
Abstract
The application of protein engineering technologies toward successfully improving antibody pharmacokinetics has been challenging due to the multiplicity of biochemical factors that influence monoclonal antibody (mAb) disposition in vivo. Physiological factors including interactions with the neonatal Fc receptor (FcRn) and specific antigen binding properties of mAbs, along with biophysical properties of the mAbs themselves play a critical role. It has become evident that applying an integrated approach to understand the relative contribution of these factors is critical to rationally guide and apply engineering strategies to optimize mAb pharmacokinetics. The study presented here evaluated the influence of unintended non-specific interactions on the disposition of mAbs whose clearance rates are governed predominantly by either non-specific (FcRn) or target-mediated processes. The pharmacokinetics of 8 mAbs representing a diverse range of these properties was evaluated in cynomolgus monkeys. Results revealed complementarity-determining region (CDR) charge patch engineering to decrease charge-related non-specific binding can have a significant impact on improving the clearance. In contrast, the influence of enhanced in vitro FcRn binding was mixed, and related to both the strength of charge interaction and the general mechanism predominant in governing the clearance of the particular mAb. Overall, improved pharmacokinetics through enhanced FcRn interactions were apparent for a CDR charge-patch normalized mAb which was affected by non-specific clearance. The findings in this report are an important demonstration that mAb pharmacokinetics requires optimization on a case-by-case basis to improve the design of molecules with increased therapeutic application.
Abbreviations
| IgGs | = | immunoglobulins |
| CDR | = | complementarity-determining region |
| FcRn | = | neonatal Fc receptor |
| KD | = | equilibrium dissociation constant |
| ELISA | = | enzyme linked immunosorbent assay |
| HRP | = | horseradish peroxidase |
| kDa | = | kilodalton |
| SPR | = | surface plasmon resonance |
| SD | = | standard deviation |
| HBSS | = | Hank's balanced salt saline solution |
| AUC | = | area under the curve |
| IV | = | intravenous |
| pI | = | isoelectric point |
| PK | = | pharmacokinetics |
| TMD | = | target mediated drug disposition |
| NSB | = | non-specific binding |
Introduction
Over the past few decades, human or humanized monoclonal antibody (mAb) pharmaceuticals have been successfully used as therapeutic modalities in a wide array of human diseases due to their target binding specificity, bivalent interaction properties, potential to have innate effector function and their in vitro and in vivo biochemical stability. The compelling efficacy of the initial generation of these molecules has stimulated a tremendous effort to improve the biopharmaceutical properties of the next generation of these therapeutics.Citation1-4 Early efforts to improve the therapeutic potency of mAbs focused extensively on altering their target binding affinity/specificity using technologies such as in vitro affinity maturation via combinatorial approaches and rational in silico computer-based modeling.Citation3,5 The application of protein engineering technologies toward successfully improving antibody pharmacokinetics on the other hand, has yielded mixed results. This is in part related to an incomplete understanding of the complexity and interdependence of the in vivo mechanisms influencing mAb disposition. In this regard, it has become increasingly apparent that a mAb's target, whether soluble or membrane associated, provides a pathway for the antibodies clearance from the peripheral circulation. With that, optimizing an antibody's properties for target interaction needs to take into account the interplay and dynamics of additional non-target related in vivo interactions and a broader view toward balancing affinity improvements, which could become liabilities with respect to in vivo disposition.
Increasingly, reports have shown non-target binding related IgG disposition mechanisms (i.e., non-specific) related to biochemical/biophysical and neonatal Fc receptor (FcRn) binding properties also play an important role in the clearance of mAbs from the blood.Citation5-9 The FcRn-mediated mAb clearance mechanism has been the most actively studied non-specific IgG clearance factor. As such, a number of laboratories have examined the effects of modulating the interaction of mAbs with the FcRn in an effort to advance improvements in mAb PK.Citation10-19 Some disclosures have reported success in reducing IgG clearance via Fc engineering strategies that improve the FcRn binding properties of mAbs.Citation12,20,21 mAbs targeting low expression soluble antigens are more likely to have their kinetics dictated by the non-specific FcRn pathway, and, as such, the influence of Fc engineering to improve FcRn interaction and pharmacokinetics is most clearly demonstrated in in vivo systems with low/no endogenous antigen.Citation11,12,17,19 An excellent example of this is the case of the Fc engineered anti-respiratory syncytial virus (RSV) mAb.Citation10 However, this situation becomes much less clear when a mAb's disposition is dictated by target type/turnover as mentioned above. For example, in a baboon pharmacokinetic study of a humanized anti-CD4 antibody with a single Fc substitution (N434H) designed to improve its binding to FcRn relative to its wild-type mAb equivalent,Citation22 the authors contended that at non-saturating concentrations, CD4 receptor-mediated internalization was the major elimination pathway for the variant mAb.Citation22 As the antibody concentration increased, the CD4 receptors became saturated and an ∼2-fold slower clearance of the variant IgG was observed compared to its wild-type counterpart.Citation22 Complementary approaches, such as engineering pH sensitivity into a mAb-receptor target interaction along with improving mAb:FcRn interactions, take into account some of the pragmatic limitations with respect to administering higher antibody concentrations to saturate antigen binding.Citation23-25 Thus, these approaches, when combined with improved FcRn interactions, have proved fruitful for improving mAb pharmacokinetics in several cases for both membrane and soluble targets.Citation26,27
There are, however, many instances that also show mixed in vivo kinetic findings based solely on the FcRn-mediated mAb clearance mechanism even in the absence of target-mediated clearance.Citation14,28 A number of recent reports have identified additional non-FcRn clearance pathways related to mAb biochemical/biophysical characteristics that can significantly affect mAb disposition.Citation7-9,21,29,30 These studies have reported that factors such as the pI/charge patches, stability/aggregation potential, post-translational modifications (glycosylation and methionine oxidation) and the characteristics of the Fab region can all have significant negative effects on antibody clearance and elimination in vivo. Such characteristics can mask or limit benefits being targeted by engineering for improved antigen binding affinity or Fc-related functions. Understanding and avoiding these negative molecular influences may allow maximal optimization of in vivo performance. Clearly, defining the relative contribution of the multiple factors affecting IgG disposition on a molecule-to-molecule basis is critical to guiding rational engineering strategies. Given these considerations, broader evaluations of the influence of both specific/target and non-specific clearance factors on the PK of mAbs in an integrated manner are warranted.
The goal of the current investigation was to integrate these concepts by characterizing the effect of charge-related non-specific cellular binding and FcRn binding affinity on the clearance of mAbs whose disposition are driven by both non-specific (FcRn) and specific or target-mediated disposition (TMD). Findings from the characterization of 2 pairs of mAbs are presented. The clearance of the non-specific clearance (FcRn) pair was influenced by charge-driven non-specific binding (NSB) interactions. The pharmacokinetics of the other IgG pair was influenced by both NSB and TMD in non-human primates. The influence of the combination of complementarity-determining region (CDR) charge patch balancing with improved FcRn binding on the pharmacokinetics of the 2 mAb pairs was assessed in cynomolgus monkeys. The previously described V308P Fc mutation, which improves FcRn binding, was studied within the context of both the parental IgGs and the CDR charge-balanced molecules for each pair of mAbs. Antibody engineering to reduce NSB was demonstrated to have a significant effect on the clearance of both pairs of mAbs, while increased FcRn interactions led to mixed pharmacokinetic findings in cynomolgus monkeys. Overall, improved pharmacokinetics through enhanced FcRn interactions were apparent for a CDR charge-patch normalized mAb, which was affected only by non-specific clearance. The results point to the importance of integrating and understanding both the characteristics of the biological target and molecular/biochemical aspects of the mAb in guiding the application of optimal engineering strategies.
Results
Description of the IgG molecules
Two previously reported pairs of humanized IgG molecules (mAb A and mAb B) and (mAb C and mAb D) developed against 2 different undisclosed targets were used in this study.Citation6 Pharmacokinetic evaluations of these molecules had shown that differential solvent-exposed patches of charge within the CDRs were connected to the in vivo clearance of the mAbs in mice.Citation6 Herein, we introduced the Fc mutation V308P into all 4 molecules to improve their FcRn binding in an effort to better characterize the role of the FcRn clearance mechanism in conjunction with the charge-driven non-specific interactions. The constructs are listed in .
Table 1. General description of the IgGsFootnote
Characterization of the FcRn binding, non-specific interactions and biophysical properties of the IgGs
Following the construction of the V308P Fc variant IgGs, we characterized the binding of each of the molecules to cynomolgus monkey FcRn (cFcRn) and their interactions with heparin to determine the role the Fc mutation has on these parameters. The binding affinities of the 4 V308P IgG molecules with immobilized cFcRn were measured using previously reported surface plasmon resonance approaches.Citation14,15 The cFcRn binding affinities (Kd) of the V308P variants at pH 6 were improved relative to the parental and ‘charge reengineered’ IgGs (). No direct binding to cFcRn at pH 7.4 was detected for any of the IgGs or V308P variant molecules (data not shown).
Table 2. Biophysical and cynomolgus monkey FcRn binding properties of the IgGs
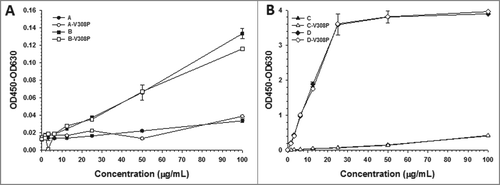
Non-specific binding of the IgGs to heparin was determined using approaches reported in earlier studies.Citation6 The V308P variants showed no differences in heparin binding relative to their wild-type mAb counterparts (, ). Greater binding to heparin-coated plates was observed for both the parental versions of the V308P IgGs (B-V308P and D-V308P) relative to their respective ‘charge reengineered’ modified V308P mAbs (A-V308P and C-V308P) (, ). Similar to mAb D, a very strong interaction of mAb D-V308P with heparin was observed at concentrations as low as 1.56 µg/ml (). In contrast, interaction of mAb C-V308P (i.e., the CDR charge balanced product of D-V308P) with heparin was only discernable at concentrations greater than 25 µg/mL (), similar to mAb C. In regards to the other mAb pair, the non-specific binding of mAb B-V308P to heparin was greater when compared to mAb A-V308P, but these interactions were much weaker than our mAb C and D pair and only discernible at concentrations above 12.5 µg/ml ().
Figure 1. CDR charge balancing reduces non-specific binding to heparin. Non-specific binding of (A) A and B and (B) C and D to heparin-coated plates.
In addition to the cynomolgus monkey FcRn binding and non-specific binding, we also measured additional properties of the mAbs in vitro that have been shown to have an influence on IgG and protein stability and clearance in vivo,Citation12,31 and compared these to each construct's parental and ‘charge-reengineered’ mAb characteristics. The pI values of the V308P variants were determined using capillary isoelectrophoresis. Our results indicated no major differences in the pI of Fc mutant molecules when compared within each platform (<0.3) (). A-V308P and B-V308P had pI values of 8.6 and 9.0, respectively, (), while the C-V308P and D-V308P molecules had pI values of 9.1 and 9.5, respectively (). The Tm of the mAbs was determined using differential scanning calorimetry (DSC). No notable differences in Tm were observed in the CH2, CH3 or Fab regions within each platform (). Using hydrophobic interaction column (HIC) chromatography, we observed no association of any of the V308P IgGs in our study to hydrophobic interaction column similar to their wild-type mAb counterparts (data not shown).
Pharmacokinetics of the parental and ‘charge reengineered’ IgGs in cynomolgus monkeys after a single intravenous administration
As mentioned above, we previously conducted murine pharmacokinetic studies for mAbs A, B, C and D, which showed the differential solvent-exposed charge was connected to the in vivo clearance in mice.Citation6 In mice, neither set of molecules had a target-mediated component to their clearance either due to low/no endogenous antigen or the lack of the cross-reactivity of the antibodies with the murine antigen. In the present work, we chose to evaluate the pharmacokinetics of our constructs in cynomolgus monkeys. In contrast to mice, we expected TMD to contribute to the overall clearance for mAbs C and D. During the course of the development of these molecules, cross-reactivity with the cynomolgus monkey version of the membrane-associated target had been observed for mAbs C and D (data not shown). The KD values of mAbs C and D for the recombinant soluble version of their cynomolgus monkey antigen were determined to be 1.76 + 0.28 nM and 2.40 + 0.72 nM, respectively, in vitro (data not shown). TMD was known to be saturated after single intravenous (IV) administrations of 10 and 30 mg/kg for mAbs C and D due to the finding of nearly dose proportional increases in exposure between these 2 levels (data not shown; unpublished findings). In the case of mAbs A and B, we did not expect target-driven clearance to influence the pharmacokinetics of these molecules as there are little/no circulating endogenous antigen in cynomolgus monkeys, similar to mice. As a consequence of the differences in TMD influencing the clearance of the IgGs in cynomolgus monkey, we used a higher dose known to nearly saturate target concentrations (data not shown) to evaluate the pharmacokinetics of mAbs C and D (10 mg/kg each) relative to mAbs A and B (2 mg/kg each).
After a single 10 mg/kg IV administration to cynomolgus monkeys, the serum concentrations for the parental and charge-reengineered molecules, mAbs C and D, respectively, showed dramatic differences in their cynomolgus monkey pharmacokinetics (). The mean ± SD clearance of mAbs C and D were 1.86 ± 0.28 mL/h/kg (N = 4 animals) and 9.7 mL/h/kg (N = 2 animals), respectively (). In the case of mAbs A and B, following a single 2 mg/kg IV administration to cynomolgus monkeys, the parent (mAb B) and charge-reengineered (mAb A) molecules were cleared from the circulation in a bi-phasic manner, showing a rapid distribution phase followed by a prolonged elimination phase characteristic of antibodies with insignificant peripheral levels of target antigen (). The serum concentrations of mAb A tended to be greater than for mAb B over the time course studied with overall mean clearance of mAb A approximately 3-fold slower than for mAb B in cynomolgus monkeys (). The mean ± SD clearance of mAbs A and B, were 0.51 ± 0.12 mL/h/kg and 1.87 ± 0.67 mL/h/kg, respectively ().
Figure 2. CDR charge balancing improves the cynomolgus monkey pharmacokinetics of mAbs. Pharmacokinetic profiles of (A) mAbs A and B and (B) C and D. mAbs A and C are CDR charge balanced from mAbs B and D, respectively. Data are the mean of 2 animals/time point for mAb D. Data are the mean ± SD (standard deviation) for 4 animals/time point for mAb C and 3 animals/time point for mAbs A and B. The pharmacokinetics were assessed following a single IV dose of 2 mg/kg for mAbs A and B and 10 mg/kg for mAb C and D.
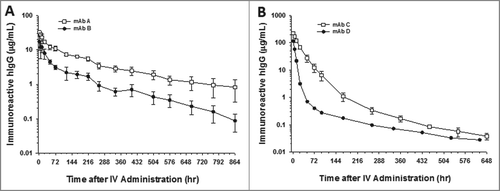
Table 3. Cynomolgus monkey pharmacokinetic parameters of the IgGs
Pharmacokinetics of the V308P Fc variant antibodies in cynomolgus monkeys
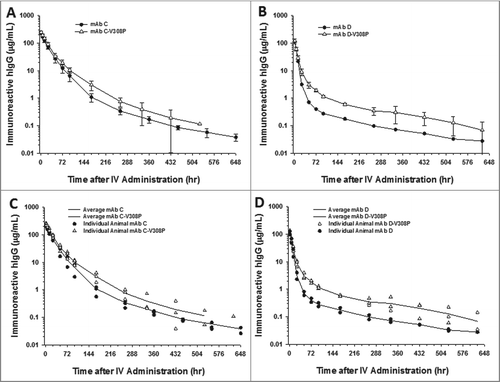
The effect of enhanced FcRn binding conferred by the V308P Fc mutation on IgG pharmacokinetics was evaluated for each of the 4 mAbs in cynomolgus monkeys. The V308P mutation significantly improved the clearance of mAb A ( and ). The mean clearance rates of mAb A improved ∼2.7-fold from 0.51 ± 0.12 mL/h/kg to 0.19 ± 0.03 mL/h/kg mL/h/kg (). Mixed pharmacokinetic findings were observed for the V308P Fc variant of mAb B ( and ). The individual animal data for the mAb B molecules () show variability in the concentration versus time profiles, wherein the pharmacokinetics of an animal in each group biases the descriptive quantitative mean clearance parameter values (). From a qualitative perspective, the pharmacokinetic profiles of the individual animal data show a longer elimination half-life is conferred by the V308P mutation to mAb B ( and ). In contrast, the V308P mutation showed no meaningful effect on the pharmacokinetics of mAbs C and D, which had both NSB and TMD clearance components ( and ).
Figure 3. The V308P Fc variant shows no improvements in the cynomolgus monkey pharmacokinetics for a mAb with charge-based NSB, but does improve the clearance of a mAb with no charge-related NSB. Mean pharmacokinetic profiles of (A) mAbs A and A-V308P, which had no charge-associated NSB, and (B) B and B-V308P, which showed binding to heparin in vitro. Individual animal pharmacokinetic profiles of (C) mAbs A and A-V308P and (D) B and B-V308P. Data are the mean ± SD (standard deviation) for 3 animals/time point for all the mAbs. The pharmacokinetics were assessed following a single IV dose of 2 mg/kg for each mAb.
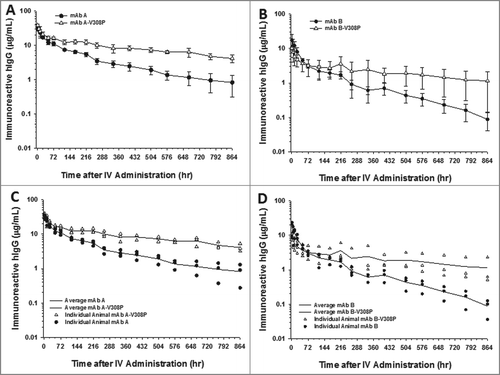
Figure 4. The V308P Fc variant shows no improvements in the cynomolgus monkey pharmacokinetics for mAb with and without charge-based NSB in the presence of a TMD clearance component. Mean pharmacokinetic profiles of (A) mAbs C and C-V308P, which had no charge-associated NSB but bind target antigen, and (B) D and D-V308P, which also bind target antigen, as well as showed binding to heparin in vitro. Individual animal pharmacokinetic profiles of (C) mAbs C and C-V308P and (D) D and D-V308P. Data are the mean ± SD (standard deviation) for 3 animals/time point for all the V308P Fc variant mAbs and 4 animals/time point for mAb C. Data are the mean of 2 animals/time point for mAb D. The pharmacokinetics were assessed following a single IV dose of 10 mg/kg for each mAb.
Discussion
In the current work, we directly examined the interplay between charge-related NSB along with the factors of target- and FcRn-mediated elimination mechanisms on the pharmacokinetics of 2 sets of mAbs in cynomolgus monkeys. Importantly, we demonstrate an antibody's NSB properties (mostly in the variable region) are perhaps the most important characteristic to take into account with regard to engineering these mAbs for improved in vivo performance. The magnitude of this interaction had an over-riding impact on the clearance of both pairs of mAbs, and is likely a major factor leading to the mixed benefit of Fc-FcRn engineering on pharmacokinetics in cynomolgus monkeys.
The A, B, C and D mAbs were described in previously published work.Citation6 Poor murine pharmacokinetics for mAbs B and D were observed and attributed to their increased charge-based non-specific binding.Citation6 In the present study, we choose to evaluate the pharmacokinetics of our constructs in cynomolgus monkeys to connect the observation between species and allow characterization of the influence of NSB in the context of mAbs cleared through TMD and non-specific (FcRn) mechanisms. Due to their well characterized physiochemical characteristics in vitro and varied target binding properties, these molecules served as reasonable tools to evaluate the relative balance of both specific (i.e., TMD) and non-specific clearance factors in an integrated matter. Employing Fc mutations that enhance the Fc-FcRn interaction allowed us to more systematically probe the inter-relation and relative contribution of NSB, TMD and FcRn recycling on the in vivo properties of these mAbs.
Similar to previous findings in mice,Citation6 the results from this study clearly demonstrate that charge-based NSB has a negative effect on pharmacokinetics in non-human primates, and that balancing charge in the solvent-exposed CDR of mAbs can improve this liability. mAbs B and D contain stretches of solvent-exposed positive charge in the CDR that increased heparin binding in vitro (), and likely contribute to increased charge-based non-specific binding in vivo. The unusually rapid clearance of the mAbs B and D (∼2 and ∼9.7 mL/h/kg, respectively) was predominantly attributed to this physiochemical property in their CDRs. The non-specific nature of the interaction in vivo is supported by the consistency of these observations across both non-human primates and rodents,Citation6 resulting in clearance rates that are well outside values generally considered ‘normal’ for well-behaved mAbs.Citation32 The charge reengineered variants of mAbs B and D (mAbs A and C, respectively) dampened the non-specific binding to heparin in vitro, which improved the cynomolgus monkey PK. As in mice,Citation33 the improved pharmacokinetics are likely related to reduced non-specific tissue uptake and subsequent catabolism. The more modest (∼2-fold) increase in exposure for mAb A relative to the ∼10-fold exposure enhancement for mAb C relative to each molecule's parental IgG was consistent with the apparently greater role of non-specific binding in influencing the clearance of mAb D vs. mAb B in cynomolgus monkeys. However, it is important to note that although charge rebalancing significantly improved the pharmacokinetics, mAb C still displays a clearance rate that is greater than 1 mL/hr/kg and is similar to the charge unbalanced mAb B (). Given the doses used to study the pharmacokinetics of mAbs C and D saturated TMD, the data suggest there may still be a component of NSB influencing mAb C clearance and thus the molecule may benefit from further charge-based engineering. Compared to the rodent findings,Citation6 the non-human primate observations also show more rapid clearance of mAbs C and D in cynomolgus monkeys. Similarly, the pharmacokinetic effect of charge rebalancing mAb B to generate mAb A trended to also show a difference across species with greater exposure increases observed in cynomolgus monkeys (∼2-fold) relative to mice (∼1.4-fold).Citation6 The findings suggest the charge-based non-specific binding may play a larger role in mAb clearance in cynomolgus monkeys relative to mice; however it cannot be fully precluded that there may be additional factors or the balance of multiple elements affect the clearance of the mAbs differentially, which lead to these observations. Nonetheless, the cynomolgus monkey pharmacokinetic findings facilitated progress toward leveraging this model system as an approach to more broadly integrate both charge-based non-specific binding and TMD (or lack thereof in the case of mAbs A and B) in the pharmacokinetic evaluations for mAbs with enhanced FcRn interactions.
Engineering of mAbs A, B, C and D to incorporate the V308P Fc mutation for enhanced FcRn-mediated recycling yielded mixed results. All four mAb V308P variants exhibited improved cynomolgus monkey FcRn binding (∼50–100 times higher affinity) relative to their native sequences.Citation33 Despite these binding improvements, mAb A-V308P is the only variant which displayed a clear clearance benefit compared to its wild-type form in cynomolgus monkeys ( and ). The apparent behavior of the V308P Fc mutation to enhance mAb A durability in non-human primates is likely related to both the in vitro and in vivo properties of this IgG. Following CDR charge balancing, mAb A showed both in vitro physiochemical properties and in vivo pharmacokinetics that are in essence characteristic of a reasonably ‘normal’ IgG. The pharmacokinetic improvement conferred by V308P Fc engineering on mAb A are not entirely surprising and consistent with previous studies of an anti-RSV, anti-hepatitis B and other Fc engineered mAbs that have insignificant target interactions and clearances expected for a mAb with well-behaved physiochemical properties.Citation10,12,18 The lack of apparent improvement in the mean in vivo half-life of V308P mAb A may be attributable to the need for a longer study duration to more adequately characterize the elimination phase. In contrast, the magnitude of pharmacokinetic influence imparted by the V308P Fc variants in the presence of moderate charge-based non-specific binding appears relatively modest. Examination of the individual animal data for mAb B and its V308P Fc variant suggests variability in the pharmacokinetic profiles across the non-human primates, raising the question of whether the non-specific binding renders FcRn affinity enhancements less effective. This may be one of the factors that explain why, when compared with mAb B, the pharmacokinetics of the V308P Fc mutant of mAb B tend to show improvement in its terminal phase half-life but no improvement in clearance ( and ). This observation may be related to increased mAb binding to endothelia via NSB interactions, followed by internalization. It may be speculated that a portion of this initially internalized mAb is diverted to a degradation pathway, with the strength of the cell surface association weak enough to allow some FcRn salvage later in the process. Taken together, the data suggest the FcRn mechanism has some potential to improve the pharmacokinetics of mAbs with weak/moderate charge-based non-specific binding, but that the characteristic of NSB is still a major over-riding factor to clearance.
In the case of the V308P Fc variant constructs for the molecules with a TMD component to their pharmacokinetics, the situation was somewhat more complicated. There was little discernable pharmacokinetic benefit imparted by V308P mutation on mAb D, which has extremely strong NSB characteristics ( and ). Consistent with the mechanism proposed above, any enhancement in FcRn recycling is masked by NSB. In this case, there is probably some TMD component over-riding this interaction as well, but this is difficult to parse out in the face of significant NSB. As mentioned above, reducing the strong charge-based NSB of mAb D yielded significantly improved clearance to mAb C ( and ). However, the data suggests that mAb C still retained enough NSB binding () to negatively influence its overall clearance (1.8 mL/h/hg), which is similar to that of non-charge balanced mAb B (). In contrast to mAb B, where there were trends of increased terminal phase half-life with improved Fc-FcRn interactions, for mAb C, improving the FcRn binding in the presence TMD did not manifest into an obvious pharmacokinetic benefit relative to the native mAb ( and ). The residual NSB for mAb C makes it difficult to clearly characterize the contribution of TMD to the lack of influence of the FcRn mutations. It is postulated that for antibodies directed to high turnover targets, the peripheral IgG clearance is driven through target binding.Citation24,27,34 The literature is generally consistent in describing the need to either saturate TMD or engineer a pH sensitivity into the receptor/target interaction to demonstrate the benefit of Fc mutations that enhance FcRn binding.Citation22,25 In the case of an anti-PCSK9 mAb series, an alternate pathway of clearance of mAb-target make engineered Fc-FcRn interactions less influential.Citation34 In this study, disengaging the formed complex was critical in allowing the mAb to be cleared through a predominantly FcRn mediated process, where Fc mutations demonstrated benefit.Citation34 This process is somewhat analogous to the charge surface interactions described here. The charge interaction is an alternate pathway or clearance mechanism that makes the mAb inaccessible to FcRn mediated disposition.
We and others previously speculated that the slower clearance of mAbs with reduced charge-related non-specific binding results from several mechanisms, including 1) decreased fluid phase endocytosis because of poorer cell binding due to charge repulsion with the negatively charged extracellular membrane, Citation6,7,31,35,36 or 2) decreased lysosomal degradation rates.Citation7,37 We postulated the reduced clearance of charge balanced mAbs is likely a consequence of both of these mechanisms, but to variable degrees as discussed elsewhere.Citation6 The current findings in non-human primate are on the whole consistent with our earlier hypotheses around the role of charge-related non-specific binding in mAb clearance. We propose, when there is no target-mediated clearance, the reduction in the non-specific binding of the charge-reengineered mAbs lessens the magnitude of the molecule's non-specific binding to cells (endothelia) and decreases its uptake into tissue. In a similar respect, the weaker interactions of Fc variants of these mAbs with membrane components may allow more efficient salvage/recycling via the FcRn mechanism and consequently improved PK properties compared with mAbs that do have a moderate charge-based pharmacokinetic liability. As the strength of the non-specific binding increases, we hypothesize the greater degree/strength of non-specific association these mAbs with membrane components leads to their increased cellular uptake independent of target. This property likely results in reduction of the ability of the FcRn to effectively salvage mAbs with these properties. From a cellular trafficking perspective, our data suggest high charge-based NSB mAbs are partitioned away from the recycling pathway and toward lysosomal degradation due to the increased charge-based interactions. In any case, to fully appreciate the benefit of Fc engineering to improve FcRn interaction, it is likely prudent to start with a molecule having desirable biophysical/physiochemical properties, including charge characteristics.
In summary, the findings in this report are an important demonstration that mAb pharmacokinetics is influenced by many factors both physiological and biochemical. There are multiple in vivo factors around the nature of the mAb (including its target/turnover/tissue distribution) that can influence disposition and elimination differentially. Careful delineation of the preponderance of these factors on a molecule-to-molecule basis, along with application of a rationally-based approach to engineer IgGs, will ultimately lead to the design of mAb molecules with increased therapeutic value for patients.
Materials and Methods
Cell culture for protein expression
293EBNA cells were maintained at 37°C under 5–8% CO2 conditions in Dulbecco's modified Eagle's medium/F-12 (Gibco) supplemented with 20 mM HEPES (Gibco), 5 μg/mL nucellin (Eli Lilly and Company), 0.4 μg/mL tropolone (Sigma Aldrich), 0.075% (w/v) F68 (Gibco) and 50 μg/mL geneticin (Sigma Aldrich).
Construction, expression and purification of recombinant proteins
The Fab regions were discovered and engineered at Eli Lilly and Company. These were cloned into mAb expression vectors to fuse with constant regions of human kappa light chain and either human IgG1 or IgG4 heavy chain with or without V308P mutation using standard molecular biology approaches and confirmed by DNA sequencing. Each of the IgG4 antibody backbones also contains a specific mutation (serine to proline) in the hinge region to inhibit heavy chain exchange.Citation38 All the IgGs were expressed in 293EBNA cells and purified from culture supernatants using Protein-A Sepharose (GE Healthcare) affinity chromatography followed by size exclusion chromatography methods.
Recombinant soluble cFcRn was expressed in 293EBNA cells transfected with plasmids encoding for the soluble portion of αFcRn and β2-microglobulin, and the protein was purified as described previously.
Evaluation of the FcRn binding affinity
The interaction of the IgG1 and IgG4 molecules with recombinant, immobilized cFcRn was monitored by SPR detection using a Biacore 3000 instrument (GE Healthcare) as described previously.Citation14 Briefly, recombinant soluble cFcRn was immobilized to flow cell 2 of a CM5 sensor chip using amine coupling chemistry (GE Healthcare). The FcRn immobilization surface density was approximately 300 RU. The first flow cell was used as a blank control surface lacking FcRn. All binding experiments were performed with compounds dissolved in running buffer phosphate buffered saline (PBS) with 0.005% Tween 20, pH 6 or PBS with 0.005% Tween 20, pH 7.4 and the samples were run at a flow rate of 100 µl/min for 30 seconds with a dissociation time of 10 minutes. PBS (pH 7.4) was used as dissociation buffer. PBS with 0.005% Tween 20, pH 6 was used as running buffer for the experiments performed to determine the affinity of IgGs to cFcRn. A concentration range of 0.00316 µM to 3.16 µM of each of the IgGs was used to estimate the association and dissociation constants. The binding data were obtained by subtracting the signal of flow cell 1 (blank flow cell not coupled with FcRn) from flow cell 2. Kinetic (association and dissociation) data were then simultaneously fit to a heterogeneous binding model for IgG-FcRn interactions (BIAevaluation, Ver. 4.1). The data curves for binding and dissociation phases of the sensorgrams for the IgGs at pH 6.0 had low residuals and low χ2 values. The mean of Kd values accounting for the greatest fraction of binding from 2 independent experiments were reported.
Evaluation of mAb isoelectric points (pIs)
The pIs of the mAbs were determined by capillary isoelectric focusing (cIEF), PA 800 plus Pharmaceutical Analysis System (Beckman Coulter). Samples were prepared by mixing 5–10 μg sample with 200 µL of 3 M urea-cIEF gel, 12.0 µL of Pharmalyte 3–10 (GE Healthcare ), 20.0 µL of cathodic stabilizer (500 mM arginine), 2.0 µL of anodic stabilizer (200 mM iminodiacetic acid), 2.0 µL of each of 5 pI markers (pI 10.0, 9.5, 7.0, 5.5, and 4.1), and vortexing for 15 seconds. The electrophoresis and data collection were performed using a focusing step voltage 25 kilovolts for 15 minutes, a chemical mobilization step voltage 30 kilovolts for 30 minutes, a UV detection at a wavelength 280 nm and a data collection rate of 2 hertz. The cartridge temperature was 20°C and the sample storage temperature was 10°C. The data were analyzed by using 32 Karat program (Beckman Coulter).
Evaluation of thermal stability (Tm)
Thermal stability of samples was measured using a TA Instruments NanoDSC equipped with an autosampler. Samples diluted to 0.5 mg/mL in PBS were heated from 20 to 110°C at a rate of 1°C/min under 45 psi of pressure. Sample scans were buffer blank subtracted, converted to molar heat capacity, and fit to a 2-state scaled model with 3 transitions representing the CH2, CH3 and Fab domain unfolding to obtain Tm.
Binding to heparin-coated plates
Heparin binding plates (BD Biosciences, Catalog no 354676) were coated overnight with 10 µg/ml of heparin (Sigma, Catalog no H3149). The plates were washed 4 times with 1X TBS Tween wash buffer (Teknova, Catalog no T0310) and blocked with PBS-casein blocking buffer (Pierce, Catalog no 37528). Again, the plates were washed and incubated with 1.56–100 µg/ml of A, B, C or D in blocking buffer at room temperature for 2 hours. The plates were washed and horseradish peroxidase conjugated mouse anti-human IgG (Southern Biotech, Catalog no. 9040–05) was added at a dilution of 1:5000. The plates were then incubated at room temperature for 1 h and washed. A freshly prepared TMB substrate (KPL, Catalog no. 50–76–01 and 50–65–00) was added to the washed plates and incubated at room temperature to develop the color. The reaction is stopped by adding stop solution and the plates were read in a Spectramax Plus microplate reader (Molecular Devices).
Cynomolgus monkey pharmacokinetic studies
A single cynomolgus monkey pharmacokinetic study was performed for mAbs A, B and their respective V308P Fc variants. In this study 3 male cynomolgus monkeys (2.8–3.8 kg) were assigned to one of 4 study groups. Each animal received a single IV dose of the mAb A, B, or V308P Fc variant IgG dissolved in PBS (pH 7.4) at 2.0 mg/kg. Blood samples were collected from femoral vein prior to dosing and at 1, 6, 12, 24, 48, 72, 120, 168, 216, 264, 336, 408, 504, 576, 672, 768 and 864 hours after administration of the dose. The blood samples were allowed to clot at ambient temperature prior to centrifugation to obtain serum.
Three independent cynomolgus monkey pharmacokinetic studies were performed for mAbs C, D and the V308P Fc variants of each molecule. In the first study, the pharmacokinetics of mAb D were evaluated in 2 male cynomolgus monkeys (2.8–3.8 kg) following an IV dose level of 10.0 mg/kg to each animal dissolved in PBS (pH 7.4). The pharmacokinetics of mAb C were examined in a separate study with 4 male cynomolgus monkeys (2.8–3.8 kg) following an IV dose of 10.0 mg/kg each dissolved in PBS (pH 7.4). In the third study, the kinetics of mAbs C-V308P and D-V308P were assessed in 3 male cynomolgus monkeys each (i.e., per compound) following an IV dose of 10.0 mg/kg each dissolved in PBS (pH 7.4). The blood samples were allowed to clot at ambient temperature prior to centrifugation to obtain serum.
Bioanalytical assays and pharmacokinetic data analysis
Concentrations of the mAbs (as either wild-type IgG or V308P) in cynomolgus monkey serum were determined using validated antigen capture ELISAs for each of 2 mAb families. The wild-type and V308P variant IgG standards were prepared in cynomolgus monkey serum using a standard curve range of 1.56 to 100 ng/mL. The lower limit of quantitation (LLOQ) was defined as 4 ng/mL for all the mAbs.
Pharmacokinetic parameters were calculated using the WinNonlin Professional (Version 3.2) software package (Pharsight Corporation, Mountain View, CA). Serum concentration-time data were calculated using a model-independent approach based on the statistical moment theory. The parameters calculated included the maximum serum concentration (Cmax), area under the curve (AUC0–∞), clearance (CL), and elimination half-life (t1/2).
Disclosure of Potential Conflicts of Interest
All authors in this report are employees of Eli Lilly and Company, Indianapolis, IN. The authors do not have any conflict of interest or financial disclosure to report.
Acknowledgments
The authors thank Robin Brown and Andrea Sperry for measuring the concentrations of the mAbs, Chi-kin Chow for purification of antibody variants, and Yu Tian for determining the pI values of the antibodies.
References
- Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20:460-70; PMID:18656541; http://dx.doi.org/10.1016/j.coi.2008.06.012
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/10.1038/nri2155
- Buss NA, Henderson SJ, McFarlane M, Shenton JM, de Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol 2012; 12:615-22; PMID:22920732; http://dx.doi.org/10.1016/j.coph.2012.08.001
- Datta-Mannan A, Wroblewski VJ. Application of FcRn binding assays to guide mab development. Drug Metab Dispos 2014; 42:1867-72; PMID:25024401; http://dx.doi.org/10.1124/dmd.114.059089
- Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A 2015; 112(19):5997-6002.
- Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015; 7:483-93; PMID:25695748; http://dx.doi.org/10.1080/19420862.2015.1016696
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; http://dx.doi.org/10.1093/protein/gzq009
- Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014; 6:1255-64; PMID:25517310; http://dx.doi.org/10.4161/mabs.29809
- Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol 2011; 48:860-6; PMID:21256596; http://dx.doi.org/10.1016/j.molimm.2010.12.009
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/10.1074/jbc.M604292200
- Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171-80; http://dx.doi.org/10.4049/jimmunol.169.9.5171
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; http://dx.doi.org/10.1124/dmd.112.045864
- Datta-Mannan A, Witcher DR, Lu J, Wroblewski VJ. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. mAbs 2012; 4:267-73; PMID:22377715; http://dx.doi.org/10.4161/mabs.4.2.19364
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 2007; 35:86-94; PMID:17050651; http://dx.doi.org/10.1124/dmd.106.011734
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem 2007; 282:1709-17; PMID:17135257; http://dx.doi.org/10.1074/jbc.M607161200
- Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil F-P, Lowman HB, Fielder PJ, Prabhu S. Pharmacokinetics of Humanized Monoclonal Anti-Tumor Necrosis Factor-α Antibody and Its Neonatal Fc Receptor Variants in Mice and Cynomolgus Monkeys. Drug Metab Dispos 2010; 38:600-5; PMID:20071453; http://dx.doi.org/10.1124/dmd.109.031310
- Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vásquez M, et al. Engineered Human IgG Antibodies with Longer Serum Half-lives in Primates. J Biol Chem 2004; 279:6213-6; PMID:14699147; http://dx.doi.org/10.1074/jbc.C300470200
- Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol 2006; 176:346-56; http://dx.doi.org/10.4049/jimmunol.176.1.346
- Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol 2009; 182:7663-71; http://dx.doi.org/10.4049/jimmunol.0804182
- Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of Neonatal FcR in Regulating the Serum Half-Life of Therapeutic Proteins Containing the Fc Domain of Human IgG1: A Comparative Study of the Affinity of Monoclonal Antibodies and Fc-Fusion Proteins to Human Neonatal FcR. J Immunol 2010; 184:1968-76; PMID:20083659; http://dx.doi.org/10.4049/jimmunol.0903296
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011; 39:1469-77; PMID:21610128; http://dx.doi.org/10.1124/dmd.111.039453
- Zheng Y, Scheerens H, Davis JC, Deng R, Fischer SK, Woods C, Fielder PJ, Stefanich EG. Translational pharmacokinetics and pharmacodynamics of an FcRn-variant anti-CD4 monoclonal antibody from preclinical model to phase I study. Clin Pharmacol Ther 2011; 89:283-90; PMID:21191378; http://dx.doi.org/10.1038/clpt.2010.311
- Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem 2012; 287:11090-7; PMID:22294692; http://dx.doi.org/10.1074/jbc.M111.319764
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203-7; PMID:20953198; http://dx.doi.org/10.1038/nbt.1691
- Igawa T, Mimoto F, Hattori K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim Biophys Acta 2014; 1844:1943-50; PMID:25125373; http://dx.doi.org/10.1016/j.bbapap.2014.08.003
- Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N, et al. Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PloS one 2013; 8:e63236; PMID:23667591; http://dx.doi.org/10.1371/journal.pone.0063236
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157-9; PMID:20081867; http://dx.doi.org/10.1038/nbt.1601
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; http://dx.doi.org/10.1016/j.molimm.2005.07.032
- Yeung YA, Wu X, Reyes AE, 2nd, Vernes JM, Lien S, Lowe J, Maia M, Forrest WF, Meng YG, Damico LA, et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res 2010; 70:3269-77; PMID:20354184; http://dx.doi.org/10.1158/0008-5472.CAN-09-4580
- Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs 2010; 2:613-24; PMID:20818176; http://dx.doi.org/10.4161/mabs.2.6.13333
- Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J 2012; 14:554-8; PMID:22610647; http://dx.doi.org/10.1208/s12248-012-9369-y
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 2014; 111:18601-6; PMID:25512516; http://dx.doi.org/10.1073/pnas.1421779112
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. mAbs 2011; 3:422-30; PMID:22048693; http://dx.doi.org/10.4161/mabs.3.5.16983
- Henne KR, Ason B, Howard M, Wang W, Sun J, Higbee J, Tang J, Matsuda KC, Xu R, Zhou L, et al. Anti-PCSK9 antibody pharmacokinetics and low-density lipoprotein-cholesterol pharmacodynamics in nonhuman primates are antigen affinity-dependent and exhibit limited sensitivity to neonatal Fc receptor-binding enhancement. J Pharmacol Exp Ther 2015; 353:119-31; PMID:25653417; http://dx.doi.org/10.1124/jpet.114.221242
- Gurbaxani B, Dostalek M, Gardner I. Are endosomal trafficking parameters better targets for improving mAb pharmacokinetics than FcRn binding affinity? Mol Immunol 2013; 56:660-74; PMID:23917469; http://dx.doi.org/10.1016/j.molimm.2013.05.008
- Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012; 4:753-60; PMID:23778268; http://dx.doi.org/10.4161/mabs.22189
- Li B, Tesar D, Boswell A, Cahaya H, Wong A, Zhang J, Gloria Meng Y, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014; 6:1255-64.
- Stubenrauch K, Wessels U, Regula JT, Kettenberger H, Schleypen J, Kohnert U. Impact of molecular processing in the hinge region of therapeutic IgG4 antibodies on disposition profiles in cynomolgus monkeys. Drug Metab Dispos 2010; 38:84-91; PMID:19850673; http://dx.doi.org/10.1124/dmd.109.029751