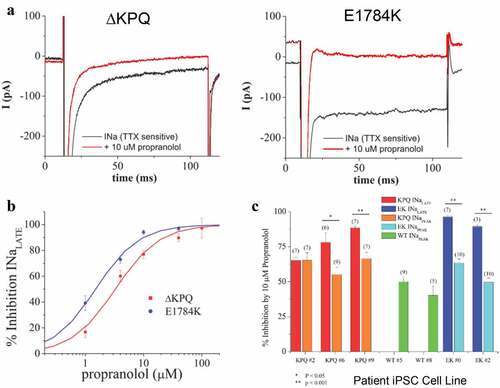
Figure 2. Propranolol modulation of INaL and INaP in patient derived ΔKPQ and E1784K IPSC-CMS. A. Representative high gain TTX-sensitive traces in the absence (black traces) and presence of 10 μM propranolol (red traces) reveal inhibition of INaL in ΔKPQ and E1784K IPSC-CMs. B. Concentration response curves for patient derived IPSC-CM total populations from three ΔKPQ patients (ΔKPQ2, ΔKPQ6, and ΔKPQ9) and two E1784K patients (EK0 and EK2). IC50 for propranolol inhibition of ΔKPQ patients’ cells combined is 3.3 μM. IC50 of propranolol inhibition of E1784K iPSC myocytes averaged is 1.5 μM. C. Propranolol (10 μM) inhibition of INaL and INaP for each cell line investigated. Currents were elicited using the protocol given in.
Statistics
Statistics were performed using in Excel 2016 Student’s t-test or Excel One Way ANOVA. When not specified, a significance p value threshold of 0.01 was used.
Data Availability
We shall share all data used in this study.
Results
INaL in cells derived from mutation-free (WT) and mutation carrying (ΔKPQ or E1784K) patients
NaV1.5 channel activity in WT and LQT3 patient-derived IPSC-CMs harboring either the NaV1.5 ΔKPQ or E1784K mutation was characterized by whole cell voltage clamp. As described in Methods, cells were held at −90 mV and pulsed to −10 mV for 100 ms () (top) with a 5s inter-pulse interval. Representative traces of current recorded at low and high gain are shown in ). Low gain records illustrate INaP measurements. For each cell studied high gain recordings are shown as insets. Note that INaL is clearly detected for ΔKPQ and E1784K expressing cells but not for the WT cell. INaL was measured as the average current (n = 9 each cell) remaining in the last 5 ms of the pulse and was determined as TTX-sensitive current. Averaged INaL was determined in each cell line as follows. We examined two WT IPSC-CM cell lines that had minimal late Na+ current (0.01 ± 0.03 and −0.11 ± 0.10 pA/pf in WT3 and WT5 IPSC-CM respectively). INaL was significantly larger than WT in all mutant expressing cells studied. For the ΔKPQ cell lines, all three had significant INaL(−0.78 ± 0.01, −0.64 ± 0.08, −0.91 ± 0.11 pA/pf ΔKPQ2, ΔKPQ6 and ΔKPQ9). In the E1784K mutant expressing IPSC-CMs, EK0 and EK2, INaL was −1.28 ± 0.24 and −2.15 ± 0.45 pA/pf, respectively. ), which illustrates the summary data for these experiments, indicates that cells expressing E1784K and ΔKPQ mutant channels had significantly more INaL than WT cells. Comparing total populations across cell lines, E1784K had significantly more INaL than the total ΔKPQ population (p = 2.4 E-04).
Propranolol inhibits INaL expressed in IPSC-CMs
We then investigated INaL inhibition by propranolol for concentrations ranging between 1 and 100 μM. Representative traces demonstrating inhibition by 10 μM propranolol for ΔKPQ and E1784K cells are shown in ). ) summarizes average percent inhibition of INaL measured in all cell lines that we studied along with best fit concentration response curves for all cell lines for both mutants. Concentration response curves were determined using a Hill equation that provided IC50 values. The fitted curves revealed the following IC50 values for inhibition of INaL: 3.36 ± 0.61 μM for cells expressing the ΔKPQ mutation, and 1.58 ± 0.04 μM for cells expressing the E1784K mutation. Inhibition of INaL ΔKPQ expressing cells was similar across all individual cell lines with the following IC50 values: 4.48 μM, 2.39 μM, and 3.21 μM, for ΔKPQ2, ΔKPQ6, and ΔKPQ9, respectively. In cells expressing E1784K mutant channels INaL in EK0 and EK2 was inhibited by propranolol with IC50 values of 1.48 μM and 1.56 μM, respectively (Please see ).
Table 1. INaL IC50 values for patient-derived IPSC-CM total populations from three ΔKPQ patients (ΔKPQ2, ΔKPQ6, and ΔKPQ9) and two E1784K patients (EK0 and EK2).
Propranolol inhibition of INaL is more potent than inhibition of INaP
We next measured the relative inhibition of INaL and INaP by propranolol applied at the high concentration of 10 μM and summarize the results for each cell line studied in ). For these recordings Na+ was reduced in the external solution (30 mM NaCl) to lower current INaP amplitude to maintain voltage control for measurement of INaP and still resolve INaL. On average, we found INaP expressed in WT, ΔKPQ, and E1785K IPSC-CMs was inhibited 46.31 ± 3.46, 61.39 ± 3.16, and 56.30 ± 2.7, respectively, by propranolol. ) illustrates the results of these experiments summarized for each cell line studied. Inhibition of INaL vs INaP was significantly greater in each of the E1784K cells lines (EK0 p = 3.65e-07 and EK2 p = 6.60e-05) and two of the three ΔKPQ IPSC-CM cell lines (p < 0.05, p = 0.02, and 0.01). However, propranolol inhibition of INaL vs INaP was not significantly greater in the ΔKPQ2 cell line.
Propranolol shifts SSI of mutant channels in the hyperpolarizing direction
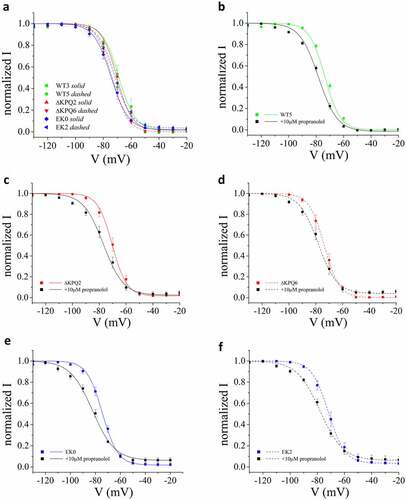
The voltage-dependence of steady state inactivation will impact the effects of propranolol on INaP as demonstrated in previous experiments in which Na+ channels were studied using heterologous expression [Citation36]. Since we found INaP to be more sensitive to propranolol inhibition in cells expressing ΔKPQ vs. E1784K Na+ channels vs. cells expressing WT channels, we next investigated the voltage dependence of steady state Na+ channel inactivation (SSI) and the effects of propranolol on SSI in each cell line studied. Interestingly, we found on average, a negative shift of about −5 mV of SSI measured in E1784K expressing cells but no shift in SSI ΔKPQ expressing cells ()). The effects of these mutations on the Voltage-dependence of NaV1.5 channel SSI in iPSC myocytes is less than the effects on SSI reported in heterologous expression systems [Citation25–27,Citation36]. As such, we replicated the observed SSI in heterologous expression to understand if it was due to changes in solutions in our study or something endogenous in iPSC myocytes (Supplemental Figure S1). These results suggest that this effect was not due to our experimental solutions. The propranolol-induced shift in SSI for WT, ΔKPQ, and E1784K cells is illustrated in panels B through F. This change in the voltage-dependence of SSI contributes to propranolol inhibition of INaP.
Figure 3. Propranolol causes a negative shift in SSI. A. SSI measured in all WT, ΔKPQ, E1784K cell lines as indicated in the figure. B-D. The influence of propranolol (10 μM) on SSI for all cell lines studied.
Propranolol inhibition of IKr is less potent than inhibition of INaL
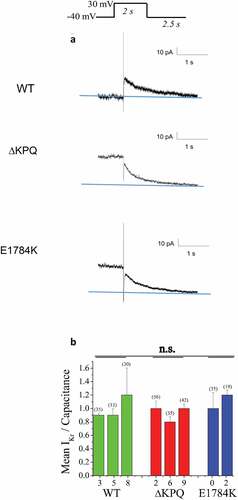
It is well established that off target block of IKr channels by multiple drugs underlies at least part of drug-induced LQT [Citation37,Citation38] and thus we investigated the effects of propranolol on IKr in iPSCs expressing E1784K and ΔKPQ channels. ) illustrates measurement of IKr in each cell line we studied and shows representative IKr current tail traces for each (WT, ΔKPQ, and E1784K cells from top-to-bottom). ) shows the summary data for peak (initial) IKr tail amplitude measured at −40 mV following 2 second activation pulses to +10 mV for each line studied. IKr expression was evident in all lines studied. Expression of this key potassium channel in iPSC myocytes is a very valuable characteristic of these cells because it allows testing for off target effects on these channels in the same cells in which drug modulation of INaL is investigated.
Figure 4. Expression of IKr in WT and LQT3 patient derived ΔKPQ and E1784K IPSC-CMS. A. Representative IKr traces in WT, ΔKPQ, and E1784K IPSC-CM. IKr was determined as E4031-sensitive current in all experiments. The holding potentials were −40 mV. Currents were elicited by pulsing from this holding potential to 30 mV for 2 s. E4031 sensitive peak tail currents following return to the holding potential after the 30 mV pulses were used for measurements and quantification. B. Bar graph summary of IKr density measured in the number of cells indicated in the figure. There is no significant difference in IKr density among the groups when cell total populations are grouped into WT, ΔKPQ, and E1784K.
We next investigated the effects of propranolol on IKr expression and have summarized our findings in . ) illustrates representative IKr tail traces recorded in the absence (black traces) and presence of 10 μM propranolol (red traces) for cells expressing WT, ΔKPQ, and E1784K channels. Inhibition by propranolol was measured as a function of reduced E4031 sensitive peak tail current as a function of propranolol concentration. ) illustrates average IKr inhibition as a function of propranolol concentration for each cell type studied. The average data were then fitted with the Hill equation yielding average IC50 values for each cell type. Propranolol inhibition IC50 values for IKr in cells expressing WT and mutant NaV1.5 channels ranged from 6.98 μM to 17.39 μM (). However, when recordings were pooled and averaged across cell lines the IC50 values extracted from the averaged IKr propranolol inhibition data were 11.35 μM propranolol for cells expressing WT channels, 10.68 μM for cells expressing ΔKPQ channels, and 13.12 μM for cells expressing E1784K channels ()).
Figure 5. Propranolol inhibits IKr in WT and LQT3 patient derived ΔKPQ and E1784K IPSC-CM cells. A. Representative IKr traces recorded as E4031-sensitive current before (black traces) and after application of 10 μM propranolol (red traces) recorded in WT, ΔKPQ, and E1784K IPSC-CMS. B. Dose response curves for total patient derived IPSC-CM populations from 2 different WT donors (WT3, WT8), 3 different LQT3 patients harboring the ΔKPQ deletion (ΔKPQ2, ΔKPQ6, and ΔKPQ9) and 2 different E1784K mutation carrying LQT3 patients (EK0 and EK2). Propranolol IC50 for IKr inhibition in combined cell populations: WT IPSC-CM = 11.35 μM; ΔKPQ IPSC-CMs = 10.68 μM; E1784K IPSC-CMs = 13.12 μM.
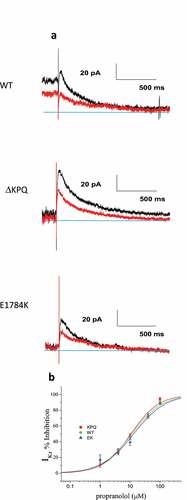
Table 2. IKr IC50 values for patient-derived IPSC-CM populations from two different WT donors (WT3, WT8), three different LQT3 patients harboring the ΔKPQ deletion (ΔKPQ2, ΔKPQ6, and ΔKPQ9), and two different E1784K mutation carrying LQT3 patients (EK0 and EK2).
Discussion
Congenital long-QT syndrome (LQTS) is now recognized as one of the most common inherited arrhythmia syndromes with 17 different genetic subtypes [Citation5,Citation33]. Beta blockers are now the primary therapy for LQTS [Citation3,Citation39]. As the genetics of congenital LQTS developed and it was clear that the risk of cardiac events for LQTS mutation carriers was dependent on the mutated gene and that for LQT3 patients, arrhythmia risk was most pronounced during bradycardia or rest [Citation16,Citation40,Citation41]. Following identification of bradycardia as a trigger in LQT3, this variant was not considered amendable to beta-blocker therapy and early clinical studies showed no clear benefit of beta blockers [Citation5].
Nonetheless, subsequent preclinical and clinical studies continued to test the antiarrhythmic activity of β-blockers in the treatment of LQT3 patients. These studies have focused on two of the most studied LQT-3 mutations: the ΔKPQ mutation and the E1784K mutation. These two Na+ channel mutations are the focus of the present study. In experiments using an established mouse model for LQT3 in which the efficacy of propranolol on preventing arrhythmias in ΔKPQ-SCN5A knock-in mice, Calvillo et al. found that β-blockade effectively prevented ventricular arrhythmias in this mouse model [Citation30]. However, the mechanism underlying this antiarrhythmic action via anti-adrenergic effects of propranolol or via propranolol inhibition of INaL was not investigated in this study [Citation31]. In addition, a major clinical study testing the effectiveness of propranolol on arrhythmias in a large multicenter study of LQT3 patients in which the largest number of mutation carriers were carriers of ΔKPQ mutation carriers (66) and E1784K mutation carriers (70). The major conclusion of this study was that β-blocker therapy reduced the risk of cardiac events in female patients with too few male patients participating with cardiac events [Citation32].
To gain further mechanistic insights into these pre-clinical and clinical investigations of the effects of propranolol on Na+ channels with ΔKPQ or E1748K mutations or patients harboring these mutations, we investigated the effects of propranolol on Na+ channels expressed in iPSC myocytes derived from patients with these LQT3 mutations. We found that propranolol indeed targets INaL with half maximal inhibitory concentrations of 3.36 μM (ΔKPQ) and 1.58 μM (E1784K). This supports its usefulness of treating LQT3 patients [Citation14]. Propranolol also causes a negative shift of Na+ channel SSI that reduces the availability of Na+ channels at diastolic potentials for the generation of impulse conduction via INaP. Interestingly, our measured IC50 for propranolol INaL inhibition in iPSC myocytes agrees well with measurement of its inhibition of INaL carried by transfected mammalian cells expressing ΔKPQ channels (IC50 = 2.4 μM) [Citation36]. Importantly, the half maximal concentrations for INaL inhibition of INaL were found to be 3.2 (E1748K) and 8.3 (E1748K) times lower than propranolol inhibition of the inwardly rectifying HERG K+ channel current IKr. These results indicate that propranolol can be effective and safe in treating LQT3 patients with these common mutations, based at least in part on its inhibition of INaL provided concentrations used in treating LQT3 patients are in the range of the IC50 values that we have measured for INaL inhibition. A similar propranaolol concentration range has been shown effective at INaL inhibition in iPSC myocytes expressing a different LQT3 mutation, SCN5A-N1774D [Citation42]. We note that these concentrations seem high compared with multiple reports of clinically used doses of propranolol. For example, in a classic study of the clinical pharmacology of propranolol, concentration ranges of up to 800 nM propranolol were reportedly [Citation43]. However, multiple studies of patients investigated in clinical studies have reported serum concentrations over broad ranges from low concentrations of 0.24 μM [Citation44] and 0.36 μM [Citation45] to moderate and high concentrations of 1.54 μM [Citation46]. In this important context Roden and Colleagues have studied concentration-response effects of propranolol on electrophysiological parameters in human subjects and found antiarrhythmic activity at propranolol concentrations of 474 ng/ml (1.83 μM) which is on the order of the IC50 values measured for propranolol INaL inhibition we report here [Citation47], a finding expanded by Ahrens-Nicklas and Clancy, using computational modeling [Citation48]. Our work thus provides experimental evidence that propranolol inhibits INaL in this concentration range, and that, in this range of concentrations, INaL inhibition is not accompanied by significant inhibition of IKr off target effects.
Supplemental Material
Download MS Word (195.8 KB)Acknowledgments
We would like to thank Barbara Corneo, PhD, Dario Sirabella, PhD, and the rest of the Columbia Stem Cell Core for producing the IPSC-CMs used in this study. We would like to thank Professor Michael R. Rosen M.D., for fruitfull discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19336950.2022.2106025
Additional information
Funding
References
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54(1):59–68.
- Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783–790.
- Ackerman MJ, Priori SG, Dubin AM, et al. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: are all beta-blockers equivalent? Heart Rhythm. 2017;14(1):e41–e44.
- Schwartz PJ, Ackerman MJ, Wilde AAM. Channelopathies as Causes of Sudden Cardiac Death. Card Electrophysiol Clin. 2017;9(4):537–549.
- Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol. 2018;4(5):569–579.
- Bohnen MS, Peng G, Robey SH, et al. Molecular pathophysiology of congenital long QT syndrome. Physiol Rev. 2017;97(1):89–134.
- Keating M. Linkage analysis and long QT syndrome. Using genetics to study cardiovascular disease. [Review]. Circulation. 1992;85(6):1973–1986. J1 - Circ.
- Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23.
- Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity [see comments]. Nat Genet. 1994;8(2):141–147.
- Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803.
- Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62(4):760–781.
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469.
- Wang Q, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805–811.
- Moreno JD, Clancy CE. Pathophysiology of the cardiac late Na current and its potential as a drug target. J Mol Cell Cardiol. 2012;52(3):608–619.
- Makielski JC. Late sodium current: a mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc Med. 2016;26(2):115–122.
- Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89–95.
- Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–2867.
- Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115(8):2018–2024.
- Papa A, Kushner J, Marx SO. Adrenergic regulation of calcium channels in the heart. Annu Rev Physiol. 2022;84(1):285–306.
- Barsheshet A, Goldenberg I, O-Uchi J, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation. 2012;125(16):1988–1996.
- Goldenberg I, Thottathil P, Lopes CM, et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm. 2012;9(1):49–56.
- Mullally J, Goldenberg I, Moss AJ, et al. Risk of life-threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm. 2013;10(3):378–382.
- Priori SG, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292(11):1341–1344.
- Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101(6):616–623.
- Wei J, Wang DW, Alings M, et al. Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na + channel. Circulation. 1999;99(24):3165–3171.
- Deschenes I, and Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46(1):55–65.
- Makita N, Behr E, Shimizu W, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118(6):2219–2229.
- Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376(6542):683–685.
- An RH, Bangalore R, Rosero SZ, et al. Lidocaine block of LQT-3 mutant human Na + channels. Circ Res. 1996;79(1):103–108.
- Calvillo L, Spazzolini C, Vullo E, et al. Propranolol prevents life-threatening arrhythmias in LQT3 transgenic mice: implications for the clinical management of LQT3 patients. Heart Rhythm. 2014;11(1):126–132.
- Fabritz L, Franz MR, Carmeliet E, et al. To the Editor–Propranolol, a beta-adrenoreceptor blocker, prevents arrhythmias also by its sodium channel blocking effect. Heart Rhythm. 2014;11(3):e1.
- Wilde AA, Moss AJ, Kaufman ES, et al. Clinical aspects of type 3 long-QT syndrome: an International multicenter study. Circulation. 2016;134(12):872–882.
- Ahn J, Kim HJ, Choi J-I, et al. Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: a meta-analysis. PLoS One. 2017;12(10):e0185680.
- Terrenoire C, Wang K, Chan Tung KW, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141(1):61–72.
- Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471(7337):230–234.
- Bankston JR, Kass RS. Molecular determinants of local anesthetic action of beta-blocking drugs: implications for therapeutic management of long QT syndrome variant 3. J Mol Cell Cardiol. 2010;48(1):246–253.
- Roden DM, Wood AJJ. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350(10):1013–1022.
- Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci. 2005;26(3):119–124.
- Mizusawa Y, Horie M, Wilde AA. Genetic and clinical advances in congenital long QT syndrome. Circ J. 2014;78(12):2827–2833.
- Clancy CE, Tateyama M, Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 2002;110(9):1251–1262.
- Schwartz PJ. Management of long QT syndrome. Nat Clin Pract Cardiovasc Med. 2005;2(7):346–351.
- Hirose S, Makiyama T, Melgari D, et al. Propranolol attenuates late sodium current in a long QT syndrome type 3-human induced pluripotent stem cell model. Front Cell Dev Biol. 2020;8:761.
- Nies AS, Shand DG. Clinical pharmacology of propranolol. Circulation. 1975;52(1):6–15.
- Salehifar E, Ebrahim S, Shiran M-R, et al. Pharmacokinetic parameters and over-responsiveness of Iranian population to propranolol. Adv Pharm Bull. 2017;7(2):195–202.
- Wojcicki J, Jaroszynska M, Droździk M, et al. Comparative pharmacokinetics and pharmacodynamics of propranolol and atenolol in normolipaemic and hyperlipidaemic obese subjects. Biopharm Drug Dispos. 2003;24(5):211–218.
- Cales P, Grasset D, Ravaud A, et al. Pharmacodynamic and pharmacokinetic study of propranolol in patients with cirrhosis and portal hypertension. Br J Clin Pharmacol. 1989;27(6):763–770.
- Duff HJ, Roden DM, Brorson L, et al. Electrophysiologic actions of high plasma concentrations of propranolol in human subjects. J Am Coll Cardiol. 1983;2(6):1134–1140.
- Ahrens-Nicklas RC, Clancy CE, and Christini DJ. Re-evaluating the efficacy of {beta}-adrenergic agonists and antagonists in long QT-3 syndrome through computational modelling. In Cardiovasc Res. 2009. Vol. 82(3). p. 439-47.