
Abstract
QBP359 is an IgG1 human monoclonal antibody that binds with high affinity to human CCL21, a chemokine hypothesized to play a role in inflammatory disease conditions through activation of resident CCR7-expressing fibroblasts/myofibroblasts. The pharmacokinetics (PK) and pharmacodynamics (PD) of QBP359 in non-human primates were characterized through an integrated approach, combining PK, PD, immunogenicity, immunohistochemistry (IHC) and tissue profiling data from single- and multiple-dose experiments in cynomolgus monkeys. When compared with regular immunoglobulin typical kinetics, faster drug clearance was observed in serum following intravenous administration of 10 mg/kg and 50 mg/kg of QBP359. We have shown by means of PK/PD modeling that clearance of mAb-ligand complex is the most likely explanation for the rapid clearance of QBP359 in cynomolgus monkey. IHC and liquid chromatography mass spectrometry data suggested a high turnover and synthesis rate of CCL21 in tissues. Although lymphoid tissue was expected to accumulate drug due to the high levels of CCL21 present, bioavailability following subcutaneous administration in monkeys was 52%. In human disease states, where CCL21 expression is believed to be expressed at 10-fold higher concentrations compared with cynomolgus monkeys, the PK/PD model of QBP359 and its binding to CCL21 suggested that very large doses requiring frequent administration of mAb would be required to maintain suppression of CCL21 in the clinical setting. This highlights the difficulty in targeting soluble proteins with high synthesis rates.
Abbreviations
| ADME | = | absorption, distribution, metabolism, and elimination |
| ABC | = | ammonium bicarbonate |
| ACN | = | acetonitrile |
| ADA | = | anti-drug antibodies |
| AUC | = | area under the curve |
| BSA | = | bovine serum albumin |
| CCL | = | chemokine (C-C) ligand |
| CCR7 | = | C-C chemokine receptor 7 |
| DOC | = | sodium deoxycholate |
| DRF | = | dose range finding |
| ELISA | = | enzyme-linked immunosorbent assay |
| FA | = | formic acid |
| FcRn | = | neonatal Fc receptor |
| FFPE | = | formalin fixed paraffin embedded |
| HRP | = | horseradish peroxidase |
| IAA | = | iodoacetamide |
| Ig | = | immunoglobulin |
| IG | = | immunogenicity |
| IHC | = | immunohistochemistry |
| IL | = | interleukin |
| LC-MS/MS | = | liquid chromatography-mass spectrometry/mass spectrometry |
| LLOQ | = | lower limit of quantification |
| MRD | = | minimal required dilution |
| PBS | = | phosphate buffered saline |
| PD | = | pharmacodynamic |
| PK | = | pharmacokinetic |
| QC | = | quality control |
| RT | = | room temperature |
| SIL-IS | = | stable isotope labeled peptide –internal standard |
| TCEP | = | triphosphine hydrochloride |
| TMB | = | 3,3′,5,5′-tetramethylbenzidine |
| ULOQ | = | upper limit of quantification |
| VH | = | variable heavy chain |
Introduction
Chemokines and their receptors are key for the recruitment of leukocytes to sites of inflammation and to the tumor microenvironment. In chronic inflammatory diseases, CCL2/CCR2, as well as other chemokines, have been reported to be important in various chronic inflammatory conditions that are associated with macrophage infiltration, such as atherosclerosis, multiple sclerosis, rheumatoid arthritis, pulmonary fibrosis, and chronic obstructive pulmonary disease.Citation1-5 Chemokines have also been demonstrated to be implicated in the regulation of tumor cell migration resulting in the development and metastasis of several adenocarcinomas, including breast and prostate.Citation6,7 Furthermore, studies in multiple myeloma have revealed a pivotal role for CCL2 in the “bone homing” phenotype of myeloma cells to the bone marrow compartment.Citation8 Thus, targeting chemokines is relevant for inflammatory diseases and cancer.
Of the same family as CCL2, the CC chemokine receptor 7 (CCR7) and its ligands, CCL19 and CCL21, have been less in the spotlight as chemokines for drug development. They play an important role in immunity, through regulating the homing of subpopulations of T cells and antigen-presenting dendritic cells (DCs) to secondary lymphoid organs. CCL19 and CCL21 are primarily produced in secondary lymphoid organs by endothelial venule cells and by fibroblastic reticular cells where their role is to direct the migration of CCR7-expressing cells.Citation9,10 CCR7/CCL21 is pivotal for lymphocyte homing to lymph nodes and is therefore a target for disease therapy of inflammation-related conditions. CCL21 can drive the recruitment of circulating fibrocytes to sites of micro-injury in the lung and contribute to the generation of the excessive fibroblast/myofibroblast population in disease.Citation11-13 Furthermore, CCL21 may also play a role in the local environment by activating resident CCR7-expressing fibroblasts/ myofibroblasts. Although CCL21 was of interest due to its role in inflammation-related indications, strategies used to suppress chemokine production and the maintenance of protein levels are of general interest from a drug development perspective.
During preclinical drug development, data is used to predict the pharmacokinetics and the pharmacological effect of the therapeutic in the clinical setting. This is often described as the pharmacokinetic – pharmacodynamics (PK/PD) relationship.Citation14 A holistic view of the target expression, drug distribution, the drug kinetics, and the pharmacological effects are integrated to better understand whether the therapeutic is going to provide the expected outcome in the clinical setting.Citation15,16 Using this knowledge, it is then possible to decide early on before entering into the clinical phase if the therapeutic will be successful. In this article, we demonstrate the use of such assessment to make a decision on whether the chemokine target, CCL21, can be suppressed in the clinical setting. Our experience has implications for drug development directed toward other chemokine targets as well due to the similarity in approaches for suppressing soluble targets with high turnover rates.
Results
General safety
QBP359 was considered well-tolerated as there were no adverse clinical signs or changes in food consumption, body weight or routine clinical pathology parameters attributable to treatment with QBP359. Wound sites remained unchanged following treatment with QBP359. In the absence of any adverse microscopic findings, minor changes in spleen and thyroid weights were considered of no toxicological significance.
QBP359 biodistribution analysis using IHC
Biodistribution analysis was performed to show the distribution of QBP359 in the main tissues with particular emphasis on lymphatic tissues. Staining with the detection antibody (goat anti-human IgG Fc fragment) that recognizes the Fc component of all human IgGs, including QBP359, was clearly evident and found in all lymph nodes (mesenteric, mandibular, accessory lymph node of the pancreas and lymph node of the injection site) and spleen () in treated animals compared to control tissue.
Figure 1. (a) (above) Staining on paraffin-embedded SP20cells (positive control material) with (A) negative detection antibody and (B) goat anti-human IgG Fc fragment specific. Figure 1b (below) Biodistribution with anti-human IgG Fc fragment specific detection antibody in Lymph Nodes of (A) control animal and (B) animal treated with QBP359 or Spleen of (C) control animal and (D) animal treated with QBT359.
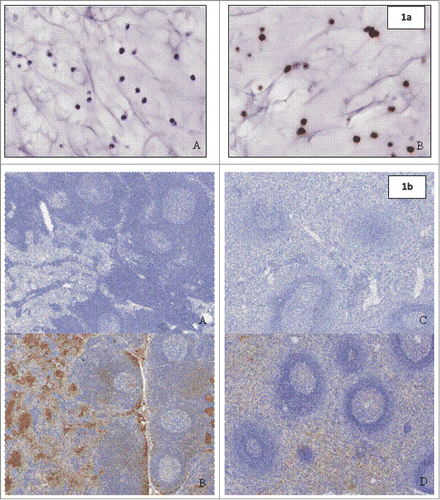
In the lymph node, staining was present in the lymphatic spaces of the medullary and subcapsular sinuses and on the membrane/cytoplasm of cells, consistent with inflammatory cells, in the parafollicular and medullary zones and the occasional germinal center. In the spleen, prominent staining was observed in the extracellular spaces and on the membrane/cytoplasm of cells, consistent with inflammatory cells, within the red pulp, the marginal zone and germinal centers of lymphoid follicles in the white pulp. Of all of the tissues examined in this study, the lymph nodes and spleen represent the primary organs of biodistribution for QBP359, which probably reflects the known high CCL21 expression levels in these tissues.Citation17
The presence of QBP359 was also confirmed through LCMS/MS analysis in the spleen and lymph nodes, as well as the absence in liver and kidney (see Figure S1 for details).
CCL21 tissue expression analysis and QBP359 blood protein binding
Tissue expression analysis in non-treated animals showed that CCL21 was expressed highly in the lymph nodes and the spleen (Fig. S2). Additional recovery analysis of QBP359 binding in whole blood indicated blood proteins did not illustrate any non-specific binding, and thus would not be responsible for the rapid clearance of QBP359 (data not shown).
Pharmacokinetics
In the dose-range-finding (DRF) study, exposure to QBP359 (as measured by AUC(0–7d) and Cmax) increased between the doses of 10 and 50 mg kg−1 as expected (). The increase in Cmax was approximately dose proportional after the first dose whereas the increase in AUC was greater than dose proportional, indicating an accumulation of QBP359. This was reflected in the comparisons between first and last dose for Cmax/Dose and AUC0-last/Dose. Mean beta half-lives were also shorter at the 10 mg/kg dose compared to the 50 mg/kg dose after both the first and the last dose. Administration of 3 μg of CCL21 to the wound site did not have any effect on the exposure. The rapid clearance of total QBP359 was the first indication that either the target (CCL21) may be affecting the clearance of the antibody or the presence of an off-target binding phenomenon.
Table 1. PK parameters for multiple dose cynomolgus monkey study
Single dose PK study
The DRF in monkeys did not provide sufficient data for a full evaluation of QBP359 and total CCL21 because the animals were sacrificed early for toxicological evaluations. A subsequent single dose PK study was performed to evaluate longer sampling times. Results of this study confirmed the rapid non-linear clearance of QBP359 and investigated the bioavailability of the drug.
Cynomolgus monkeys were administered 10 mg kg−1 intravenous (i.v.) or subcutaneous (s.c.) or 50 mg kg−1 i.v. doses of QBP359. Sampling was followed up to 56 days, and data was reported for all values above the LLOQ of the total monoclonal antibody (mAb) assay. PK profiles for both s.c. and i.v. administration of 10 mg kg−1 were similar except for the absorption phase, and, as expected, an increase in drug levels was observed for 50 mg kg−1 (Fig. S3).
Animals from all treatment groups showed an increase in total CCL21 starting from 0.5 h following drug administration (Fig. S3). Concentration levels of CCL21 were maintained at a plateau up to 21 d following a single administration of 50 mg kg−1 QBP359 whereas the 10 mg kg−1 dose decreased 4 d following drug administration. The maximum level of CCL21 reached was 210 nM and 343 nM after i.v. administration of QBP359 of 10 mg kg−1 and 50 mg kg−1, respectively. There was a small increase in AUC/D, but not in Cmax of QBP359 between doses 10 and 50 mg kg−1 ().
Table 2. Mean PK parameters from single dose study in cynomolgus monkeys
The beta t1/2 values after the QBP359 i.v. doses were 4.6 and 5.1 d for the 10 mg kg−1 dose and 50 mg kg−1 doses, respectively, and averaged 6.5 d after the 10 mg kg−1 s.c. dose. Other PK parameters were evaluated, as shown in . Bioavailability was calculated by using the 10 mg kg−1 dose as follows: Fsc= AUC(0-last)s.c./AUC(0-last)i.v. * 100% to give 52%. This demonstrates that the tissue absorption of the drug is within the range of a normal IgG biotherapeutic.
Pharmacodynamics and immunogenicity
PK/PD analysis of the DRF study was explored to better understand CCL21 suppression upon repeat QBP359 administration. Total concentration levels of CCL21 showed complementary profiles to the PK profiles (). Concentration levels of total CCL21 increased after drug administration of 10 and 50 mg kg−1 up to 96 hours after the first dose administration. A gradual decrease in total CCL21 levels was observed for the low dose group before the second injection on Day 8, and trough levels steadily increased after subsequent dose administrations. For the high doses, total CCL21 appeared to reach a maximum level after a single dose, and this level remained constant after each weekly injection with a small accumulation over time. Overall, an accumulation between first and fourth dose was observed to go from 158 to 282 nM and from 199 to 494 nM after administration of 10 mg kg−1 and 50 mg kg−1 (both high dose groups excluding one animal with immunogenicity), respectively.
Figure 2. (a) PK/PD profiles following weekly i.v. administration (shown with arrows) of QBP359 in Cynomolgus monkeys dosed at 10 mg kg−1 and 50 mg kg−1 expressed in nM (n = 4). Standard deviations are shown with the error bars. (b) PK/PD profiles following weekly i.v. administration of QBP359 in Cynomolgus monkeys dosed at 50 mg kg−1 expressed in nM for one animal with high levels of immunogenicity after the third dose administration.
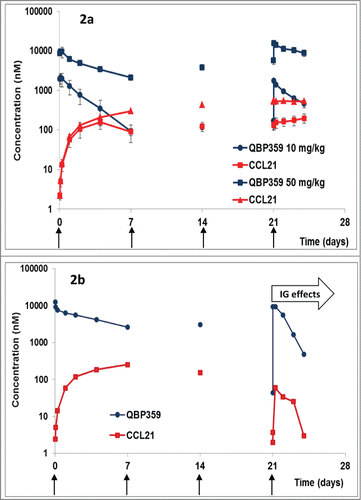
Plots of total QBP359 and total CCL21 () suggested that the antibody was not able to saturate CCL21 in the 10 mg kg−1 cohort. This data demonstrates that CCL21 was present at very high steady state concentrations in monkeys, since the total CCL21concentrations were nearly equal to QBP359 at the PK trough values for the 10 mg kg−1 cohort. However, in the 50 mg kg−1 cohort, the plots suggest that QBP359 had likely saturated the target and was present at concentrations in excess of the target ().
One animal of Group IV (50 mg kg−1) displayed a marked decrease in total CCL21 after the fourth weekly injection, which mirrored the decline in pharmacokinetics in this animal and a decline in CCL21. This was most likely due to the production of anti-QBP359 antibodies as the animal tested positive for immunogenicity (). This animal was not included in the mean group calculations following the fourth dose.
One other animal (10 mg kg−1) tested weakly positive for anti-QBP359 antibodies after the fourth dose. However, anti-QBP359 antibody activity appeared to be marginal and PK/PD parameters were not affected.
PK/PD modeling
In order to further understand the kinetics of QBP359 and CCL21 capture, a PK-binding model was used to characterize the non-linear PK and total CCL21 PD data from the 4-week DRF toxicity study (). Synthesis of CCL21 was specified in the peripheral tissue compartment; this was further confirmed by gene expression and direct measurement of CCL21 protein levels in lymph nodes and spleen. As IHC of tissue cross-sections demonstrated that the antibody accumulated in the peripheral lymph nodes, antibody-target binding was allowed to occur in both serum and tissue compartments.
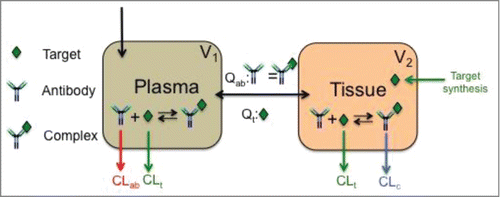
Figure 3. QBP359 – CCL21 PK- binding model structure. A two compartmental binding model structure was utilized. Compartment (volume, V1) represents the serum compartment and compartment (volume, V2) represents the peripheral tissue compartment. CCL21 was assumed to be synthesized in the peripheral tissue compartment at the rate of RLIt. QBP359 – CCL21 binding was assumed to occur in the both the serum and tissue compartment. Clearance of QBP359 (CLab), CCL21 (CLt) and complex (CLc) were assumed to be different. Inter-compartmental transfer rates for QBP359 and complex were assumed to be equal (Qab), but were assumed to be different from CCL21 (Qt).
Both fitting the data to the model (Fig. 4) and the diagnostics (Supplemental Material) demonstrates that the model fits the PK and total CCL21 data adequately. Estimated model parameters (, Supplemental Material) indicated that CCL21 had a high-turnover with a high synthesis rate and rapid clearance. The estimated model parameters also highlighted that the complex of QBP359 and CCL21 was cleared more rapidly than the free antibody.
Figure 4. PK- binding model fits to total QBP359 (top) and total CCL21 (bottom) from 4 week dose-range finding data. Lines represent model predictions, and x represents data.
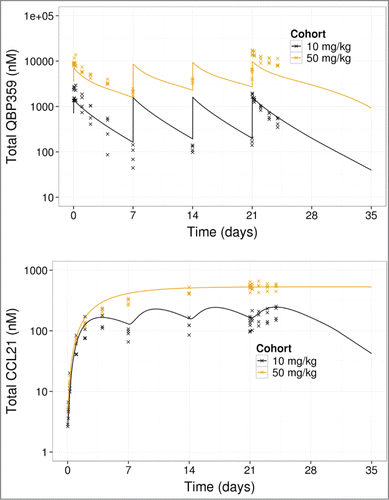
Table 3. Parameters for cynomolgous monkey QBP359 – CCL21 model
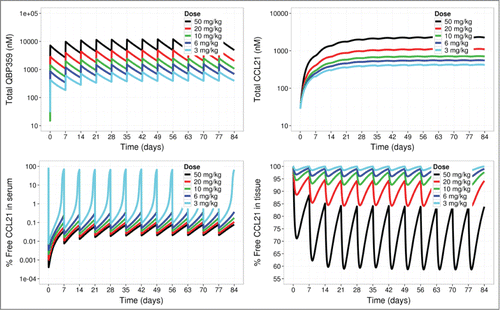
In order to perform a clinical feasibility analysis for QBP359 targeting CCL21, the PK-binding model was scaled up from monkeys to pulmonary fibrosis patients, accounting for ∼10-fold higher CCL21 levels in patients than in the monkey. Citation18 The scaled model for patients was then used to simulate CCL21 neutralization for weekly i.v. infusions of QBP359. Model simulations suggested that nearly 6 mg kg−1 of QBP359 would be required to maintain >90% neutralization of CCL21 in the serum (). In contrast, simulations showed that even very high weekly doses of 50 mg kg−1 could not completely neutralize CCL21 in the peripheral tissue compartment (maximum of 60% neutralization) throughout the duration of the therapy. Together, these model predictions suggested that QBP359 would be unlikely to neutralize CCL21 effectively in the bone marrow, lung and other tissues of patients.
Figure 5. Predictions of PK- binding model based on a typical pulmonary fibrosis patient. Once-a-week intravenous infusions of QBP359 (3 mg kg−1, 6 mg kg−1, 10 mg kg−1, 20 mg kg−1 and 50 mg kg−1) were applied to the model. Simulations of total QBP359 in serum (top left), total CCL21 (top right),% free CCL21 in serum (bottom left) and % free CCL21 in peripheral tissue (bottom right) are shown.
Discussion
In human disease states, where CCL21 expression is believed to be expressed at much higher concentrations compared to baseline levels, high frequent doses of mAb would be required to maintain suppression of CCL21 in the clinical setting, thus emphasizing the difficulty in targeting soluble proteins with high synthesis rates. The excessive production and release of chemokines appears to be a common characteristic, and new strategies are needed to suppress chemokine levels more effectively.
The high total target levels consist of free antibody and antibody-chemokine complexes that are thought to be maintained through the high synthesis rate and the release of extracellular stores bound to GAG or potentially scavenger receptors.Citation19-22 Chemokines are present at high concentrations locally and are known to be produced rapidly as part of the innate immune response. It is not only the production of chemokine, but the maintenance of circulating complexes that leads to high levels of total chemokine. The complexes may be multimer complexes with more than one antibody–chemokine pair per complex. This can be observed if the chemokine is present as a homodimer where each arm of an antibody binds one end of the dimer and forms multi-antibody-chemokine complexes. Such theories are currently being explored as a strategy for enhancing chemokine removal.Citation23 Bioengineers can modify the binding affinity of biotherapeutic mAbs to bind with a higher affinity to the FcγRIIb receptors to enhance complex-mediated clearance and reduce the amount of circulating chemokine complexes. This approach has yet to show its ability to reduce the doses required in a clinical setting.
Another related approach for reducing soluble target levels is to enhance the dissociation of the target from the mAb to allow FcRn recycling for further target binding. Dissociation of the target chemokine from the antibody during normal FcRn-protected endosomal recycling has been described for sIL6-R and PCSK9.Citation24,25 In this approach, the antibody is engineered with a lower binding affinity to the target ligand at pH6.0, which results in dissociation of the antibody-ligand complex during normal endosomal recycling. The antibody is protected from degradation via tight binding to FcRn at pH6.0, whereas the target ligand is released and destroyed. Enhanced affinity in the FcRn region of the antibody can also affect antibody recycling. This “antigen sweeping” of antibody-ligand complex releases the antibody to bind more ligand, and can reduce the dose and dosing frequency required for effective ligand suppression.
High CCL21 turnover rates appear to be the most likely reason why drug-target complex levels increased upon dose administration and upon repeat dosing. The 50 mg kg−1 dose level was capable of saturating free CCL21 since the levels of total drug versus total CCL21 did not match until drug levels dropped to equivalent molar ratios, indicating significant levels of unbound QBP359. Starting from 3 weeks, both total drug and total target levels were cleared at similar rates. It is interesting that the curve shape of total CCL21 levels contrasts with the 10 mg/kg dose levels and those observed for MCP-1,Citation26 where bell-shaped curves with a well-defined Cmax are evident. Here, we observed that the levels of CCL21 are maintained at a constant maximal level of 353 nM for 2 weeks after administration of 50 mg kg−1, and thus Cmax levels remain constant rather than peaking at a single time. It would appear that this level represents the maximum amount of CCL21 that is synthesized and released from intracellular stores or from hypothetical extracellular deposits. This supports the notion that the drug saturates CCL21 at 50 mg kg−1 and the maximum turnover levels are reached. Curiously, multiple dosing showed that CCL21 levels increased from 158 to 282 nM and from 199 to 494 nM after 4 doses of 10 mg kg−1 and 50 mg kg−1, respectively. This would indicate that CCL21 complexes can accumulate after multiple dosing on a weekly basis, where the production rate is faster than its elimination in a one-week period.
In addition to elimination through the FcRn pathway, drug-target complexes can be eliminated through alternative pathways. Unbound drug demonstrated normal mAb kinetics, as shown by the high dose administration (50 mg kg−1) of QBP359 during the first 21 d until reaching equivalent molar ratios of drug and target. Once bound to target, increased clearance was observed, which was demonstrated through modeling. Clearance of the drug –target was estimated to be 0.11102 L day−1, which is considerably faster than a typical IgG, ranging from 0.015 to 0.036 L day−1.Citation27 Xiao et al. reported a study of hepcidin kinetics where total hepcidin levels increased with increasing doses of Ab12B9m.Citation28 The authors developed a model where both the FcRn and the complex-mediated elimination pathways play a role in the PK of Ab12B9m. It was demonstrated that the rate of elimination through complex-mediated pathways was faster than the FcRn pathway, and thus contributed most strongly to complex elimination. Also, hepcidin was found to be saturable at 300 mg kg−1 in contrast to CCL21, which was saturable at 50 mg kg−1. Additional PK/PD models exist for other drugs such as carlumab, that also focus on complex-mediated clearance.Citation29
Many similarities of the PD behavior of CCL21 can be seen with CCL2 (MCP-1). Studies with ABN912 for treatment of rheumatoid arthritis patients showed that CCL2 levels increased with dose.Citation26 This result is similar to what was observed with QBP359 after i.v. administration, although only 2 dose levels were explored. A similar effect, which was described to be due to high target turnover of CCL2, was observed with carlumab (CNTO 888) for the treatment of solid tumors.Citation30 Although carlumab was able to suppress CCL2 levels below baseline initially, it rapidly increased above baseline levels and demonstrated an accumulation of inactive complexes. In both cases, anti-CCL2 treatment was terminated because the drugs did not suppress CCL2 sufficiently.
Due to the complexity of our target, an integrated approach was necessary to describe the PK and PD behavior of QBP359. We found that the rapid clearance of mAb-ligand complex was the most likely explanation for the rapid clearance of QBP359 in cynomolgus monkey, and that the target levels would not be saturable at a reasonable antibody dose following i.v. administration, due to high CCL21 turnover. From modeling, we were able to discern the expected higher turnover rates of CCL21 in man and in the target disease population, suggesting that high and frequent doses would be required to maintain effective target suppression, with a weekly dose of 50 mg kg−1 to achieve about 60% target suppression in peripheral tissues. Our study demonstrates the difficulty that may be associated with chemokine suppression and the necessity for newer strategies to overcome this. These complexities in targeting a soluble target should be taken into account when taking the decision to target a soluble protein vs. its receptor.
Methods
Biotherapeutic
QBP359 is a human mAb (IgG1, VH3/Vgamma3) that binds specifically to human CCL21 (approximately 14.6 kDa) with high affinity (30 pM as measured on BIAcore) and neutralizes its biological activity in human cell-based assays. It is cross-reactive to cynomolgus monkey. The molecular weight of QBP359 is approximately 143 kDa, and the antibody is soluble up to 50 mg mL−1 in 50 mM citrate buffer, 140 mM NaCl, pH 5.0.
In vivo study design
Animals
Female cynomolgus (Macaca fascicularis) monkeys were from Harlan UK Ltd, Bicester (originating from Bioculture Ltd, and Biodia Co. Ltd, Mauritius) and the studies were performed at Covance Harrogate, UK. Cages conformed to the ‘Code of practice for the housing and care of animals used in scientific procedures’ (Home Office, London, 1989).
Cynomolgus monkey dose-range finding study
A 4-week DRF study was performed using i.v. bolus administration of QBP359 weekly in monkeys with excisional/ incisional wounds to promote recruitment of fibrocytes from the bone marrow to the wound sites. The wounds in this study were surgical incisions performed and sutured under general anaesthetic by a veterinary surgeon. Four females were allocated to each group and each group was designed as follows: Group I Control group, Group II Low dose group (10 mg kg−1), Group III High dose group (50 mg kg−1), and Group IV High dose group (50 mg kg-1) with injection of 3 µg CCL21 into the wound sites to promote fibrocyte recruitment. Animals were dosed on Day 1, Day 8, Day 15 and Day 22. A full PK profile was taken after the first dose (Day 1) with sampling times at 0, 0.5, 2, 6, 24, 48, and 96 hours post-dose and after the last dose (Day 22) with sampling times at 0, 0.5, 2, 6, 24, 48, and 72 hours post dose. Trough values were obtained at Day 8 and Day 15. Initial PK analysis was performed using compartmental analysis of data with Phoenix software version 6.2 (Pharsight Inc., St. Louis, MI, USA)
Single dose cynomolgus monkey PK study
A single dose PK study was performed to acquire additional recovery data and to evaluate the bioavailability of QBP359. Three animals were allocated per dose group and the groups consisted of: Group I 10 mg kg−1 of QBP359 administered i.v., Group II 10 mg kg−1 of QBP359 administered s.c. and Group III 50 mg kg−1 of QBP359 administered i.v.. Samples were taken at 0, 0.5, and 6 hours and at 1, 2, 4, 7, 14, 21, and 28 d post-dose.
Immunoassay measurements
Total mAb assay
Total QBP359 in serum was measured using a sandwich immunoassay with an in-house mouse anti-human IgG mAb capture antibody and an HRP-conjugated goat anti-human IgG detection antibody (Jackson ImmunoResearch, 1 µg mL−1). The lower limit of quantification (LLOQ) of the assay was 300 ng mL−1 and the upper limit of quantification of the assay was 7500 ng mL−1.
Total ligand assay
Total CCL21 in serum consisting of unbound CCL21 and QBP359-bound CCL21 was measured using a sandwich immunoassay. A biotin-conjugated rabbit anti-human CCL21 antibody (Lifespan LS-C42953, 1 µg mL−1) was used to capture CCL21 on a streptavidin plate and detection was performed by adding a saturating level of QBP359 (to ensure the capture of CCL21) followed by the addition of an in-house mouse anti-human antibody, and then, the addition of a goat anti-mouse IgG conjugated to sulfotag (Meso Scale Discovery, MSD-R32AC, 3 nM). The plate was read on a Sector Imager 2400 from Meso Scale Discovery. The LLOQ of the assay was 2.5 ng mL−1 and the ULOQ was 500 ng mL−1.
Immunogenicity measurements
A BIAcore T100 instrument (BIAcore AB, Uppsala, Sweden) was used to test for the presence of anti-QBP359 antibodies. Briefly, QBP359 was covalently immobilized on one flow cell on one chip sensor surface using amine coupling to ensure that QBP359 is randomly distributed on the surface and each region of the molecule is accessible. Samples were injected and binding of anti-drug antibodies to QBP359 was measured in real time. Relative signal levels were calculated by subtracting the blank flow cell from the sample flow cell and compared with an anti-idiotypic anti-QBP359 antibody as positive control.
Immunohistochemistry of QBP359 in tissues
A panel of tissues was collected at necropsy at the end of the DRF study. Tissues were dissected, formalin fixed and embedded in paraffin wax (FFPE). A selection of tissues (adrenal, brain, caecum, ileum, kidney, liver, gall bladder, lung, lymph nodes (mesenteric, mandibular and injection site), pancreas, salivary gland, spleen, and thymus) was sectioned and investigated by IHC for distribution of QBP359.
SP2/0 cells producing recombinant human monoclonal IgG1 antibody was selected as positive control for demonstration of positive immunohistochemical staining with the selected detection antibody, goat anti-human IgG Fc fragment (Jackson ImmunoResearch Laboratories, Suffolk, UK).
PK/PD model structure, data fitting and parameter value estimation
A two-compartment PK-binding model was used to describe the observed QBP359 (PK) and total CCL21 (PD) data (, Supplemental Material). In this model the antibody binds CCL21 in both compartments and the model structure is essentially the same as previously used for canakinumab and interleukin-1beta. <τβρ>Citation31,32
Intravenous bolus doses of QBP359 were applied to the central compartment. Because CCL21 is primarily found in the secondary lymphoid organs in high endothelial venules,Citation33 CCL21 is assumed to be synthesized in the peripheral compartment. The model also specified that QBP359, CCL21 and the QBP359-CCL21 complex have different clearance rates. QBP359 is assumed to clear from the central compartment, free CCL21 is assumed to be clear from both central and peripheral compartments, and QBP359-CCL21 complex is assumed to be cleared only from the peripheral compartment. QBP359 is assumed to clear from the central compartment, free CCL21 is assumed to be clear from both central and peripheral compartments, and QBP359-CCL21 complex is assumed to be clear only from the peripheral compartment. Inter-compartmental serum-peripheral tissue transfer rates for QBP359 and the QBP359-CCL21 complex are assumed to be equal. Other assumptions and detailed derivation of model equations can be found in the Supplementary Methods.
Model fitting to data and estimation of PK-binding model parameters was performed using NONMEM (Version VI, 2.0 Intermediate) with the first-order conditional estimation method (METHOD = FOCE). Berkeley Madonna 8.0.1 was used for simulating the model for pulmonary fibrosis patients; Tibco Spotfire S-Plus (version 8.1) was used for graphical output.
Disclosure of Potential Conflicts of Interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no authors had support for the submitted work; and all had financial relationships with Novartis Pharma AG as principal employer during the period of the studies presented in the paper over the previous 3 y. In some cases, the co-authors are employed with different employers and this involves a new financial relationship. All authors claim no other relationships or activities that could appear to have influenced the submitted work.
Supplemental_Material.zip
Download Zip (558.2 KB)Acknowledgments
The authors would like to thank LeeAnne McLean for providing the material and methods for the tissue expression analysis and Denise Sickert for the development of the immunogenicity assay used for sample analysis measurements. Also, the support of Novartis Biologics Center for drug characterization and providing affinity measurement data for modeling and simulation.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Namiki M, Kawashima S, Yamashita T, Ozaki M, Hirase T, Ishida T, Inoue N, Hirata K, Matsukawa A, Morishita R, et al. Local overexpression of monocyte chemoattractant protein-1 at vessel wall induces infiltration of macrophages and formation of atherosclerotic lesion: synergism with hypercholesterolemia. Arterioscler Thromb Vasc Biol 2002; 22:115-20; PMID:11788470; http://dx.doi.org/10.1161/hq0102.102278
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 2000; 192:1075-80; PMID:11015448; http://dx.doi.org/10.1084/jem.192.7.1075
- Shahrara S, Proudfoot AE, Park CC, Volin MV, Haines GK, Woods JM, Aikens CH, Handel TM, Pope RM. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol 2008; 180:3447-56; PMID:18292571; http://dx.doi.org/10.4049/jimmunol.180.5.3447
- Moore BB, Paine R, 3rd, Christensen PJ, Moore TA, Sitterding S, Ngan R, Wilke CA, Kuziel WA, Toews GB. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol 2001; 167:4368-77; PMID:11591761; http://dx.doi.org/10.4049/jimmunol.167.8.4368
- Chung KF. Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy 2005; 4:619-25; PMID:17305518; http://dx.doi.org/10.2174/156801005774912806
- Youngs SJ, Ali SA, Taub DD, Rees RC. Chemokines induce migrational responses in human breast carcinoma cell lines. Int J Cancer 1997; 71:257-66; PMID:9139852; http://dx.doi.org/10.1002/(SICI)1097-0215(19970410)71:2%3c257::AID-IJC22%3e3.0.CO;2-D
- Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, Pienta KJ, Taichman RS. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem 2003; 89:462-73; PMID:12761880; http://dx.doi.org/10.1002/jcb.10522
- Vanderkerken K, Vande Broek I, Eizirik DL, Van Valckenborgh E, Asosingh K, Van Riet I, Van Camp B. Monocyte chemoattractant protein-1 (MCP-1), secreted by bone marrow endothelial cells, induces chemoattraction of 5T multiple myeloma cells. Clin Exp Metastasis 2002; 19:87-90; PMID:11918087; http://dx.doi.org/10.1023/A:1013891205989
- Bai Z, Hayasaka H, Kobayashi M, Li W, Guo Z, Jang MH, Kondo A, Choi BI, Iwakura Y, Miyasaka M. CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer's patches. J Immunol 2009; 182:1287-95; PMID:19155474; http://dx.doi.org/10.4049/jimmunol.182.3.1287
- Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 2008; 8:362-71; PMID:18379575; http://dx.doi.org/10.1038/nri2297
- Choi ES, Pierce EM, Jakubzick C, Carpenter KJ, Kunkel SL, Evanoff H, Martinez FJ, Flaherty KR, Moore BB, Toews GB, et al. Focal interstitial CC chemokine receptor 7 (CCR7) expression in idiopathic interstitial pneumonia. J Clin Pathol 2006; 59:28-39; PMID:16394278; http://dx.doi.org/10.1136/jcp.2005.026872
- Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Evanoff H, Flaherty KR, Martinez FJ, Toews GB, Hogaboam CM. Idiopathic pulmonary fibrosis fibroblasts migrate and proliferate to CC chemokine ligand 21. Eur Respir J 2007; 29:1082-93; PMID:17331965; http://dx.doi.org/10.1183/09031936.00122806
- Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Flaherty KR, Martinez FJ, Hogaboam CM. Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol 2007; 170:1152-64; PMID:17392156; http://dx.doi.org/10.2353/ajpath.2007.060649
- Peck CC, Barr WH, Benet LZ, Collins J, Desjardins RE, Furst DE, Harter JG, Levy G, Ludden T, Rodman JH, et al. Opportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development. J Pharm Sci 1992; 81:605-10; PMID:1355792; http://dx.doi.org/10.1002/jps.2600810630
- Vugmeyster Y, Harrold J, Xu X. Absorption, distribution, metabolism, and excretion (ADME) studies of biotherapeutics for autoimmune and inflammatory conditions. Aaps J 2012; 14:714-27; PMID:22798020; http://dx.doi.org/10.1208/s12248-012-9385-y
- Xu X, Vugmeyster Y. Challenges and opportunities in absorption, distribution, metabolism, and excretion studies of therapeutic biologics. Aaps J 2012; 14:781-91; PMID:22864668; http://dx.doi.org/10.1208/s12248-012-9388-8
- Comerford I, Nibbs RJ, Litchfield W, Bunting M, Harata-Lee Y, Haylock-Jacobs S, Forrow S, Korner H, McColl SR. The atypical chemokine receptor CCX-CKR scavenges homeostatic chemokines in circulation and tissues and suppresses Th17 responses. Blood 2010; 116:4130-40; PMID:20562329; http://dx.doi.org/10.1182/blood-2010-01-264390
- Nureki S, Miyazaki E, Ishi T, Ito T, Takenaka R, Ando M, Kumamoto T. Elevated concentrations of CCR7 ligands in patients with eosinophilic pneumonia. Allergy 2013; 68:1387-95; PMID:24111618; http://dx.doi.org/10.1111/all.12243
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 2006; 354:610-21; PMID:16467548; http://dx.doi.org/10.1056/NEJMra052723
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol 2001; 2:108-15; PMID:11175802; http://dx.doi.org/10.1038/84209
- Harada K, Nakanuma Y. Innate immunity in the pathogenesis of cholangiopathy: a recent update. Inflamm Allergy Drug Targets 2012; 11:478-83; PMID:22920631; http://dx.doi.org/10.2174/187152812803589976
- Feng LY, Ou ZL, Wu FY, Shen ZZ, Shao ZM. Involvement of a novel chemokine decoy receptor CCX-CKR in breast cancer growth, metastasis and patient survival. Clin Cancer Res 2009; 15:2962-70; PMID:19383822; http://dx.doi.org/10.1158/1078-0432.CCR-08-2495
- Igawa T, Mimoto F, Hattori K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim Biophys Acta 2014; 1844:1943-50; PMID:25125373; http://dx.doi.org/10.1016/j.bbapap.2014.08.003
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203-7; PMID:20953198; http://dx.doi.org/10.1038/nbt.1691
- Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem 2012; 287:11090-7; PMID:22294692; http://dx.doi.org/10.1074/jbc.M111.319764
- Haringman JJ, Gerlag DM, Smeets TJ, Baeten D, van den Bosch F, Bresnihan B, Breedveld FC, Dinant HJ, Legay F, Gram H, et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum 2006; 54:2387-92; PMID:16869001; http://dx.doi.org/10.1002/art.21975
- Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 2011; 3:61-6; PMID:20962582; http://dx.doi.org/10.4161/mabs.3.1.13799
- Xiao JJ, Krzyzanski W, Wang YM, Li H, Rose MJ, Ma M, Wu Y, Hinkle B, Perez-Ruixo JJ. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. Aaps J 2010; 12:646-57; PMID:20737261; http://dx.doi.org/10.1208/s12248-010-9222-0
- Fetterly GJ, Aras U, Meholick PD, Takimoto C, Seetharam S, McIntosh T, de Bono JS, Sandhu SK, Tolcher A, Davis HM, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2 and carlumab (CNTO 888) concentration time data. J Clin Pharmacol 2013; 53:1020-7; PMID:23878055; http://dx.doi.org/10.1002/jcph.140
- Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol 2013; 71:1041-50; PMID:23385782; http://dx.doi.org/10.1007/s00280-013-2099-8
- Chakraborty A, Tannenbaum S, Rordorf C, Lowe PJ, Floch D, Gram H, Roy S. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1beta monoclonal antibody. Clin Pharmacokinet 2012; 51:e1-18; PMID:22550964; http://dx.doi.org/10.2165/11599820-000000000-00000
- Lachmann HJ, Lowe P, Felix SD, Rordorf C, Leslie K, Madhoo S, Wittkowski H, Bek S, Hartmann N, Bosset S, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J Exp Med 2009; 206:1029-36; PMID:19364880; http://dx.doi.org/10.1084/jem.20082481
- Carlsen HS, Haraldsen G, Brandtzaeg P, Baekkevold ES. Disparate lymphoid chemokine expression in mice and men: no evidence of CCL21 synthesis by human high endothelial venules. Blood 2005; 106:444-6; PMID:15863780; http://dx.doi.org/10.1182/blood-2004-11-4353