
Abstract
Engineered antibody domains (eAds) are promising candidate therapeutics but their half-life is relatively short partly due to weak or absent binding to the neonatal Fc receptor (FcRn). We developed a novel approach to increase the eAd binding to FcRn based on a combination of structure-based design, computational modeling and phage display methodologies. By using this approach, we identified 2 IgG1 CH2-derived eAds fused to a short FcRn-binding motif derived from IgG1 CH3 that exhibited greatly enhanced FcRn binding with strict pH dependency. Importantly, the increased affinity resulted in significantly enhanced FcRn-mediated epithelial transcytosis and prolonged elimination half-life (mean 44.1 hours) in cynomolgus macaques. These results demonstrate for the first time that the half-life of isolated eAds can be prolonged (optimized) by increasing their binding to FcRn while maintaining their small size, which has important implications for development of therapeutics, including eAd-drug conjugates with enhanced penetration in solid tissues.
Introduction
Monoclonal antibodies (mAbs) have become the fastest growing class of new therapeutic molecules. Fifty mAbs are currently approved for clinical use, and hundreds are in clinical trials for treatment of various diseases.Citation1 However, the large size of full-length mAbs (∼150 kDa), which could prevent them from accessing sterically restricted epitopes and efficiently penetrating into tissues, remains a substantial concern. Consequently, a variety of smaller engineered antibody domains (eAds) and fragments are under development.Citation2,3 The small size (12–50 kDa) of eAds results in better penetration into normal tissues and solid tumors and the ability to bind into cavities or other sterically occluded epitopes that cannot be accessed by full-length mAbs. These features could be particularly important for targeting tumor antigens and rapidly mutating viruses, e.g., HIV-1, in which steric shielding of surface epitopes is often observed.Citation3,4 Furthermore, the small-size eAds can be produced by using bacterial expression systems and have higher molar quantities per gram of product, which provides a significant increase in potency per dose and reduction in overall manufacturing cost. Despite these advantages, to date the eAds have limited therapeutic applications, mainly due to their short elimination half-life (typically less than one hour), which greatly decreases their efficacy in vivo in many cases.Citation3 Several approaches, such as PEGylationCitation5 and fusion to albumin,Citation6,7 have been shown to increase the half-life of antibody domains, but this resulted in a dramatic increase in molecular size, and thus the loss of advantages conferred by the small size of the parent molecule. Therefore, the development of an effective strategy to extend the eAd serum half-life, while still maintaining their small size, is highly desirable.
IgG1 has an exceptionally long circulation half-life of up to 3 weeks, mostly because of its pH-dependent association with the neonatal Fc receptor (FcRn).Citation8,9 FcRn binds the Fc portion of IgG1 in the acidic environment of the endosome after the IgG1 internalization; the IgG1 is then recycled to the cell surface and released back into the circulation. This process rescues IgG1 from degradation and increases its serum half-life. Additionally, FcRn can drive the transport process of IgG1 across the epithelium of various tissues, such as intestine, lung, liver and kidney, by FcRn-mediated transcytosis.Citation9-13 This pathway is poorly understood, but could be important for drug absorption, transport and delivery. In this regard, engineering approaches to incorporate suitable FcRn binding sites into small-size eAds could lead to long-acting therapeutics.
Engineering full-length mAbs with enhanced binding to FcRn has been reported to correlate with prolonged serum half-life and improved bioactivity.Citation14-18 However, some IgG mutants with significantly increased binding affinity to FcRn have not shown improved half-life compared with the unmutated molecule.Citation19-22 An alternative approach has been to fuse a short FcRn-binding peptide to therapeutic proteins.Citation23 The peptide fusion proteins were found to have improved transport across FcRn-expressing cells, but their half-life was not measured or reported.Citation23 These and other observations indicate that, in addition to binding affinity, other factors, such as the strict pH-dependency, and the dissociation rate (koff) of the IgG-FcRn interaction, contribute significantly to in vivo half-life. Notably, it is also possible that the eAds, even when engineered to possess effective FcRn binding, could still be rapidly cleared by kidney filtration because their size is below the threshold for renal elimination (50 to 60 kDa), making the FcRn-mediated engineering strategies irrelevant. To our knowledge there are no reported data that describe how increased binding to FcRn with preserved size can affect eAd elimination half-life in vivo. The clarification of these issues, therefore, is of great importance to guide future directions for improving the pharmacokinetics (PK) of eAds.
We developed a novel eAd based on the CH2 domain of IgG1 by removing the unstructured N-terminal residues and introducing an additional disulfide bond into CH2.Citation24 The resulting eAd, named m01s, exhibits significantly increased stability compared to the isolated native CH2. By grafting the heavy chain complementary-determining regions (CDRs) onto the m01s framework, we were able to generate small and stable binders against therapeutic targets, e.g., to an HIV-1 neutralizing epitope.Citation25 However, m01s contains only a portion of the FcRn binding site of the IgG1 and exhibits only modest binding to FcRn. This feature along with the advantages mentioned above makes m01s an ideal model to explore whether the increase in the FcRn binding of small-size antibody domains correlates with prolonged in vivo half-life.
Here, we present a strategy for improving the FcRn binding capability of the m01s scaffold by grafting a 5-residue FcRn-binding motif from the IgG1 CH3 domain. A short randomized peptide, designed to modulate the orientation of this CH3 motif, was also introduced, and a panning/screening-based approach was devised to only select binders that have native-like FcRn binding modes. Two binders, 2C10 and a2b, were identified using this strategy. They have small size (16 kDa) and high thermal stability, can be efficiently expressed in E. coli, and exhibited greatly enhanced FcRn binding abilities compared to m01s. Importantly, we further demonstrated that 2C10 has both significantly enhanced FcRn-mediated epithelial transcytosis and prolonged in vivo half-life. This proof-of-concept study provides the first evidence, to the best of our knowledge, that the serum half-life of the small-size eAds could be effectively prolonged by the FcRn-mediated engineering strategies and has important implications for the development of novel types of candidate therapeutics with improved efficacy and safety.
Results
Identification of pH-dependent FcRn binders from a rationally designed library
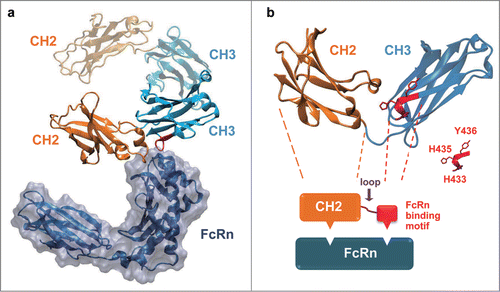
The eAd m01s, derived from IgG1 CH2 domain, only exhibited modest FcRn binding.Citation24 Both the CH2 and CH3 domains of IgG Fc contribute to the binding of FcRnCitation26 (). We found that all the residues involved in the FcRn interaction in CH3 are in a small C-terminal loop, and that this loop is spatially close to the CH2 domain (). Importantly, His433 and His435 in this loop are considered to participate in the formation of titratable salt bridges between Fc and FcRn, which are likely to confer pH dependence to this interaction.Citation27,28 Thus, we hypothesized that the FcRn binding of m01s could be enhanced by connecting this FcRn binding motif to the C-terminal of m01s using a peptide linker.
Figure 1. The native-like FcRn binding could be re-built by transplanting a short FcRn binding motif of IgG1 CH3 onto IgG1 CH2 domain. (a) The complex structure of rat FcRn with Fc (PDB entry 1FRT). The Fc CH2 domain was colored in orange and the Fc CH3 domain was colored in cyan. (b) A schematic diagram to describe the in silico design of CH2-CH3 hybrids. One chain of IgG1 Fc dimer compromising one CH2 and one CH3 domain was shown, and the FcRn binding motif in the CH3 domain was colored red. The CH2 domain and the FcRn binding motif of CH3 were joined by a randomized peptide loop. The residues considered to be important for FcRn binding (H433, H435 and Y436) in CH3 were highlighted.
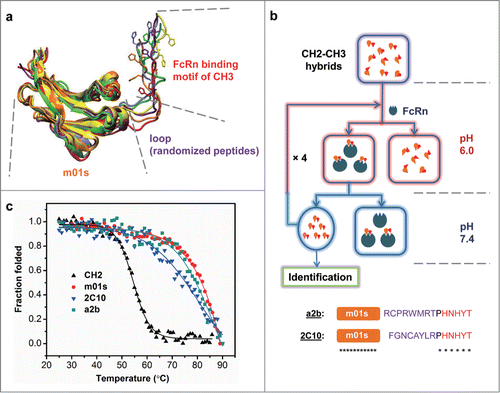
A critical requirement for the development of such “CH2-CH3 hybrids” with native-like FcRn binding pattern is that the CH3 motif in hybrids must be similarly positioned and oriented relative to the CH2 domain as it is in wild-type Fc. To achieve this, we performed a series of molecular dynamics simulations to generate energy-minimized structures of CH2-CH3 hybrids. We found that structures of hybrids that have 8-mer peptide linkers between m01s and the FcRn binding motif of CH3 superimposed well with the structure of Fc. We also found that a proline residue following the linker can confer rigidity to the backbone of CH3 motif. Thus, a large phage display library (∼3×1010 size) was constructed by joining m01s and the FcRn binding motif of CH3 together with an up to 8-mer peptide linker followed by a proline. The linker was randomized by NNB codons that encode the full amino acid repertoire. Notably, the CH3 motif of random selected hybrid models exhibited distinct folding, position and orientation, providing a molecular basis for this in silico design ().
Figure 2. Schematic representation of the engineering strategy to generate pH-dependent FcRn binders. (a) The overlapping view of 6 modeling structures of CH2-CH3 hybrids that possess randomly selected peptide linkers between m01s and FcRn binding motif of CH3. The molecule with a 4-mer linker was colored red, with a 6-mer linker was colored orange while all others have 8-mer linkers. Two histidines in the CH3 motif were drawn as bonds, showing different position and orientation of this motif with relative to m01s. (b) schematic representation of the panning method for the enrichment of only pH-dependent FcRn binders, as described in Experimental Procedures. (c) Plots of the change in fraction folded (calculated from CD molar ellipticity at 216 nm) for the isolated CH2 domain, 2C10, a2b and m01s.
Initially, this library was panned against human single-chain soluble FcRn at pH 6.0 using a standard biopanning procedure. We obtained some FcRn binders, but found they also bound to FcRn at neutral pH. It has been reported that the loss of pH-dependency enhanced IgG clearance in vivo.Citation19 Thus, we developed an improved panning procedure for the identification of pH-dependent binders (). The binders from the library that bound to biotin-labeled FcRn were captured by streptavidin beads at acidic environment, and unbound phage were washed away. The incubation mixture was then adjusted to neutral pH, and carefully eluted phage instead of beads were used for amplification by infecting TG1 cells. Such panning resulted in the enrichment of pH-dependent binders that cannot bind FcRn at neutral pH. The library was further panned for 4 rounds and the clones were identified by monoclonal phage ELISA at both pH 6.0 and 7.4.
Using this procedure 2 binders, a2b and 2C10, were identified (). Both of them contain a 9-mer linker between m01s and the FcRn binding motif of CH3. Although the linkers have distinct mutations, both, a2b and 2C10, were expressed in E. coli with high efficiency, with yields of more than 15 mg purified proteins per liter bacterial culture.
2C10 and a2b have high thermal stability
The thermal stability of 2C10 and a2b were determined by measuring their CD ellipticity at 216 nm as a function of temperature, and the results were compared with that of an isolated CH2 domain and m01s (). It is evident that CH2 is significantly less stable than 2C10, a2b and m01s. The CH2 unfolding started at 46°C and was completed at 62°C. Its melting temperature (Tm) was calculated to be 54.4 ± 0.2°C. In contrast, the Tm of 2C10, a2b and m01s were 76.9°C, 80.3°C, and 82.2°C, respectively. Notably, although 2C10 is slightly less stable than a2b and m01s, its stability is still comparable to that of an isolated Fc fragment (Tm = 78.9°C), which is considered to be extremely stable partly due to the CH3 dimerization effect.
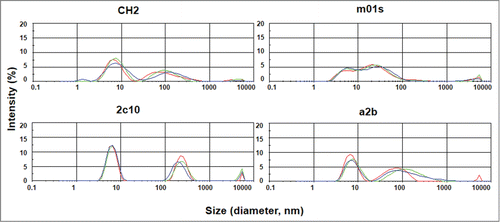
We also used dynamic light scattering (DLS) to measure the aggregation tendencies of CH2, m01s, 2C10 and a2b. We have previously found that the IgG1 Fc and Fc-derived molecules, although eluted as a single peak in size exclusion chromatography, typically have 2 peaks in the DLS measurements indicating possible oligomer formation after purification.Citation26 Similarly, we found that CH2, 2C10 and a2b have a major peak at a smaller size followed by another peak at a larger size indicating the predominance of monomeric species with the lesser extent of soluble oligomers (). The m01s displayed a relatively smaller monomer peak but it seems that there is no critical difference among the 4 proteins. These results suggest that the grafting of FcRn binding motif did not lead to an increase in aggregation tendency. One should note that the DLS measurements require relatively high (0.25 mg/ml) concentration. Dilutions at physiologically relevant concentrations are likely to reduce or eliminate aggregation.
Figure 3. Measurement of aggregation for IgG1 CH2, m01s, 2C10 and a2b by dynamic light scattering with the X-axis denoting protein particle size (diameter, nanometer) and the Y-axis intensity (%). Three different colors represent 3 measurements of the same protein.
2C10 and a2b have greatly enhanced FcRn binding
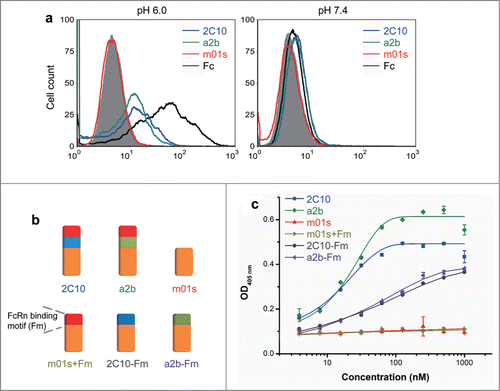
We next examined whether 2C10 and a2b could bind to FcRn in a pH-dependent manner. Surface plasmon resonance (SPR) experiments were first used to validate the pH-dependent FcRn binding (). We have previously found that CH2 and m01s that were over-expressed on the yeast cell surface have only very weak binding to FcRn.Citation24 Similarly, in this analysis with FcRn immobilized, m01s only had a low level of binding at pH 6 and 7.4 (). In contrast, both 2C10 and a2b displayed greatly enhanced binding at pH 6.0 () and negligible binding at pH 7.4 (). Interestingly, a2b exhibited a fast-on and fast-off kinetic profile, while 2C10 exhibited a relatively slow-on and slow-off binding kinetics, suggesting that they have different modes of binding to FcRn. Both of them can be fitted based on an one-to-one binding model. The determined binding affinity (KD) of a2b was 2.2 µM and 2C10 was 1.1 µM. After the measurements, the FcRn chip was treated by pH 8.0 buffer and the measurements of binding at 500 nM were repeated. We found that the second 500 nM binding curves of a2b and 2C10 overlapped well with the first 500 nM curves, indicating that they were efficiently removed by pH 8.0 buffer. Taken together, these data support the notion that 2C10 and a2b bind to human FcRn in a pH-dependent manner. This binding pattern was also confirmed by flow cytometry and ELISA (). 2C10 and a2b can bind FcRn-expressing cells at pH 6 but not at pH 7.4, while m01s did not show detectable binding. We also generated constructs with the FcRn binding motif or the conformational linker removed from 2C10 or a2b (), and found all of them displayed greatly reduced FcRn binding (), indicating that both the CH3 motif and the 9-mer linker are essential for proper binding to FcRn.
Figure 4. FcRn binding of 2C10, a2b and m01s measured by BIAcore.
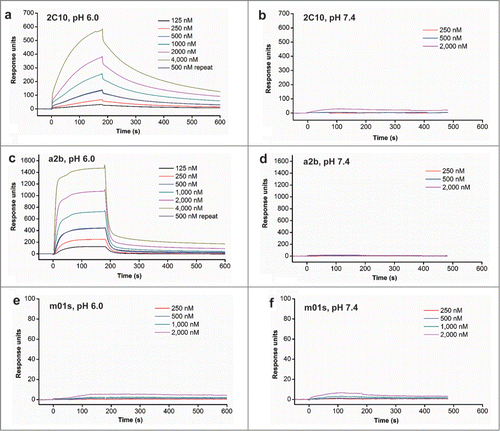
Figure 5. FcRn binding of measured by flow cytometry and ELISA. (a) Binding of MDCK-FcRn cells to 2C10, a2b, m01s and wild-type Fc (1 µM) at pH 6.0 (left) and 7.4 (right) analyzed by flow cytometry. (b) Schematic representation of the constructs with the FcRn binding motif or the conformational linker removed from 2C10 or a2b. (c) Binding of different antibody domain constructs to human single-chain soluble FcRn measured by ELISA at pH 6.0.
2C10 and a2b exhibited greatly enhanced epithelial transcytosis
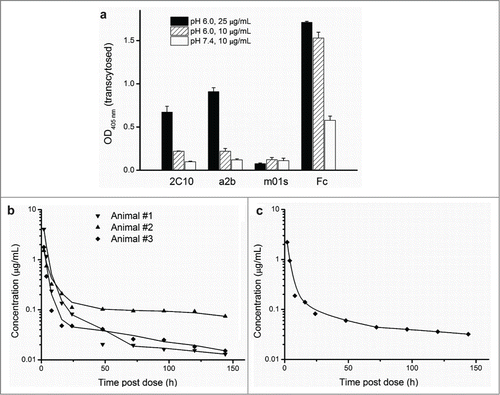
To investigate FcRn-mediated transepithelial transport, MDCK cells stably expressing FcRn and β2m were cultured on transwell filters, and the ability of 2C10 and a2b to transport across polarized MDCK-FcRn monolayers was investigated (). The medium in basolateral side was buffer at neutral pH, so proteins transported from the apical side were released and used for quantitative analysis by ELISA. As expected, m01s did not show detectable transcytosis at pH 6.0 or 7.4, due to its ineffective binding to FcRn. Notably, both 2C10 and a2b exhibited enhanced transcytosis at pH 6.0. Increasing protein concentration led to more effective transcytosis. No active transport was observed for 2C10 or a2b at pH 7.4. These results indicate that engineering small-size eAds with FcRn binding can confer significantly enhanced FcRn-mediated epithelial transcytosis.
Figure 6. The improved FcRn binding correlates with significantly enhanced transcytosis and prolonged in vivo half-life. (a) In vitro transepithelial transport of m01s, a2b, 2C10 and Fc at 10, 25 ug/mL (pH 6.0) and 10 ug/mL (pH 7.4). (b) Pharmacokinetics of 2C10 dosed to individual primates numbered 1, 2 and 3. (c) Serum concentration-time course plot of pooled serum samples of the same 3 animals.
Pharmacokinetics of 2C10 in cynomolgus monkeys
We previously found that the IgG1 CH2-derived eAd m01s has an elimination half-life of 11.4 h in cynomolgus monkeys.Citation29 To investigate whether the significantly enhanced FcRn binding of 2C10 would result in prolonged in vivo half-life, we performed PK experiments in cynomolgus macaques for 2C10, which has slightly higher FcRn affinity than a2b. The plasma-concentration profiles of 2C10 are shown in . Using individual animal data from 3 monkeys, the mean elimination half-life for 2C10 was 44.1 h (). The elimination half-life calculated from pooled plasma samples was 40.9 h (). These results indicated that the circulation half-life of 2C10 is significantly longer than m01s, which possesses only a portion of the FcRn binding site of the IgG1 and modest binding to FcRn.
Discussion
A major finding of this study is that improving the FcRn binding ability of small-size antibody domains enhances not only their transport across FcRn-expressing cells, but also their in vivo half-life. Although the size of a2b and 2C10 (16 kDa) was far below the renal clearance threshold of 50 to 60 kDa, the hybrids still exhibited relatively long half-life. Previous studies found that engineering improved FcRn binding in full-length mAbs correlated with prolonged serum half-life, but this approach for eAds has not been demonstrated to result in increased half-life.Citation14,23 We hypothesized that 2 major limitations to enhancing half-life of antibody domains were failure to: 1) fully mimic the interactions of IgG with FcRn; and 2) exhibit the strict pH-dependency inherent in these interactions.
We devised a novel strategy to engineer FcRn binding into small-size antibody domains to explore whether their enhanced FcRn binding would correlate with prolonged in vivo half-life.Citation30-32 Our strategy used a combination of in silico and screening-based rational design to overcome the 2 limitations described above. Using molecular dynamics simulations, we found that CH2-CH3 hybrid structures with 8-mer peptide linkers between m01s and the CH3 FcRn-binding motif superimposed with the structure of Fc, and that a proline residue following the linker improved the backbone rigidity of the CH3 motif. Based on these in silico findings, we constructed a large phage display library with CH2-CH3 hybrids with an up to 8-mer randomized peptide linker followed by a proline residue, which would improve the probability of identifying hybrids with the proper orientation, position and folding to facilitate interactions with FcRn. To pan/screen for CH2-CH3 hybrids from this library, we also developed a novel panning/screening-based approach to select binders with native-like FcRn binding modes. CH2-CH3 hybrids were first panned for binding to FcRn at pH 6.0. After unbound hybrids were washed away, bound hybrids were eluted by adjusting the incubation mixture to pH 7.4. Those eluted at pH 7.4 were further amplified and panned for 4 more rounds. This panning procedure resulted in the identification of the CH2-CH3 hybrid proteins a2b and 2C10.
The effectiveness of our strategy was confirmed through further analysis of these identified CH2-CH3 hybrids. Both a2b and 2C10 contained a 9-mer linker between m01s and the CH3 FcRn-binding motif, which supports the molecular dynamics simulations that showed a superimposition of the CH2-CH3 hybrids linked by an 8-mer linker followed by proline with the Fc structure. Additionally, both of these hybrids, similar to other hybrids identified from our panning/screening procedure described above, bind in a pH-dependent manner, i.e., they can bind FcRn at endosomal pH, but not at neutral pH. We also found that the binding affinity of a2b and 2C10 were relatively high (2.2 uM and 1.1 uM, respectively). In the future, this type of strategy could be potentially used in identifying more small-size eAds with desired properties, such as FcRn binding and other effector functions, and improving functions and characteristics of protein therapeutics in general.
In addition to strong, pH-dependent FcRn binding, a2b and 2C10 possess other characteristics that would make them promising candidate scaffolds and fusion partners. Their small sizes (˜16 kDa) could allow better tissue penetration and, in the case of fusion proteins, reduced steric hindrance for therapeutic activity. Additionally, a2b and 2C10 exhibit significantly higher thermal stability (76.9°C and 80.3°C) compared to the isolated CH2 domain; it is comparable to or higher than that of an isolated Fc fragment.Citation33 They are also highly expressed in E. coli, with yields of over 10 mg per liter of bacterial culture. Although immunogenicity is a potential problem for such engineered proteins, it is likely that the immunogenicity of a2b and 2C10 could be relatively low compared with similar foreign proteins, considering that both the engineered CH2 domain and the FcRn binding motif are of human origin, and that they have been engineered to mimic the native conformation of IgG1 Fc to effectively bind FcRn. Compared to other methods to extend the half-life of protein therapeutics, such as PEGylation or fusion to Fc domains or albumin,Citation5,6,34 our CH2-CH3 hybrids offer advantages in terms of size, stability and production.
This proof-of-concept study provides the first evidence, to the best of our knowledge, that the half-life of small-size eAds can be effectively prolonged by FcRn-dependent engineering strategies. The identified eAds exhibited not only high, pH-dependent binding affinity to FcRn in vitro but also significantly increased transcytosis and half-life in vivo. Due to these properties and their small size (16 kDa), high thermal stability and efficient expression in E. coli, a2b and 2C10 could be promising candidate scaffolds and fusion partners. Our strategy, which effectively combined in silico and panning/screening-based rational design, could be used to engineer improved FcRn binding and other desirable properties in antibody domains and other protein therapeutics. Furthermore, our study demonstrates the potential for the use of antibody domains with enhanced FcRn binding properties, which possess both small size and long half-life, in candidate therapeutics.
Methods
Library construction and selection of CH2-CH3 hybrid clones
A very large phage display library (∼3 × 1010 individuals) was constructed by joining m01s and the FcRn binding motif of CH3 with a randomly mutated loop. This loop contains 1–8 residues, all randomized by NNB codons, which encode the full amino acid repertoire. A proline was also inserted between the loop and the CH3 motif to give the backbone rigidity. Amplified libraries with 10Citation12 CH2-CH3 hybrids were used in the biopanning against FcRn. Human FcRn, containing both β and α chains in a 1:1 molar ratio, was expressed in mammalian cells and purified as a soluble protein as previously described.Citation35 Libraries were adjusted to pH 6.0 and incubated with biotin labeled FcRn conjugated to magnetic beads (Invitrogen, Carlsbad, CA) for 2 h at room temperature. After incubation, the beads were washed extensively with phosphate-buffered saline (PBS; pH 6.0) containing 0.05% Tween 20. The bound phages were incubated with PBS (pH 7.4), and the supernatants were amplified by infecting TG1 cells along with helper phage M13KO7 (Invitrogen, Carlsbad, CA). Clones that bound to FcRn at pH 6.0, but did not bind at pH 7.4, were identified from the fourth and fifth round of biopanning by using monoclonal phage ELISA.
Expression and purification of CH2‒CH3 hybrid proteins
The plasmids extracted from the identified CH2-CH3 hybrid clones, 2C10 and a2b, were used to transform HB2151 cells. A single and freshly transformed colony was inoculated into 200 mL SB medium with 100 µg/mL ampicillin, and incubated at 37°C with vigorous shaking at 250 rpm. When optical density of the culture at 600 nm reached around 0.6, expression was induced by the addition of isopropyl-1-thio-β-D-galactopyranoside to a final concentration of 1 mM, and the culture was further incubated at 30°C for 6 h. Cells were then harvested by centrifugation at 6,000 rpm for 15 min and resuspended in PBS buffer. Polymixin B (Sigma-Aldrich) (0.5 mu/mL) was added to the suspension (1:1000). After 30 min incubation with rotation at 50 rpm at room temperature, the culture was centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was used for further purification by Ni-NTA resin (Qiagen, Valencia, CA) according to the manufacturer's protocols. Protein purity was estimated as >95% by SDS-PAGE and protein concentration was measured spectrophotometrically (NanoVue, GE Healthcare).
Circular dichroism (CD)
The circular dichroism spectra were collected with an AVIV Model 202 spectropolarimeter (Aviv Biomedical). The protein samples were dissolved in PBS, pH 7.4 at a final concentration of 0.4 mg/mL. Spectra were first recorded at 25°C for native structure measurements. For evaluation of refolding, the samples were heated slowly to 90°C (1°C/min), kept at 90°C for 10 min, then rapidly cooled down to 25°C (10°/min), and the spectra were recorded again. For evaluation of thermal stability, CD signals at 216 nm were recorded and the instrument was programmed to acquire spectra at 1°C intervals over the range 25–90°C.
Dynamic light scattering
For comparison of aggregation tendency, the proteins were filtered through a 0.22-μm filter (Millipore, Bedford, MA, USA) and adjusted to the same concentration (0.25 mg/mL). Measurements were conducted using a Zetasizer Nano ZS ZEN3600 (Malvern Instruments Limited, Westborough, MA, USA) to determine the size of the protein particles. The samples were transferred to polystyrene cuvettes with the path length of 1 cm. Each sample was measured 3 times with 7 subruns of 10 s.
Binding ELISA
Antigens were coated on ELISA plate wells at 50 ng per well in PBS overnight at 4°C and blocked with protein-free blocking buffer for 1 h at 37°C. The human single-chain soluble FcRn, mouse anti-human IgG (CH2 specific, AbDSerotec, Oxford, UK), and anti-FLAG M2 antibody (Sigma-Aldrich) were used as coating antigens for FcRn binding, transcytosis and PK studies, respectively. Serially diluted protein was added and incubated at 37°C for 2 h. The plates were washed with PBS with Tween®20 (PBST; pH 6.0 or 7.4), and secondary antibody in PBS (pH 6.0 or 7.4) was incubated with wells for 1 h at 37°C. The HRP-conjugated anti-FLAG antibody (Sigma-Aldrich) was used for FcRn binding and transcytosis studies, and HisDetector Nickel-HRP (KPL, Gaithersburg, MD) was used for PK studies. After extensive washes with PBST (pH 6.0 or 7.4), binding was detected with the addition of ABTS substrate, and signals were read at 405 nm.
Surface plasmon resonance experiments
The interaction of proteins with soluble human FcRn was analyzed by SPR method using a BIAcore X100 instrument (GE Healthcare). Purified human FcRn was diluted in 10 mM Na-acetate buffer, pH 5.0, and immobilized on a CM5 biosensor chip using an amine coupling kit. The running buffer was PBS with 0.005% Tween 20 for binding at pH 7.4 or PBS pH 6.0 with 0.005% Tween 20 for binding at pH 6.0. The proteins diluted with running buffer were allowed to flow through the cells, at concentrations 125, 250, 500, 1000, 2000, and 4000 nM. After 10 min of dissociation, the chip was regenerated with pH 8.0 buffer (100 mM Tris, pH 8.0, 50 mM NaCl). For each analysis after the binding of 4000 nM, the binding of the analyte at 500 nM was repeated to monitor the regeneration efficiency.
Simulation of molecular structures
The initial coordinates of the hybrid proteins were created based on the crystal structure of an isolated CH2 domain (pdb entry: 3DJ9). Random residues were inserted between m01s and the FcRn binding motif of CH3 (pdb entry: 2WAH) with both the VMD program and Swiss-Model homology-modeling server (http://swissmodel.expasy.org). The structures were further simulated with CHARMM force field using the NAMD program. Water molecules were retained in the simulation. The structures of hybrid proteins were first minimized for 5000 steps with the conjugate gradient method, and equilibrated for 10 ps with the time step of 1 fs, then were further minimized for 100,000 steps for analysis with the VMD program.
Flow cytometry
To measure the interactions of proteins with FcRn, aliquots of MDCK cells co-transfected with human FcRn and β2m were incubated with 1 µM proteins in 100 μL PBS (pH 6.0 or 7.4) plus 0.5% bovine serum albumin (BSA) for 1 h on ice. Unbound antibodies were washed away with the same buffer. The secondary antibody FITC-conjugated goat F(ab')2 anti-human IgG (Fc-specific) antibody (Sigma-Aldrich) was incubated with cells for 45 min. Cells were washed and resuspended in PBS plus 0.5% BSA for flow cytometry on FACSCalibur (Becton Dickinson).
Transcytosis studies
Transcytosis studies used Transwell plates (Costar) inoculated with MDCK cells co-transfected with human FcRn and β2m. The cell monolayers were formed after 2 days' culturing at 37°C. Before the transcytosis tests, cells were washed and then starved with HBSS medium (pH 6.0 or 7.4) at 37°C for 1 h to remove surface-bound proteins. The outer chambers were filled with HBSS (pH 7.4), and proteins in HBSS medium (pH 6.0 or 7.4) were added into the inserts. After incubated overnight at 37°C, solution in the outer chambers were collected and analyzed by ELISA to measure the concentration. The same proteins with serially diluted concentrations in HBSS medium were used to produce the standard curves.
Pharmacokinetics in cynomolgus monkeys
The PK study protocol was approved by the Institutional Animal Care and Use Committee at Shin Nippon Biomedical Laboratories USA, Ltd. (Everett, WA) and conducted at Shin Nippon Biomedical Laboratories USA. Three female cynomolgus monkeys (age 7.5 ∼ 12 years; average weight 3.9 kg) with acceptable results in physical examinations were acclimated to the study room for 8 days. They were given a single intravenous bolus injection of 2 mg/kg endotoxin-removed 2C10 proteins in PBS, followed by 1 ml sterile flush. Blood samples (∼2 ml) were taken from all animals at baseline (t0), 2, 4, 8, 16, 24, 48, 72, 96, 120 and 144 h after test article administration. Plasma was recovered and stored at −80°C until analysis. Plasma was analyzed by quantitative ELISA, and the material used for injection was used to make the standard curves. ELISAs were performed with both individual animal samples and pooled samples. All PK analyses were performed using ELISA concentration/time point data running the PK Solutions 2.0, noncompartmental PK data analysis software from Summit Research Services.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
TY and DSD designed research; TY, YW, YF, RG, LW, KC performed experiments; and TY and DSD analyzed the data and wrote the paper.
Acknowledgements
We thank Tina W. Ju of Stanford University for help.
Funding
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, Federal funds from the NIH, National Cancer Institute, under Contract No. N01-CO-12400, and the Shanghai Pujiang Talent Program.
References
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010; 9:767-74; PMID:20811384; http://dx.doi.org/10.1038/nrd3229
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23:1126-36; PMID:16151406; http://dx.doi.org/10.1038/nbt1142
- Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol 2009; 27:331-7; PMID:19352366; http://dx.doi.org/10.1038/nbt0409-331
- Chen W, Gong R, Ying T, Prabakaran P, Zhu Z, Feng Y, Dimitrov DS. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Curr Drug Discov Technol 2014; 11:28-40; PMID:23863097; http://dx.doi.org/10.2174/15701638113109990032
- Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Dev 2008; 11:242-50; PMID:18283612
- Sleep D, Cameron J, Evans LR. Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta 2013; 1830:5526-34; PMID:23639804; http://dx.doi.org/10.1016/j.bbagen.2013.04.023
- Andersen JT, Cameron J, Plumridge A, Evans L, Sleep D, Sandlie I. Single-chain variable fragment albumin fusions bind the neonatal Fc receptor (FcRn) in a species-dependent manner: implications for in vivo half-life evaluation of albumin fusion therapeutics. J Biol Chem 2013; 288:24277-85; PMID:23818524; http://dx.doi.org/10.1074/jbc.M113.463000
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. mAbs 2011; 3:422-30; PMID:22048693; http://dx.doi.org/10.4161/mabs.3.5.16983
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/10.1038/nri2155
- Rath T, Kuo TT, Baker K, Qiao SW, Kobayashi K, Yoshida M, Roopenian D, Fiebiger E, Lencer WI, Blumberg RS. The immunologic functions of the neonatal Fc receptor for IgG. J Clin Immunol 2013; 33(Suppl 1):S9-17; PMID:22948741; http://dx.doi.org/10.1007/s10875-012-9768-y
- Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, Blumberg RS. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol 2010; 30:777-89; PMID:20886282; http://dx.doi.org/10.1007/s10875-010-9468-4
- Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci Transl Med 2013; 5:213ra167; PMID:24285486; http://dx.doi.org/10.1126/scitranslmed.3007049
- Gupta S, Gach JS, Becerra JC, Phan TB, Pudney J, Moldoveanu Z, Joseph SB, Landucci G, Supnet MJ, Ping LH, et al. The Neonatal Fc receptor (FcRn) enhances human immunodeficiency virus type 1 (HIV-1) transcytosis across epithelial cells. PLoS Pathog 2013; 9:e1003776; PMID:24278022; http://dx.doi.org/10.1371/journal.ppat.1003776
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157-9; PMID:20081867; http://dx.doi.org/10.1038/nbt.1601
- Ko SY, Pegu A, Rudicell RS, Yang ZY, Joyce MG, Chen X, Wang K, Bao S, Kraemer TD, Rath T, et al. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature 2014; 514:642-5; PMID:25119033; http://dx.doi.org/10.1038/nature13612
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/10.1074/jbc.M604292200
- Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vasquez M, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem 2004; 279:6213-6; PMID:14699147; http://dx.doi.org/10.1074/jbc.C300470200
- Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol 2006; 176:346-56; PMID:16365427; http://dx.doi.org/10.4049/jimmunol.176.1.346
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol 2005; 23:1283-8; PMID:16186811; http://dx.doi.org/10.1038/nbt1143
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; http://dx.doi.org/10.1124/dmd.112.045864
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 2007; 35:86-94; PMID:17050651; http://dx.doi.org/10.1124/dmd.106.011734
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; http://dx.doi.org/10.1016/j.molimm.2005.07.032
- Sockolosky JT, Tiffany MR, Szoka FC. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci USA 2012; 109:16095-100; PMID:22991460; http://dx.doi.org/10.1073/pnas.1208857109
- Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. J Biol Chem 2011; 286:27288-93; PMID:21669873; http://dx.doi.org/10.1074/jbc.M111.254219
- Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (Nanoantibodies) that interact mnoncompetitively with an HIV-1 neutralizing epitope and FcRn. PLoS One 2012; 7:e42288; PMID:22879932; http://dx.doi.org/10.1371/journal.pone.0042288
- Ying T, Ju TW, Wang Y, Prabakaran P, Dimitrov DS. Interactions of IgG1 CH2 and CH3 Domains with FcRn. Front Immunol 2014; 5:146; PMID:24765095; http://dx.doi.org/10.3389/fimmu.2014.00146
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994; 372:379-83; PMID:7969498; http://dx.doi.org/10.1038/372379a0
- Martin WL, West AP, Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell 2001; 7:867-77; PMID:11336709; http://dx.doi.org/10.1016/S1097-2765(01)00230-1
- Gehlsen K, Gong R, Bramhill D, Wiersma D, Kirkpatrick S, Wang Y, Feng Y, Dimitrov DS. Pharmacokinetics of engineered human monomeric and dimeric CH2 domains. MAbs 2012; 4:466-74; PMID:22699277; http://dx.doi.org/10.4161/mabs.20652
- Ying T, Chen W, Feng Y, Wang Y, Gong R, Dimitrov DS. Engineered soluble monomeric IgG1 CH3 domain: generation, mechanisms of function, and implications for design of biological therapeutics. J Biol Chem 2013; 288:25154-64; PMID:23867459; http://dx.doi.org/10.1074/jbc.M113.484154
- Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. J Biol Chem 2012; 287:19399-408; PMID:22518843; http://dx.doi.org/10.1074/jbc.M112.368647
- Ying T, Gong R, Ju TW, Prabakaran P, Dimitrov DS. Engineered Fc based antibody domains and fragments as novel scaffolds. Biochim Biophys Acta 2014; 1844:1977-82; PMID:24792384; http://dx.doi.org/10.1016/j.bbapap.2014.04.018
- Ying T, Feng Y, Wang Y, Chen W, Dimitrov DS. Monomeric IgG1 Fc molecules displaying unique Fc receptor interactions that are exploitable to treat inflammation-mediated diseases. mAbs 2014; 6:1201-10; PMID:25517305; http://dx.doi.org/10.4161/mabs.29835
- Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol 2013; PMID:24156398
- Feng Y, Gong R, Dimitrov DS. Design, expression and characterization of a soluble single-chain functional human neonatal Fc receptor. Protein Expre Purif 2011; 79:66-71; PMID:21453773; http://dx.doi.org/10.1016/j.pep.2011.03.012