
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Knowledge of the 3-dimensional structure of the antigen-binding region of antibodies enables numerous useful applications regarding the design and development of antibody-based drugs. We present a knowledge-based antibody structure prediction methodology that incorporates concepts that have arisen from an applied antibody engineering environment. The protocol exploits the rich and continuously growing supply of experimentally derived antibody structures available to predict CDR loop conformations and the packing of heavy and light chain quickly and without user intervention. The homology models are refined by a novel antibody-specific approach to adapt and rearrange sidechains based on their chemical environment. The method achieves very competitive all-atom root mean square deviation values in the order of 1.5 Å on different evaluation datasets consisting of both known and previously unpublished antibody crystal structures.
Abbreviations
| RMSD | = | root mean square deviation |
| AMA | = | Antibody Modeling Assessment |
Introduction
Antibodies are proteins that form an integral part of the specific immune response due to their ability to engage extracellular pathogenic structures (or antigens) with high affinity and specificity. Based on the recombination of a limited repertoire of different variable region germline genes, followed by somatic hypermutation and B cell-mediated antigen-dependent affinity maturation, evolution has brought forth an ingenious mechanism that enables the organism to generate a practically unlimited number of different specificities in answer to almost any antigen that the immune system is confronted with.Citation1-3 The unique properties of antibodies have made them an important tool in diagnostic and biotechnological applications,Citation4 and, last but not least, an increasingly sought-after therapeutic modality, in particular in the field of oncology.Citation5-7
In order to fulfill the desired drug profile, therapeutic antibodies are often heavily engineered. For example, modifications may be made to modulate target binding and pharmacokinetic properties, increase the degree of humanness, or address developability issues that might affect drug safety and shelf life.Citation8-11 Many of those tasks are enabled, or at least greatly facilitated, by structure-based considerations on the atomic level.Citation12 The large number of publicly available antibody crystal structures that have been solved during the last 25 y is an indicator for the relevance of this class of proteins. As of April 2015, the structural antibody database SAbDabCitation13 counts 2025 entries, a number that is steadily increasing.
The demand to accelerate and facilitate the rational design and engineering of antibody-based therapeutics has brought forth a multitude of computational methods aimed at supporting those tasks.Citation12-16 Among these computational methods is a number of homology modeling applications that have been tailored to exclusively generate atomistic models of the antigen-binding (Fv) region of antibodies,Citation17-19 a functionality that is meant to complement and relieve experimental structure elucidation by crystallography.
Given the large number of available crystal structures that can serve as templates, antibody Fv homology modeling is justifiably considered a less challenging problem than general homology modeling of arbitrary proteins, where often only a single template structure of low homology is available.Citation20-22 Considering that the overall structure of the Fv region is well understood and characterized,Citation23 the challenge in antibody homology modeling is to create a model that is sufficiently precise to form a sound basis for structure-based engineering and design, at a level of resolution that rivals that of a crystal structure. The main obstacle in this regard is the fact that the specificity of the antibody is mediated by 6 antigen-binding loops (complementarity-determining regions, CDRs) that are hyper-variable in sequence and length.Citation24,25 While 5 of these CDRs are prone to adopt canonical conformationsCitation26-28 that to a certain degree can be predicted directly from sequence, the third loop on the variable region of the heavy chain, CDR-H3, is known to form a unique conformation that, in the majority of the known complex structures, constitutes the key contributor to the antibody-antigen interaction. Due to the fact that CDR-H3 is by far the most variable of the 6 CDR loops,Citation29 a template loop structure of acceptable homology is often not available. Predicting the conformation of CDR-H3 with satisfactory accuracy is thus the key challenge in antibody homology modeling.Citation19 The situation is complicated further because the Fv region is composed of 2 separate immunoglobulin domains, the variable region of the heavy chain and the variable region of the light chain (VH and VL), with 3 CDRs distributed on each of the 2. The relative orientation of the 2 domains exhibits a notable variability over the known repertoire of antibody crystal structures,Citation30-32 and it has been suggested that VH-VL orientation, in addition to length and sequence of the CDRs, is a co-modulator of antigen specificity and affinity.Citation12 The parameters of VH-VL orientation determine how the CDRs are presented to the antigen, and thus contribute to the shape of the paratope (the antibody counterpart of the antigen's epitope). It seems likely that the variability in VH-VL orientation is a necessary means to accommodate the diverse antigenic shapes that antibodies are confronted with,Citation33 ranging from spacious conformational epitopes on globular proteins to linear peptides and even small molecule-like haptens. In summary, another challenge of antibody homology modeling lies in finding the correct relative orientation of the 2 variable domains.Citation19
The state of current antibody modeling technology was benchmarked in the 2011 and 2014 Antibody Modeling Assessment studies (AMA1 and AMA2),Citation17-19 where sets of previously unpublished crystal structures were compared to their blindly modeled counterparts in terms of backbone coordinate accuracy (root-mean-square deviation, RMSD) and structure quality (e.g., number of steric clashes, percentage of backbone conformations in the favored Ramachandran regionCitation34). The 2014 AMA iteration added the analysis of the models' VH-VL orientation parameters to the set of criteria to be assessed. Not surprisingly, the models submitted by the participants, comprising structures generated both with and without expert intervention, tend to achieve the highest accuracy in the conserved framework regions (typically below 1.0 Å backbone RMSD), while the largest deviations from the crystal structure are occurring in CDR-H3 (often above 3.0 Å backbone RMSD). We showed recently that the contribution of VH-VL misorientation to the overall RMSD of the AMA2 dataset models is approximately 0.5 Å.Citation35 The different methodologies differ mainly in the approach of the initial template selection, the extent of de novo modeling in CDR-H3, and the preference for either knowledge-based or forcefield-based model refinement techniques.Citation36-41 The latter also applies to the problem of VH-VL orientation, which some try to tackle by protein-protein docking-like approachesCitation36 and sophisticated energy-based refinement,Citation37 while others are relying more on identifying a suitable VH-VL orientation template structure.Citation38-41
Here, we present a novel antibody homology methodology that incorporates experiences and ideas that have arisen from applied antibody engineering in an industry environment. Our protocol MoFvAb (“Modeling the Fv region of Antibodies”) is primarily knowledge-based and exploits the large number of available antibody template structures to the best extent possible. MoFvAb is built around the WolfGuyCitation35 antibody numbering scheme that assigns a unique index to every conserved position in the Fv, identifies CDR loop tips and discriminates between ascending and descending loop segments. The initial WolfGuy numbering of the input sequences, the equivalent of performing a sequence alignment with the available antibody template structures, forms the basis for template selection, VH-VL orientation adjustment and model refinement. Unlike other published antibody modeling protocols, MoFvAb selects framework templates not per VH and VL or per Fv, but for every framework segment separately to minimize the number of necessary amino acid exchanges. After the raw model has been assembled from different template structures for framework and CDR regions, each residue is examined with regard to its (altered) chemical neighborhood formed by certain types of sidechains in its vicinity. Based on a conserved neighborhood definition for each position in the Fv, the sidechain (and to a certain extent also backbone) conformation of a given residue are adopted from matching known chemical neighborhood constellations that can be looked up in the template database. Finally, we pursue an active approach of adjusting VH-VL orientation that is based on first predicting the absolute parameters of VH-VL orientation from the amino acid types of certain key residues at the domain interface, followed by a coordinate transformation that applies the predicted orientation parameters to the model.Citation35
Although MoFvAb allows inserting de novo segments into CDR-H3 to tackle cases where a template loop of sufficient homology is not available, it does not include sophisticated loop modeling algorithmsCitation42,43 and in general tries to reduce the use of forcefield-based methods to the very last step. The philosophy is to generate a solid model quickly and without expert intervention that, depending on its purpose, can serve as the basis for further refinement. In spirit, MoFvAb is thus related to the popular PIGS algorithmCitation41 that was able to impress with very good results in the past AMA studies. In the following, we evaluate the quality of MoFvAb models by remodeling the antibodies of the AMA1 and AMA2 studies, as well as a set of 42 antibody structures crystallized in the complexed form (Table SI1). The latter set consists of 41 antibody crystal structures selected from the Protein Data Bank (PDB) for their high resolution and completeness, and one novel anti-theophylline antibody crystal structure described in this article. While the AMA1 and AMA2 studies focus on the backbone (or backbone carbonyl) RMSD to assess model accuracy, we include all heavy atoms into RMSD calculation to better monitor the effect of our novel approach of neighborhood-based sidechain refinement.
Results
High-resolution complex structure test set
shows the averaged all-atom RMSD and values for the 42 structure high-resolution complex test set modeled with different variants of MoFvAb. The listed variants differ in the energy minimization approach and the absence or presence of neighborhood-based sidechain refinement. The average all-atom RMSD for the complete Fv is in the order of 1.5 Å for the unrefined structures (Dreiding-r, CHARMm-r, CHARMm), and improves to approximately 1.41 Å for the neighborhood-refined structures (NR-Dreiding-r, NR-CHARMm-r, NR-CHARMm). The lowest RMSD values are achieved in the β-sheet core regions of VH and VL, and, after neighborhood-refinement, in CDR-L2. Not surprisingly, the largest deviations are occurring in CDR-H3, and cannot be improved beyond a value of 3.33 Å.
Table 1. Averaged all-atom RMSD and values for the 42 structure high-resolution complex test set. VHc and VLc refer to the β-sheet core of VH and VL, respectively. Dreiding-r and CHARMm-r refer to structures minimized with restraints, fixing all residues that were not modified in the modeling process. The prefix “NR” indicates structures that were processed with neighborhood-based sidechain refinement
Interestingly, the energy minimization methodology, either the computationally inexpensive restrained geometry relaxation with a Dreiding-like forcefield (Dreiding-r), the restrained CHARMm minimization or the fully flexible CHARMm minimization (CHARMm-r or CHARMm, both in combination with the GBSW implicit water model), only has a minor effect on the overall RMSD values. However, switching off the position restraints during CHARMm minimization enables the VH and VL domain to reorient according to the gradient of the forcefield, and thus to adapt different orientation parameters than the ones that were predicted and applied to the model. In this case, this leads to a minor increase in both and RMSD with regard to the respective crystal structures.
The difference between the energy minimization approaches becomes more apparent by comparing the MolProbity structure quality metrics listed in .
Table 2. Averaged MolProbity structure quality metrics for the 42 structure high-resolution complex test set and the reference set of crystal structures (“X-ray”)
The models relaxed with the restrained Dreiding-like forcefield (Dreiding-r, NR-Dreiding-r) suffer from a high percentage of Ramachandran outliers, and perform badly in terms of clash score and Cβ deviations. The conformation of the protein backbone is not remodeled properly, and a high number of steric clashes remains unresolved. Given that it takes only fractions of a second, this type of geometry relaxation may be useful to examine an initial choice of template structures, but it does not produce a valid protein structure. The restrained CHARMm minimization (CHARMm-r, NR-CHARMm-r) decreases the percentage of Ramachandran outliers and the extent of Cβ deviations, but the clash score remains high. The clash score, in turn, can be reduced to crystal structure standards and below by using the unrestrained CHARMm minimization routine (CHARMm, NR-CHARMm). The latter also leads to MolProbity scores comparable to those of the reference crystal structures. Therefore, despite the fact that the models are assembled rather coarsely from a large number of different template structures 1) by picking multiple framework templates per VH and VL, 2) by optionally using separate templates for ascending and descending CDR loop portions, and 3) by exchanging complete residues based on their sidechain neighborhood, it appears that the major issues revealed by MolProbity can be tackled by a single round of unrestrained energy minimization. The effect of neighborhood-based sidechain refinement is visible in terms of a higher amount of Cβ deviations and a better clash score than in the unrefined models, with almost no effect on the overall MolProbity score. The fraction of rotamer outliers is above crystal structure level for all MoFvAb models, and gets slightly worse when the models are minimized without restraints.
The effect of neighborhood-based sidechain refinement on the model structures is illustrated with a number of examples in .
Figure 1. CDR-H1 residue W197/H33 (WolfGuyCitation35/ChothiaCitation44 index) and its immediate surroundings (residues R251/H50, D253/H52 and Y353/H97) in the crystal structure with PDB ID 1OAU (cyan) in comparison to the unrefined MoFvAb model (orange) and the neighborhood-refined MoFvAb model (green).
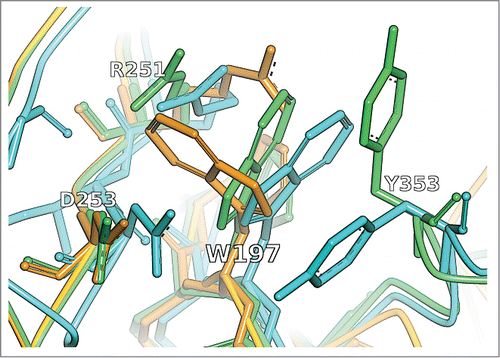
Figure 2. CDR-H1 of the crystal structure with PDB ID 1NBY (cyan) in comparison to the unrefined MoFvAb model (orange) and the neighborhood-refined MoFvAb model (green). The WolfGuy indices 151, 152, 153, 154, 155, 156, 196, 197, 198 and 199 correspond to Chothia indices H26, H27, H28, H29, H30, H31, H32, H33, H34 and H35, respectively.
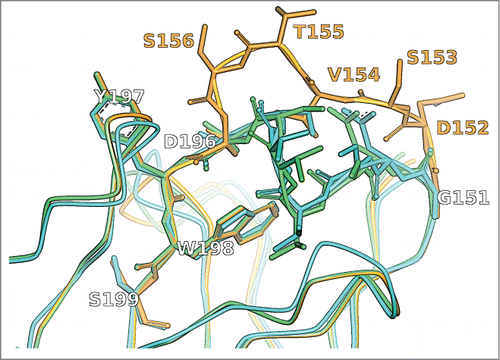
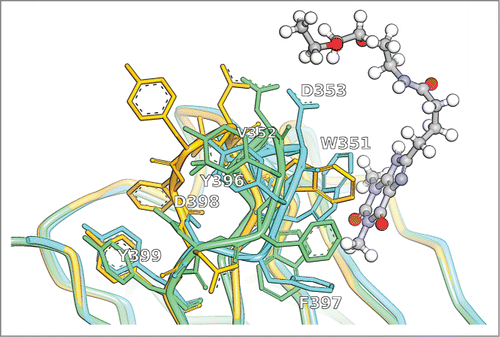
Figure 3. CDR-H3 of the crystal structure with PDB ID 5BMF (cyan) in comparison to the unrefined MoFvAb model (orange) and the neighborhood-refined MoFvAb model (green). The theophylline molecule binding to the antibody is shown in ball and stick representation. The WolfGuy indices 351, 352, 353, 396, 397, 398, and 399 correspond to Chothia indices H95, H96, H97, H99, H101 and H102, respectively.
shows the sidechain placement of CDR-H1 residue W197/H33 (WolfGuyCitation35/ChothiaCitation44 index) for the MoFvAb models of the crystal structure with PDB ID 1OAU. The bulky aromatic sidechains of tryptophan and tyrosine residues often constitute important players in antibody-antigen binding motifs.Citation45-47 Therefore, the correct placement of these residues at key positions is crucial to obtain a precise antibody homology model. In this example, the tryptophan W197/H33 from the original template 1ngz_B (the CDR-H1 loop of 1ngz_B has 100% identity to the query sequence) is replaced by a tyrosine from a different structure, based on the neighborhood query ‘W|HRDPXGKMWYY’, matched by Y197/H33 with neighborhood string ‘Y|HRDPXNKFGYY’ in template 1j05_H. After the exchange, the tyrosine adopted from template 1j05_H is automatically remodeled to a tryptophan residue, with the result that its conformation is more accurate than that of the original template. also documents an improvement of the sidechain conformation of R251/H50 in the vicinity of W197/H33. Although the sidechain of residue Y353/H97 (CDR-H3) is clearly misplaced, one can perceive an improvement of the course of the CDR-H3 backbone that in the unrefined model takes a completely different route, divergent from the crystal structure.
Another example of how neighborhood-based refinement can reroute CDR loops is given in .
The CDR-H1 loop with sequence GDSVTSDYWS shown in was originally modeled from the 2 template structures 3gkz_A using the ascent [GDSVTS]GYWS (residues 151/H26 to 156/H31) and 3d9a_H using the descent GDSITS[DYWS] (residues 196/H32 to 199/H35). Interestingly, the ascending part of the loop takes a completely different route than the one in the crystal structure, leading to a large deviation in the unrefined model. In the course of neighborhood refinement, residues 151/H26 to 196/H32 are exchanged by their counterparts taken from the template structures 1ndm_B, 1uac_H, 1uac_H, 3a67_H, 4hs6_B, 3a6c_H and 1ic5_H, respectively. There is no neighborhood-based match for Y197/H33, so it is retained. The best neighborhood matches for residues W198/H34 and S199/H35 point to template structure 3d9a_H, which is already in place. The resulting “patchwork” loop is remarkably close to the crystal structure. Notably, model residue V154/H29 (neighborhood query ‘V|VDDWYRTNY’) is a reconstructed isoleucine adopted from template 3a67_H that was chosen based on a perfect neighborhood match (neighborhood string ‘I|VDDWYRTNY’).
The limits of neighborhood-based refinement are illustrated in . The CDR-H3 of crystal structure 5BMF forms one side of a hydrophobic cage for its antigen, the small molecule theophylline, in this case prolonged by a linker for covalent coupling.Citation48 Among the key players of the interaction are W351/H95, which realizes π-stacking interactions with the xanthine moiety of theophylline, and F397/H99, which contributes to the bottom of the cage (Fig. SI5). Neighborhood refinement of the CDR-H3 loop results in a rerouting of the backbone, and a relatively accurate placement of the sidechains of D398/H101, Y396/H98, D353/H97 and V352/H96. Unfortunately, residue W351/H95 by mistake is flipped downwards, bringing it to the level of the (original) F397/H99, while the model's residue F397/H99 is shifted sideways, so that the important π-stacking interaction between W351/H95 and theophylline cannot be reproduced. Hence, the MoFvAb model would not be sufficiently accurate to perform meaningful docking experiments, despite the fact that the length of CDR-H3 in this case should be manageable. A more detailed comment on the difficulty to place tryptophan (and other aromatic residues) at position 351/H95 can be found in the Supplemental Information, Figure SI3. We speculate that an additional user-directed modeling step for reviewing alternative conformations identified in neighborhood-based hits might help to detect such errors in the future.
AMA1 and AMA2 datasets
shows the averaged all-atom RMSD and values for the 9 structures of the AMA1 dataset modeled with different variants of MoFvAb. For comparison with the original study, the table also states the average value over all AMA1 participants (AMA mean), as well as the average value achieved by the best AMA1 participant (AMA best). To put the direct comparison into perspective, one should keep in mind that the original AMA participant models were generated under different conditions, with a lesser set of available template structures, and with a focus on backbone rather than on sidechain accuracy.
Table 3. Averaged all-atom RMSD and values for the AMA1 dataset: Green cells indicate values better than or equal to the best AMA1 participant structure, yellow cells indicate values better than or equal to the AMA1 participant average, and red cells indicate values worse than the AMA1 participant average
The best average all-atom RMSD for the AMA1 dataset is achieved by the neighborhood-refined and CHARMm-minimized variants (NR-CHARMm-r and NR-CHARMm). Similar to the 42 structure complex test set, the gain by neighborhood refinement is approximately 0.09 Å RMSD on the whole Fv. MoFvAb's best all-atom RMSD of 1.49 Å for the Fv does not reach the level of the high-resolution complex structure test set (1.39 Å), but is still able to exceed the best AMA1 participant (1.66 Å). Segment-wise, MoFvAb is able to perform best or better than average, with the striking exception of CDR-L3, where the RMSD value falls short of the AMA1 participant average by about 0.5 Å. The extent of VH-VL misorientation in terms of is similar to the values of the large test set. Again, unrestrained CHARMm minimization leads to a perceptible increase of
.
summarizes the model-wise all-atom RMSD and values for the 9 structures of the AMA1 dataset modeled with the NR-CHARMm-r variant of MoFvAb.
Table 4. Model-wise all-atom RMSD and values for the AMA1 dataset: Green cells indicate values better than or equal to the best AMA1 participant structure, yellow cells indicate values better than or equal to the AMA1 participant average, and red cells indicate values worse than the AMA1 participant average. “MoFvAb” refers to the NR-CHARMm-r models
The issues with CDR-L3 arise mainly from the 2 models ama1ab06 (C706, PDB ID 3MCL) and ama1ab07 (2507, PDB ID 3QPX). C706 is known to have a unique CDR-L3 conformation that does not match the canonical conformation suggested by its sequence [QNWRSSPT] due to specific interactions with CDR-H3. For C706, MoFvAb uses the 2 template structures 2fat_L [QQWN] and 3w9e_B [SSPT] to model the loop, which results in a largely canonical CDR-L3 loop conformation and thus explains the unfavorable RMSD. Given that the value is still distinctly worse than the AMA1 average, it can be argued that CDR-L3 does not respond very well to choosing separate templates for ascending and descending loop. Interestingly, the C706 model ama1ab06 also suffers from a rather poorly predicted VH-VL orientation. By contrast, antibody 2507 does not exhibit an unusual CDR-L3 conformation; however, rather unfortunately, the automatic template selection routine chooses to model the loop [QQYNSRDT] from the 2 templates 2fat_L [QQWN] and 3mcl_B [SSPT], the latter being exactly the “exceptional” antibody C706. This shows that MoFvAb can be susceptible for errors caused by atypical template structures that have not been identified and labeled as such.
shows the averaged all-atom RMSD and values for the 11 structures of the AMA2 dataset modeled with different variants of MoFvAb, once more in comparison to the average value over all AMA2 participants, and the average value achieved by the best AMA2 participant.
Table 5. Averaged all-atom RMSD and values for the AMA2 dataset: Green cells indicate values better than or equal to the best AMA2 participant structure, yellow cells indicate values better than or equal to the AMA2 participant average, and red cells indicate values worse than the AMA2 participant average
The best average all-atom RMSD of 1.52 Å for the AMA2 dataset is achieved by the neighborhood-refined and CHARMm-minimized variants of MoFvAb (NR-CHARMm-r and NR-CHARMm). Again, the improvement by neighborhood-refinement on the all-atom RMSD for the whole Fv is approximately 0.09 Å. On the AMA2 dataset, there seems to be no particular problem with the modeling of CDR-L3, although the RMSD value of the best original participant (1.82 Å) cannot be met. The average error in CDR-H3 is above 4 Å, slightly worse than for the other 2 datasets. The same holds for VH-VL misorientation in terms of , which in this case is predicted slightly less accurate. Unlike in the other 2 cases, unrestrained energy minimization with the CHARMm forcefield is able to correct the predicted orientation values by a small margin. Notably, although the AMA2 crystal structure set shows more diversity in VH-VL orientation than the AMA1 crystal structures (data not shown), the AMA2 participant models achieve distinctly better accuracy with regard to VH-VL orientation (average
5.96) than the AMA1 models (average
7.3, cp. ), showing that the problem is now actively addressed by the different antibody modeling methodologies.
summarizes the model-wise all-atom RMSD and values for the 11 structures of the AMA2 dataset modeled with the NR-CHARMm-r variant of MoFvAb.
Table 6. Model-wise all-atom RMSD and values for the AMA2 dataset: Green cells indicate values better than or equal to the best AMA2 participant structure, yellow cells indicate values better than or equal to the AMA2 participant average, and red cells indicate values worse than the AMA2 participant average. “MoFvAb” refers to the NR-CHARMm-r models
In the case of the AMA2 antibody models generated by MoFvAb, no clear pattern for poor (i.e., below AMA2 average) choices of template structures is discernible. One of the worst results (1.7 Å in terms of whole Fv all-atom RMSD) is found for model ama2ab05, which suffers from incorrectly predicted VH-VL orientation parameters. Model ama2ab05 also has the highest average VH-VL misorientation among the original AMA2 participant models (average 8.52). The respective antibody (PDB ID 4M6M) is phage display-derived and shows a rather extreme value for the ABangle HL torsion angle (−50.52°). It is the only antibody among the AMA2 structures with a λ type light chain, which, due to the relatively low number of crystal structures available for this class, might pose a larger problem for our machine learning-based approach.
Model ama2ab01 of a rabbit antibody (PDB ID 4MA3) has the highest all-atom RMSD among the MoFvAb models (1.76 Å for variant NR-CHARMm-r), which is largely due to the very inaccurate modeling of CDR-H3 (6.99 Å RMSD). The overall RMSD for this model still exceeds the best AMA2 participant (2.45 Å) by a large margin because the MoFvAb model repository contains a number of proprietary rabbit antibody structures that complement the rather sparse supply of publicly available rabbit antibody structures, and thus help to model, for instance, the characteristic deletions in Framework 3 of the heavy chain.
Model ama2ab06 of the antibody crystal structure with PDB ID 4M6O features the longest CDR-H3 of the AMA2 dataset. In this model, the loop of length 14 [VYSSGWHVSDYFDY] is modeled from the 2 separate template structures 3efd_H and 3ifp_H, which both have a CDR-H3 that is one residue shorter than the target loop (ascent 3efd_H: [SWEAYW]RWSAMDY, descent 3ifp_H: RRIIY[DVEDYFDY]). The resulting CDR model achieves an all-atom RMSD of 5.41 Å, which is better than the AMA2 average of 5.91 Å, but quite obviously in need of improvement. We assume that neighborhood-based sidechain refinement works best near the stem of the loop, where a conserved neighborhood can still be defined, while the modeling of long and exposed loops, as in the case of structure 4M6O, will remain the domain of more sophisticated de novo loop modeling approaches.
In summary, the AMA1 and AMA2 crystal structures proved to be a harder test for MoFvAb than the 42 structure high-resolution complex test set, which shows in the all-atom RMSD values for the whole Fv (1.39 Å compared to 1.49 Å for AMA1, and 1.52 Å for AMA2). We attribute this mainly to the more challenging distribution of CDR-H3 length (the high-resolution complex structure test set is limited to CDR-H3 lengths of 13 and below), and the fact that the AMA1 and AMA2 crystal structures have been crystallized in the unbound state, which makes them less well predictable in terms of VH-VL orientation. It was shown that the intrinsic variability in VH-VL orientation of sequence-identical Fv structures is less for antibodies crystallized in the complexed form.Citation32 As a consequence, the random forest model is able to predict the VH-VL orientation parameters more accurately, which translates into favorable RMSD values.
The AMA1 and AMA2 datasets also provide us with the valuable opportunity to compare the MoFvAb models to those delivered by the other antibody modeling methodologies. While it is not always clear which templates were used or how much expert intervention (if any) was involved, the RMSD values for a given model segment provide a benchmark of what is achievable in terms of model quality. What must follow now is a more thorough analysis of the cases where MoFvAb fell short of these standards to provide hints on how to improve the algorithm in the future. In general, considering that the MoFvAb models were created without user intervention, we find that the overall agreement with regard to the crystal structures is very competitive.
Run-time
The time required per Fv model is 3.36 s for template look-up and automatic selection, and 6.44 s for model assembly, neighborhood-based sidechain refinement and VH-VL orientation prediction and adjustment. The values are averaged over 3 runs of the 42 structure high-resolution complex test set. Unrestrained CHARMm-minimization of a single Fv with the GBSW implicit water model can take up to 10 minutes with our current settings (use of 2 cores for the forcefield evaluation), but should be able to benefit significantly by further parallelization.
Discussion
Here, we introduced and evaluated the MoFvAb antibody homology modeling protocol. The most fundamental step of the algorithm is the annotation of the input sequences with the WolfGuy antibody numbering scheme. WolfGuy is used to discriminate between framework and CDR segments and to define ascending and descending loop sections, but it also enables the characterization of conserved amino acid neighborhoods, as well as the VH-VL interface residue fingerprint that is used to predict VH-VL orientation.
The template selection routine of MoFvAb is extremely simplistic in the regard that it chooses the highest homology, best resolution template structure for each framework and (ascending and descending) CDR segment in VH and VL, regardless of the numerous complex interactions that are occurring between these functional units. The latter is tackled by: 1) predicting and adjusting VH-VL orientation based on the VH-VL interface fingerprint, and 2) performing neighborhood-based sidechain (and to a certain degree also loop) refinement. The first methodology aims at adjusting the “macroscopic” geometry of the VH and VL, and the second one at tuning and adapting the individual amino acids in the Fv region, adopted from a number of different template structures, with regard to their new chemical environment.
Both of these approaches were demonstrated to perform well, which shows in terms of an improvement of the all-atom RMSD after neighborhood-based refinement, as well as in comparatively low VH-VL misorientation values (), on all 3 of our evaluation datasets. MoFvAb is a knowledge-based methodology, and as such it is susceptible to lack of template structures, or to templates that are faulty, or exceptional (e.g., CDR loops that are warped due to antigen contacts). We feel that more work is needed to properly identify such “hazardous” template structures, e.g., by comparing the individual CDR loop geometries in our template database with averaged or idealized canonical loop structures. We are also planning to ascertain that, whenever a CDR loop is reconstructed from multiple template structures, the selection routine does not accidentally make its pick from different, non-matching canonical classes of loops; an issue that we believe might explain some of the high-RMSD spikes in CDR-L3.
Another challenge we see for the future is how to define a reliable measure that might tell us whenever the currently available information in terms of loop, neighborhood or VH-VL orientation template structures is not sufficient to achieve a meaningful result, and thus to fall back to de novo (forcefield and dynamics-based) modeling instead.
Furthermore, we would be interested in evaluating if and how the idea of neighborhood-based sidechain (or model) refinement could be combined with current de novo CDR-H3 loop modeling methodologies. The problem in CDR-H3 loop modeling is typically to efficiently select the “right” candidate from a large ensemble of (in principle valid) loop geometries that have been generated. Possibly, a neighborhood-based model of the framework-proximal CDR loop stems might provide a computationally inexpensive funnel that could help to narrow down the selection of legitimate geometries, and thus to arrive at better results even for long CDR-H3.
Materials and Methods
Template structure repository
The template structure repository for MoFvAb is based on the Roche in-house antibody structure database RAB3D. RAB3D contains almost all of the publicly available antibody structures from the PDB (http://www.rcsb.org/pdb/), and is enriched by a number of unpublished in-house antibody crystal structures. RAB3D, in turn, uses the SabDabCitation13 antibody database engine to identify and add novel antibody structures from the PDB on a regular basis.
From each confirmed antibody structure, all paired heavy and light chain variable domains (VH and VL) are extracted and annotated with the WolfGuy antibody sequence numbering scheme. WolfGuy defines CDR regions as the set union of the KabatCitation49 and ChothiaCitation44 definition, and annotates CDR loop tips based on CDR length (and partly based on sequence) so that the index of a CDR position indicates if a CDR residue is part of the ascending or the descending loop. Similar to the Honegger-PlückthunCitation50 numbering scheme, WolfGuy is designed such that residues that are very similar in terms of conserved spatial localization in the Fv structure are numbered with equivalent indices as far as possible. A WolfGuy-based alignment of all VH and VL sequences involved in this study can be found in the Supplemental Information (Figs. SI1 and SI2).
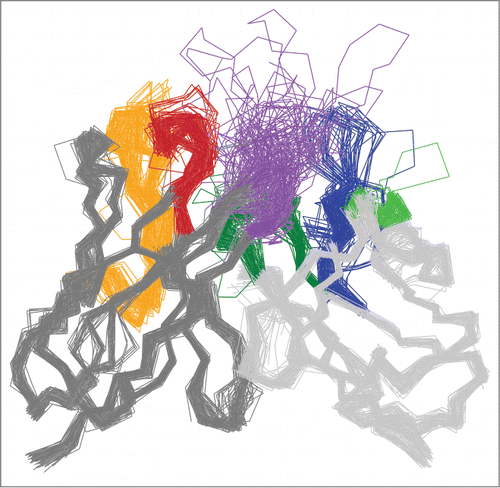
For use with MoFvAb, the annotated variable domain structures are positioned in a common coordinate system. The VH domain is superimposed on a VH consensus framework using all VH framework Cα atoms sharing the same WolfGuy index, and the VL domain is superimposed on a VL consensus framework (divided further into κ and λ subtype) following the same procedure. The VH and VL consensus structures, in turn, are aligned on a consensus Fv structure, leading to a spatial prearrangement of the template structures that already approximates an idealized antibody Fv region ().
Figure 4. Cα trace representation of 100 randomly chosen VH and VL MoFvAb template structures. The spatial distribution in the framework regions (dark gray for VH and light gray for VL) and the CDR regions that are prone to adopt canonical conformations is typically very compact. Framework and CDR classification follows WolfGuy nomenclature. CDR color coding follows IMGT/Collier-de-PerlesCitation51 conventions: CDR-H1 is colored red, CDR-H2 orange, CDR-H3 purple, CDR-L1 blue, CDR-L2 light green, and CDR-L3 dark green. Despite the relatively low number of structures shown, the exceptional structural diversity of CDR-H3 is clearly visible.
All consensus structures are calculated using a set of high-resolution Fv structures from RAB3D. During the template processing phase, we carefully monitor the VH and VL framework alignment RMSD, as high RMSD values tend to indicate misnumbered, atypical or warped structures that would make poor templates for homology modeling. Whenever a crystal structure contains multiple copies of the same Fv in a single asymmetric unit, we preferably add the structure with the lowest consensus alignment RMSD to the template repository, given it has the same completeness as the other copies. For fast lookup of the available VH and VL template structures, we store the sequences of the 7 functional segments, i.e., Framework 1–4 and CDR 1–3 (designation according to WolfGuy numbering) in a hash table.
Template selection
MoFvAb is implemented as an Accelrys Pipeline Pilot 9.1Citation52 protocol relying mainly on the Accelrys Discovery Studio 4.0Citation53 application programming interface (Perl). MoFvAb can be run from a web browser, either for a single pair of heavy or light chain sequences, or in batch mode. Each input sequence is annotated following WolfGuy and reduced to its variable domain. Both VH and VL are divided into the 7 functional framework and CDR segments as listed in the previous section. In contrast to other published antibody homology modeling protocols, we do not pick a common framework template per variable domain or per Fv, but look up every segment independently based on sequence homology. Thus, in principle, a single MoFvAb model can be assembled from 14 different template structures per Fv. This number can increase even further as we allow selecting the ascending and descending section of a single CDR loop from 2 different structures, given the best template for the complete CDR loop scores worse. This concept is illustrated in .
Table 7. Example template hits for query CDR-H3 sequence EGSNNNALAY from crystal structure with PDB ID 3MCL. The best hit for the complete segment is shown in light gray. The best hits for ascending and descending loop are shown in dark gray. The designated CDR loop tip position is indicated by the ˜ character
In the given example, the assembly of the query CDR-H3 loop EGSNNNALAY from 2 different template structures (3kym_F and 4leo_A, meaning chains F and A from the crystal structures with PDB ID 3KYM and 4LEO, respectively) leads to an improved homology score in comparison to the best complete segment hit (3dur_D). To account for total loop length, the ascent and descent hit score is penalized if the total length of the hit loop does not match the total length of the query loop, as is shown here for the ascent score (analogous for the descent score):
In general, the template hits for each segment are ranked in the following order: 1) Sequence similarity (BLOSUM62 scoreCitation54), 2) number of incomplete sidechains, 3) resolution of the template structure, and 4) alignment RMSD of the template structure with regard to the VH or VL consensus framework. While MoFvAb's template selection algorithm works automatically, it is possible to review and modify the initial template selection if so desired. Furthermore, the interactive mode allows augmentation (or even replacement in whole) of the available template selection for CDR-H3 by a de novo segment, an option that may be preferable if even the best available templates offer only very poor homology. The MoFvAb models evaluated in this article were generated without expert intervention in fully automatic mode. As all template sequences have been split into the respective framework and CDR segments a priori, they can be looked up in a hash without the need for BLASTp searchCitation55 or other alignment-based approaches during run time. For the CDR segments of variable length, we prepare prefix and suffix alignments of the ascending (aligned from left) and the descending (aligned from the right) loop portions, respectively. The average template structure look-up and selection time is less than 5 seconds per VH-VL pair.
Model assembly
As all VH and VL template structures have been pre-aligned onto a common consensus framework, the template coordinates for each segment can be transferred to the raw model without further adjustments. Afterwards, non-homologous sidechains are exchanged, and incomplete template sidechains, if any, are remodeled using the standard methods provided in Accelrys Discovery Studio 4.0.Citation53 Due to the fact that each variable domain segment is picked independently, the number of required sidechain adaptations per model is typically manageable.
Neighborhood-based sidechain refinement
One major (and to the best of our knowledge unique) feature of MoFvAb is the knowledge-based refinement of Fv sidechain conformations based on their chemical neighborhood. Due to the high degree of homology within the Fv region and the large number of available crystal structures, it is possible to characterize, for each position in the Fv, the spatially conserved neighborhood. In other words, it is possible to state that the sidechain of the amino acid at position A is surrounded by the sidechains of the amino acids at positions X, Y and Z in most of the available experimentally derived Fv structures.
Based on the antibody structures stored in RAB3D, we determined the spatially conserved neighborhood for each position in the Fv based on a 90% neighboring frequency threshold and a 4.0 Å distance cut-off. This means that we perceive a position X as being a conserved neighbor of position A if the sidechain atoms of the amino acid at position X are in contact (of equal to or below 4.0 Å) with the sidechain atoms of the amino acid at position A in more than 90% of the available structures. Not surprisingly, framework positions typically have relatively compact and conserved neighborhoods, while CDR loop positions can have larger and very diverse neighborhoods. For positions near the top of long CDR-H3 loops, the notion of a conserved neighborhood disappears completely, as their sidechains are either solvent exposed or in contact with antigen residues. An overview of the neighborhood variants per fingerprint position can be found in Table SI2.
Once the conserved neighborhood for each Fv position has been defined, it is possible to query the database for structures that feature a certain neighborhood constellation at a certain position, e.g., a template where position A is an arginine and its neighbors X, Y and Z are glycine, leucine and proline. A real example is given in . If a match is found, the sidechain conformation of X in the model can be adapted accordingly. We do this by replacing the original template coordinates with the coordinates of the template with the best neighborhood match. Again, the template coordinates (both backbone and sidechain) are copied to the raw model without modification.
Figure 5. The conserved neighborhood of position 199 is formed by the 8 residues 197, 202, 212, 251, 331, 351, 397 and 798. In the example structure shown here (PDB ID 3PP4), the neighborhood constellation for the asparagine at position 199 can be fully characterized by the string ‘N|WVWRANLY’, i.e., the one letter amino acid code for the amino acid residues at the given conserved neighborhood positions, or the letter X if the given antibody does not have the respective residues. Using this compact neighborhood notation, the database can be queried quickly for known structures with equal or similar neighborhoods from which the conformation for the central asparagine might be adapted. Color coding as described in .
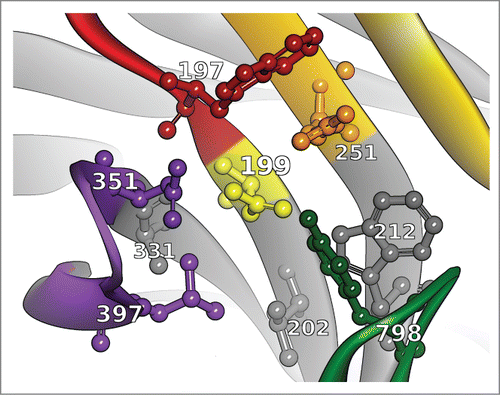
The neighborhood hits are ranked based on: 1) neighborhood amino acid similarity using a modified BLOSUM62 score, 2) resolution of the neighborhood template structure and 3) coordinate displacement with regard to the template currently in place. The BLOSUM62 score is modified such that the “match” of amino acid X with amino acid X (where an X in the neighborhood notation means that the residue is not present in the given antibody) is not penalized.
The coordinate displacement criterion is a double-edged sword: If a residue within our model is replaced, we would want it to fit the backbone of the structural template that is already in place. Adapting the sidechain conformation should not lead to additional error in the backbone conformation. On the other hand, correcting the amino acid sidechain conformation based on its chemical neighborhood might also involve justified changes to the course of the protein backbone. Within certain margins, neighborhood-based sidechain refinement should be able to correct the course of a misplaced loop. We are therefore pursuing 2 slightly different approaches of how coordinate displacement is handled, depending on where the individual residue is situated in the Fv. In the first case, depicted in , panels a) and b), neighborhood hits are ranked based on Cα distance with regard to the residue that is already in place. This promotes neighborhood-based exchanges that are largely retaining the backbone placement of the original template choice. In the second case, depicted in , panels c) and d), neighborhood hits are ranked based on the distance to the preceding residue in the chain, so that a neighborhood-based residue exchange is allowed to reroute the course of the backbone if it connects well to the backbone part already in place. In this case, the ascending and the descending part of the loop are treated separately, and closure at the tip of the loop is (at that time) not checked or enforced.
Figure 6. Schematic comparison of the 2 neighborhood-based sidechain refinement strategies. The situation prior to refinement is shown on the left. In the first approach, shown in panels a and b, a residue (white) is only replaced by another residue with a matching neighborhood (gray) if the Cα distance with regard to the residue that is already in place is low (the dashed red circle indicates the Cα distance check). This enables rearrangements of the sidechains (b) with little effect on the original loop structure (a). In the second approach, shown in panels c and d, a residue can be exchanged based on its neighborhood if it connects well to the backbone of its predecessor (the dashed blue and red lines indicate the backbone distance check) while moving from the N- and C-terminal edges of the CDR to the loop tip. The latter approach allows for larger corrections (d) to the course of the original loop (c) and is applied in CDR-H1 and CDR-H3.
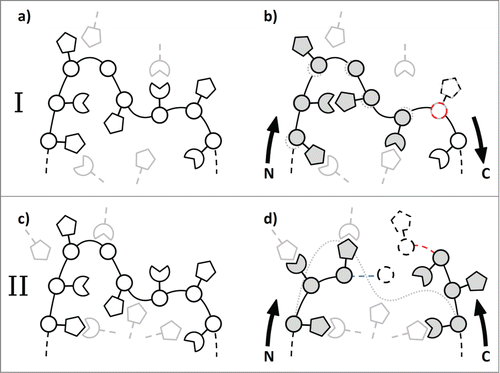
We obtained the best results when using the first approach (Cα distance) for CDR-H2, CDR-L2, and certain (in most cases CDR-proximal) parts of the framework, and the second approach (backbone distance to predecessor) for CDR-H1 and CDR-H3. Neither of the 2 approaches is applied to CDR-L1 and CDR-L3, where we could not detect a positive effect after neighborhood-based sidechain-refinement (data not shown). In the case of CDR-L1, this can probably be explained by the high number of distinct and non-overlapping canonical variants that are hard to differentiate by a single conserved neighborhood definition per residue.
For residues within the framework regions, database hits for structures with equal or very similar neighborhood are often abundant. Within the CDRs, and certainly within CDR-H3, neighborhood hits with sufficient homology are becoming very scarce (see Table SI2 for the number of known distinct neighborhood variants for a given position). To increase the number of possible hits for certain types of amino acids, we allow to exchange a residue based on a homologous neighborhood even if the hit sidechain is not equal, but only chemically similar to the query sidechain. This requires that the sidechain is restored to its original type after the exchange, under the constraint that conformation and placement of the sidechain are retained as far as possible. A list of the tolerated non-equal root sidechain exchanges is given in .
Table 8. List of tolerated sidechain exchanges between different but chemically similar amino acid types during neighborhood-based sidechain refinement
VH-VL orientation adjustment
After the raw models of VH and VL have been assembled, we use a knowledge-based approach to determine how the 2 domains will arrange with regard to each other. In this context, we quantify the degrees of freedom of VH-VL orientation in terms of the 6 ABangle parameters derived by Dunbar et al.,Citation32 consisting of one torsion angle, 4 bend angles (2 for each domain) and one distance. Earlier, we were able to show that the ABangle parameters can be predicted reasonably well based on a number of key residues at the interface of the 2 domains.Citation35 For a given VH-VL pair, we derive the 54 residue orientation fingerprint and use random forest model predictors learned using the Fv structures stored in RAB3D in order to obtain a prediction for each of the 6 ABangle parameters. Due to the fact that VH-VL orientation is largely determined by the complex interplay of a number of amino acids at key positions of the domain interface, the machine learning approach leads to more accurate results than deducing VH-VL orientation parameters merely from overall Fv sequence similarity. Finally, the VH and VL domain of the raw model are reoriented so as to match the predicted ABangle parameters.Citation35
ABangle parameter prediction and model reorientation are computationally inexpensive, in particular when compared to forcefield and dynamics-based approaches that would be the most obvious alternative for obtaining a realistic VH-VL orientation. Given that VH-VL orientation is subject to 6 different degrees of freedom that cover ranges of up to 25 degrees and are dependent on a high number of flexible and interacting sidechains, a single dynamics trajectory (and even less a single energy minimization gradient) might not be sufficient to identify the preferred orientation. By contrast, our approach is limited by the fact that predictions for Fv regions with a very uncommon VH-VL interface and rather “extreme” ABangle parameters tend to be inaccurate.
Clash removal and energy minimization
After VH-VL orientation has been adjusted, the model is scanned for steric clashes. Wherever steric overlap is detected, the involved sidechains are processed by rotamer search using the Oldfield rotamer library,Citation56 starting from the residues positioned at the CDR loop tips and moving toward the framework region (i.e., antigen or solvent exposed sidechains of residues at the top of the CDR loop have to give way before the less mobile sidechains of framework or framework-proximal residues). During this process, we pick the rotamer that minimizes steric clashes and maximizes predicted intramolecular hydrogen bonding, following a standard Accelrys Discovery StudioCitation53 protocol.
The processed model is parameterized for the CHARMm forcefieldCitation57 and minimized using the ‘Generalized Born with a simple Switching’ (GBSW)Citation58 implicit water model, first by the steepest descent and then by the conjugate gradient method.Citation59 To preserve the conformation of the template structures as far as possible during the course of energy minimization, we by default restrain the coordinates of all residues except those that are situated at fragment edges (with adjacent residues originating from different template structures) and those that have been adapted (change of amino acid type), remodeled (reconstruction of missing coordinates), or exchanged (in the course of neighborhood-based sidechain refinement).
Because all template coordinates are copied to the raw model without further adaptation, MoFvAb models tend to suffer from long or short bonds prior to energy minimization. This mainly affects CDR loops that have been assembled from 2 separate template structures for the ascending and the descending loop segment, but also regions where multiple residues have been exchanged based on their chemical neighborhood. To obtain a more robust starting conformation for energy minimization with CHARMm, we scan the model for very long bond and very short bonds between the amide carbon and the amide nitrogen of the protein backbone and relax affected CDR loops with a fast Dreiding-like forcefieldCitation60 while restraining the remainder of the model.
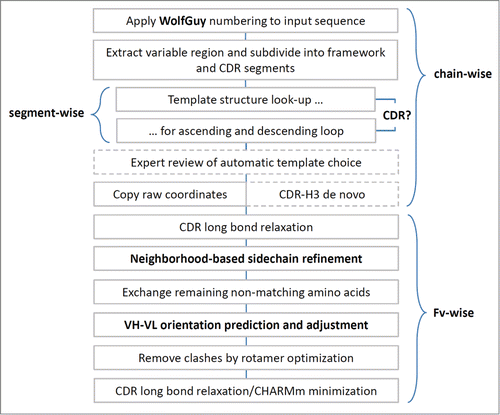
After the final energy minimization, the model is complete. The MoFvAb algorithm as presented here is summarized in .
Figure 7. Flow chart of the MoFvAb algorithm (production version). Boxes delineated with dashed lines indicate optional steps that require user intervention.
Excluded templates and evaluation datasets
To evaluate the performance of the algorithm, we compare the models generated by MoFvAb with a number of antibody crystal structures with regard to all-atom RMSD, deviation in ABangle VH-VL orientation space, and overall structural quality as described in the next sections. To simulate a situation where the structure to be build is not known, the use of any template structure that has equal to or more than 95% sequence identity in the CDRs of the respective variable domain is automatically prohibited, both during the initial template selection as well as during neighborhood-based sidechain refinement. This can be interpreted as a slightly more rigorous leave-one-out evaluation approach, as we not only exclude the original crystal structure from being used as a template, but also any sequence-identical antibodies (e.g., the same antibody in the complexed form) or variants with single point mutations (e.g., humanized variants of a murine antibody). The identification of sequence-identical template structures to exclude from model building is performed using the software CD-HIT.Citation61,62
To evaluate how MoFvAb compares to state-of-the-art antibody modeling software, we downloaded the crystal structures and participant models of the 2011 and 2014 AMA1 and AMA2Citation17-19 blind modeling studies from http://www.3dabmod.com. AMA1 Despite the fact that we try to mimic a blind modeling simulation by template exclusion, we, at the time of model building, had access to more template structures than the original AMA participants (in particular with regard to AMA1 in 2011) so that the direct comparison in terms of all-atom RMSD is not altogether fair, but possibly still indicative. The nine and 11 antibody structures of AMA1 and AMA2, respectively, have all been crystallized in the unbound form. To complement the evaluation set with a number of antibodies crystallized in the complexed form, we selected all publicly available antibody complex structures with a resolution of 1.8 Å or higher, and then discarded structures that were incomplete (having missing or incomplete residues in the Fv region) or had a CDR-H3 length of more than 13 residues. After making the dataset non-redundant with regard to Fv sequence, 41 distinct antibody crystal structures remained. We complemented this “high-resolution complex structure test set” with a single previously unpublished crystal structure of a theophylline-binding antibody that is introduced in the Supplemental Information of this article (Table SI3 and Fig. SI5). The coordinates of this antibody have been deposited in the PDB under the PDB ID 5BMF. See Table SI1 for an overview of the crystal structures of the evaluation datasets.
All-atom RMSD and 
To calculate the all-atom RMSD values for a given model-crystal structure pair, we first superimpose the VH domains to obtain the VH-specific values, then the VL domains to obtain the VL specific values, and finally both domain types simultaneously to obtain the all-atom RMSD for the complete Fv. In any case, the structures are superimposed using the Cα atoms of the respective β-sheet core (or cores) with the ‘SuperimposeProtein’ method provided in Accelrys Discovery Studio.Citation53
The all-atom RMSD is then calculated as the deviation of all heavy (i.e., non-hydrogen) atoms of the model with regard to the respective atoms in the crystal structure, given that the coordinates have been resolved. During the all-atom RMSD calculation, we correct for possible symmetry variants of the sidechains of arginine, aspartate, glutamate, leucine, phenylalanine, tyrosine and valine. For the sake of consistency with AMA2, we use the β-sheet core and CDR loop definition of Teplyakov et al.Citation19 as highlighted in Figures SI1 and SI2.
To quantify distances in the ABangle VH-VL orientation space, we define a set of ABangle parameters as the tuple The Euclidean distance between 2 sets of ABangle parameters is then
(See also ref. Citation35.)
As mingles angular (HL, HC1, LC1, HC2, LC2) with linear (dc) distance measures, it cannot be interpreted in terms of a unit of measure such as degrees. For calculating ABangle orientation parameters, we use the program code available at http://www.stats.ox.ac.uk/˜dunbar/abangle/ in a slightly modified version that works on WolfGuy-numbered structures.
MolProbity analysis
Model quality was assessed with MolProbityCitation63 as included in PHENIX version 1.9.Citation64 From the different structure validation metrics that MolProbity provides, we state the percentage of backbone conformations inside and outside of the favored Ramachandran region, the percentage of sidechain conformations classified as rotamer outliers, the Cβ deviations measure that is sensitive to incompatibilities between sidechain and backbone,Citation65 the clash score that is defined as the number of unfavorable all-atom steric overlaps larger or equal to 0.4 Å per 1000 atoms,Citation66 and finally the MolProbity score that merges clash score, the percentage of rotamer outliers, and the percentage of backbone conformations outside of the favored Ramachandran region into a single metric (smaller is better). If not already present, explicit hydrogen atoms were added to the structures prior to the analysis using Accelrys Discovery Studio.Citation53 All crystal structures were truncated to the Fv region to provide for better comparability with the MoFvAb Fv models.
Disclosure of Potential Conflicts of Interest
AB, AF, CQ, SK and GG are under paid employment by Roche Diagnostics GmbH. JB is under paid employment by F. Hoffmann-La Roche AG.
Supplemental_Material.docx
Download MS Word (2.2 MB)Acknowledgments
AB would like to thank Markus Rudolph for his help regarding MolProbity analysis, and James Dunbar for his valuable input regarding VH-VL orientation analysis and adjustment. A first prototype of MoFvAb was implemented in 2012 with the help of experts from Accelrys.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
AB is funded by the Roche Postdoc Fellowship Program.
References
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V (D) J recombination. Cell 2002; 109: S45-S55; PMID:11983152; http://dx.doi.org/10.1016/S0092-8674(02)00675-X
- Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev 2004; 18: 1-11; PMID:14724175; http://dx.doi.org/10.1101/gad.1161904
- Schatz DG, Ji Y. Recombination centres and the orchestration of V (D) J recombination. Nat Rev Immunol 2011; 11: 251-63; PMID:21394103; http://dx.doi.org/10.1038/nri2941
- Borrebaeck CA. Antibodies in diagnostics – from immunoassays to protein chips. Immunol Today 2000; 21: 379-82; PMID:10916140; http://dx.doi.org/10.1016/S0167-5699(00)01683-2
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2003; 2: 52-62; PMID:12509759; http://dx.doi.org/10.1038/nrd984
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10: 345-52; PMID:20414207; http://dx.doi.org/10.1038/nri2747
- Reichert JM. Marketed therapeutic antibodies compendium. mAbs 2012; 4: 413-5; PMID:22531442; http://dx.doi.org/10.4161/mabs.19931
- Hayden MS, Gilliland LK, Ledbetter JA. Antibody engineering. Curr Opin Immunol 1997; 9: 201-12; PMID:9099794; http://dx.doi.org/10.1016/S0952-7915(97)80136-7
- Kim SJ, Park Y, Hong HJ. Antibody engineering for the development of therapeutic antibodies. Mol Cells 2005; 20: 17-29; PMID:16258237
- Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Delivery Rev 2006; 58: 640-56; http://dx.doi.org/10.1016/j.addr.2006.01.026
- Niwa R, Satoh M. The current status and prospects of antibody engineering for therapeutic use: focus on glycoengineering technology. J Pharm Sci 2015; 104(3):930-41; PMID:25583555
- Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel 2012; 25(10):507-21; gzs024; PMID:22661385
- Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, Shi J, Deane CM. SAbDab: the structural antibody database. Nucleic Acids Res 2014; 42: D1140-6; PMID:24214988; http://dx.doi.org/10.1093/nar/gkt1043
- Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol 2007; 25: 1171-6; PMID:17891135; http://dx.doi.org/10.1038/nbt1336
- Pedotti M, Simonelli L, Livoti E, Varani L. Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin. Int J Mol Sci 2011; 12: 226-51; PMID:21339984; http://dx.doi.org/10.3390/ijms12010226
- Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci 2012; 101: 102-15; PMID:21935950; http://dx.doi.org/10.1002/jps.22758
- Almagro JC, Beavers MP, Hernandez-Guzman F, Maier J, Shaulsky J, Butenhof K, Labute P, Thorsteinson N, Kelly K, Teplyakov A, Luo J, Sweet R, Gilliland GL. Antibody modeling assessment. Proteins 2011; 79: 3050-66; PMID:21935986; http://dx.doi.org/10.1002/prot.23130
- Almagro JC, Teplyakov A, Luo J, Sweet RW, Kodangattil S, Hernandez-Guzman F, Gilliland GL. Second antibody modeling assessment (AMA-II). Proteins 2014; 82: 1553-62; PMID:24668560; http://dx.doi.org/10.1002/prot.24567
- Teplyakov A, Luo J, Obmolova G, Malia TJ, Sweet R, Stanfield RL, Kodangattil S, Almagro JC, Gilliland GL. Antibody modeling assessment II. Structures and models. Proteins 2014; 82: 1563-82; PMID:24633955; http://dx.doi.org/10.1002/prot.24554
- Tramontano A, Morea V. Assessment of homology-based predictions in CASP5. Proteins 2003; 53: 352-68; PMID:14579324; http://dx.doi.org/10.1002/prot.10543
- Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struc Biol 2005; 15: 285-9; http://dx.doi.org/10.1016/j.sbi.2005.05.011
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006; 22: 195-201; PMID:16301204; http://dx.doi.org/10.1093/bioinformatics/bti770
- Padlan EA. Anatomy of the antibody molecule. Mol Immunol 1994; 31: 169-217; PMID:8114766; http://dx.doi.org/10.1016/0161-5890(94)90001-9
- Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 1970; 132: 211-50; PMID:5508247; http://dx.doi.org/10.1084/jem.132.2.211
- Mariuzza RA, Phillips SEV, Poljak RJ. The structural basis of antigen-antibody recognition. Annu Rev Biophys Bio 1987; 16: 139-59; http://dx.doi.org/10.1146/annurev.bb.16.060187.001035
- Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342: 877-83; PMID:2687698; http://dx.doi.org/10.1038/342877a0
- Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol 1996; 263: 800-15; PMID:8947577; http://dx.doi.org/10.1006/jmbi.1996.0617
- North B, Lehmann A, Dunbrack RL. A new clustering of antibody CDR loop conformations. J Mol Biol 2011; 406: 228-56; PMID:21035459; http://dx.doi.org/10.1016/j.jmb.2010.10.030
- Weitzner BD, Dunbrack RL, Gray JJ. The Origin of CDR H3 Structural Diversity. Structure 2015; 23(2):302-11; PMID:25579815
- Abhinandan KR, Martin AC. Analysis and prediction of VH/VL packing in antibodies. Protein Eng Des Sel 2010; 23: 689-97; PMID:20591902; http://dx.doi.org/10.1093/protein/gzq043
- Chailyan A, Marcatili P, Tramontano A. The association of heavy and light chain variable domains in antibodies: implications for antigen specificity. FEBS J 2011; 278: 2858-66; PMID:21651726; http://dx.doi.org/10.1111/j.1742-4658.2011.08207.x
- Dunbar J, Fuchs A, Shi J, Deane CM. ABangle: characterising the VH-VL orientation in antibodies. Protein Eng Des Sel 2013; 26: 611-20; PMID:23708320; http://dx.doi.org/10.1093/protein/gzt020
- Dunbar J, Knapp B, Fuchs A, Shi J, Deane CM. Examining Variable Domain Orientations in Antigen Receptors Gives Insight into TCR-Like Antibody Design. PLoS Comput Biol 2014; 10: e1003852; PMID:25233457; http://dx.doi.org/10.1371/journal.pcbi.1003852
- Ramachandran GN. Conformation of polypeptides proteins. Adv Protein Chem 1968; 23: 283; PMID:4882249; http://dx.doi.org/10.1016/S0065-3233(08)60402-7
- Bujotzek A, Dunbar J, Lipsmeier F, Schäfer W, Antes I, Deane CM, Georges G. Prediction of VH-VL domain orientation for antibody variable domain modeling. Proteins 2015; 83(4):681-95; PMID:25641019
- Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 2014; 82: 1611-23; PMID:24519881; http://dx.doi.org/10.1002/prot.24534
- Zhu K, Day T, Warshaviak D, Murrett C, Friesner R, Pearlman D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014; 82: 1646-55; PMID:24619874; http://dx.doi.org/10.1002/prot.24551
- Maier JK, Labute P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014; 82: 1599-610; PMID:24715627; http://dx.doi.org/10.1002/prot.24576
- Berrondo M, Kaufmann S, Berrondo M. Automated Aufbau of antibody structures from given sequences using Macromoltek's SmrtMolAntibody. Proteins 2014; 82: 1636-45; PMID:24777752; http://dx.doi.org/10.1002/prot.24595
- Fasnacht M, Butenhof K, Goupil-Lamy A, Hernandez-Guzman F, Huang H, Yan L. Automated antibody structure prediction using Accelrys tools: Results and best practices. Proteins 2014; 82: 1583-98; PMID:24833271; http://dx.doi.org/10.1002/prot.24604
- Marcatili P, Olimpieri PP, Chailyan A, Tramontano A. Antibody modeling using the Prediction of ImmunoGlobulin Structure (PIGS) web server. Nature Protoc 2014; 9: 2771-83; http://dx.doi.org/10.1038/nprot.2014.189
- Canutescu AA, Dunbrack RL. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci 2003; 12: 963-72; PMID:12717019; http://dx.doi.org/10.1110/ps.0242703
- Choi Y, Deane CM. FREAD revisited: accurate loop structure prediction using a database search algorithm. Proteins 2010; 78: 1431-40; PMID:20034110
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196: 901-17; PMID:3681981; http://dx.doi.org/10.1016/0022-2836(87)90412-8
- Robin G, Sato Y, Desplancq D, Rochel N, Weiss E, Martineau P. Restricted diversity of antigen binding residues of antibodies revealed by computational alanine scanning of 227 antibody-antigen complexes. J Mol Biol 2014; 426: 3729-43; PMID:25174334; http://dx.doi.org/10.1016/j.jmb.2014.08.013
- Ramaraj T, Angel T, Dratz EA, Jesaitis AJ, Mumey B. Antigen-antibody interface properties: Composition, residue interactions, and features of 53 non-redundant structures. Biochim Biophys Acta Proteins Proteom 2012; 1824: 520-32; http://dx.doi.org/10.1016/j.bbapap.2011.12.007
- Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J Mol Biol 2008; 377: 1518-28; PMID:18336836; http://dx.doi.org/10.1016/j.jmb.2008.01.093
- Dengl S, Hoffmann E, Grote M, Wagner C, Mundigl O, Georges G, Thorey I, Stubenrauch KG, Bujotzek A, Josel HP, et al. Hapten-directed spontaneous disulfide shuffling: a universal technology for site-directed covalent coupling of payloads to antibodies. FASEB J 2015; 83(4):681-95; fj–14; PMID:25670234
- Kabat EA, Te Wu T, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. Darby, PA: DIANE Publishing; 1992
- Honegger A, Plückthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol 2001; 309: 657-70; PMID:11397087; http://dx.doi.org/10.1006/jmbi.2001.4662
- Kaas Q, Ehrenmann F, Lefranc MP. IG, TR and IgSF, MHC and MhcSF: what do we learn from the IMGT Colliers de Perles? Brief Funct Genomics Proteom 2007; 6: 253-64; http://dx.doi.org/10.1093/bfgp/elm032
- Accelrys Software Inc. Pipeline Pilot, Release 9.1.0.13, San Diego: 2013
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 4.0.0.13259, San Diego: 2013
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. P Natl Acad Sci U S A 1992; 89: 10915-9; http://dx.doi.org/10.1073/pnas.89.22.10915
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389-402; PMID:9254694; http://dx.doi.org/10.1093/nar/25.17.3389
- Oldfield T. Pattern-recognition methods to identify secondary structure within X-ray crystallographic electron-density maps. Acta Crystallogr D 2002; 58: 487-93; PMID:11856835; http://dx.doi.org/10.1107/S0907444902000525
- Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem 2009; 30: 1545-614; PMID:19444816; http://dx.doi.org/10.1002/jcc.21287
- Im W, Lee MS, Brooks CL. Generalized born model with a simple smoothing function. J Comput Chem 2003; 24: 1691-702; PMID:12964188; http://dx.doi.org/10.1002/jcc.10321
- Luenberger DG. Introduction to linear and nonlinear programming. Reading, MA: Addison-Wesley Reading; 1973
- Mayo SL, Olafson BD, Goddard WA. DREIDING: a generic force field for molecular simulations. J Phys Chem 1990; 94: 8897-909; http://dx.doi.org/10.1021/j100389a010
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22: 1658-9; PMID:16731699; http://dx.doi.org/10.1093/bioinformatics/btl158
- Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012; 28: 3150-2; PMID:23060610; http://dx.doi.org/10.1093/bioinformatics/bts565
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 2009; 66: 12-21; PMID:20057044; http://dx.doi.org/10.1107/S0907444909042073
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 2010; 66: 213-21; PMID:20124702; http://dx.doi.org/10.1107/S0907444909052925
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 2003; 50: 437-50; PMID:12557186; http://dx.doi.org/10.1002/prot.10286
- Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol 1999; 285: 1711-33; PMID:9917407; http://dx.doi.org/10.1006/jmbi.1998.2400