
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The Fc (fragment crystallizable) is a common structural region in immunoglobulin gamma (IgG) proteins, IgG-based multi-specific platforms, and Fc-fusion platform technologies. Changes in conformational stability, protein-protein interactions, and aggregation of NS0-produced human Fc1 were quantified experimentally as a function of pH (4 to 6) and temperature (30 to 77°C), using a combination of differential scanning calorimetry, laser light scattering, size-exclusion chromatography, and capillary electrophoresis. The Fc1 was O-glycosylated at position 3 (threonine), and confirmed to correspond to the intact IgG1 by comparison with Fc1 produced by cleavage of the parent IgG1. Changing the pH caused large effects for thermal unfolding transitions, but it caused surprisingly smaller effects for electrostatic protein-protein interactions. The aggregation behavior was qualitatively similar across different solution conditions, with soluble dimers and larger oligomers formed in most cases. Aggregation rates spanned approximately 5 orders of magnitude and could be divided into 2 regimes: (i) Arrhenius, unfolding-limited aggregation at temperatures near or above the midpoint-unfolding temperature of the CH2 domain; (ii) a non-Arrhenius regime at lower temperatures, presumably as a result of the temperature dependence of the unfolding enthalpy for the CH2 domain. The non-Arrhenius regime was most pronounced for lower temperatures. Together with the weak protein-protein repulsions, these highlight challenges that are expected for maintaining long-term stability of biotechnology products that are based on human Fc constructs.
Abbreviations
| CE | = | capillary zone electrophoresis |
| DSC | = | differential scanning calorimetry |
| ETD | = | electron transfer dissociation |
| Fc | = | fragment crystallizable |
| HMW | = | high molecular-weight |
| IEX | = | ion-exchange chromatography |
| IgG | = | immunoglobulin gamma |
| KB | = | Kirkwood Buff |
| LS | = | light scattering |
| MALS | = | multi-angle laser light scattering |
| MS | = | mass spectrometry |
| Mw | = | weigh-average molecular weight |
| SEC | = | size-exclusion chromatography |
| SLS | = | static light scattering |
Introduction
Biological drugs grew to be among the largest-selling medicines from 2011 to 2014.Citation1,2 They also represent one of the largest growing classes of drugs in development by the biopharmaceutical industry.Citation2,3 Formats in development include canonical immunoglobulin gamma (IgG) molecules, IgG-like multi-specific platforms such as Triomab, kih IgG, ortho-Fab IgG, DVD-Ig, DART-Fc, IgG-scFv and scFv2-Fc,Citation4 and the fragment crystallizable (Fc) fusion platform.Citation5,6 The Fc region of IgG proteins is common to many new platforms; it facilitates purification and can also contribute to improved solubility and stability.Citation4,7 The presence of an Fc region has consequences for Fc-mediated effector functions that might be desirable for therapeutic applications, such as antibody-dependent cell-mediated cytotoxicity, complement fixation, and long serum half-life resulting from their larger size and FcRn-mediated recycling processes.Citation8
Non-native aggregates (hereafter simply termed aggregates) formed during in vivo expression, purification and processing, as well as upon storage and administration, are one of the major concerns associated with successful delivery of protein therapeutics to patients. Aggregation in formulated therapeutic products can cause unwanted immune responses and loss of biological activity.Citation9,10,11 The aggregates formed are often irreversible under typical solution conditions for the product and upon administration to patients. IgGs and some of the newer therapeutic platforms are multi-domain proteins; as such, they might be expected to have multiple regions within their sequences and domains that could be inherently aggregation-prone. A previous studyCitation12 of IgG1 aggregation showed that both Fab and Fc were aggregation-prone, and a pH-dependent competition determined which domain dominates the aggregation behavior of the full IgG1. This report focuses on quantifying and characterizing the pH and temperature dependence of the aggregation rates and mechanism(s) for the Fc of human IgG1 (termed Fc1 in what follows), as well as how they relate to unfolding of the two domains within the Fc1, and the protein net charge and protein-protein interactions. The results provide a basis for what to expect for aggregation mediated by partial unfolding of the Fc1 when dealing with a range of different Fc-based platforms, as well as for IgG proteins that have aggregation-resistant Fab designs.Citation13,14
The protein used in this study is the NS0-produced Fc1 that has the same sequence as the Fc1 fragment produced by enzymatic cleavage of the native anti-CD40 IgG1.Citation12 Unfolding transitions were characterized with differential scanning calorimetry (DSC). Inter-protein interactions of folded Fc1 were characterized using the protein-protein Kirkwood-Buff (KB) integral (G22) measured with static laser light scattering (SLS). Aggregation kinetics and mechanism(s) were assessed using size-exclusion chromatography with inline multi-angle laser light scattering (SEC-MALS). The results across a range of solution pH and temperatures illustrate changes in aggregation mechanisms, the importance of CH2 unfolding in mediating Fc1 aggregation, and, to the best of our knowledge, provide the most extensive report of aggregation rates for Fc1 across a broad range of temperature and pH.
Results
Production and purification of Fc1
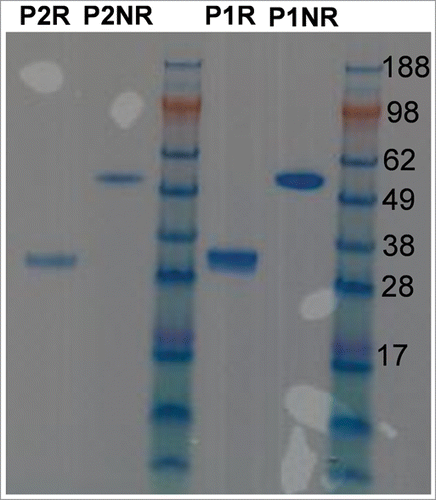
The Fc1 was purified via protein A affinity chromatography, followed by ion-exchange chromatography (IEX). It is well known (Boehringer Ingelheim internal data not shown) that IgG or Fc-fusion proteins produced by NS0 cells have heterogeneous glycosylation patterns. The IEX purification step was applied to separate species potentially carrying different charges that resulted from different types of glycosylation. There were two peaks observed during IEX elution, and the fractions in the middle of each peak were collected and combined to form one pool. Pool 1(P1) was from the first peak and pool 2 (P2) was from the second peak. shows illustrative sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of purified Fc1 expressed in NS0 cells. There is a single band around 58 kDa in the non-reduced (NR) SDS-PAGE for P1 and P2. One band around 29 kDa in the reduced SDS-PAGE of P2 corresponds to one heavy chain (half-Fc) from Fc1. However, there were consistently two bands, one major band and one minor band with different mobilities, in reduced SDS-PAGE of P1 samples. The observed bands in reduced and non-reduced SDS-PAGE confirm the identity and purity of the Fc1; however, heterogeneity of the half-Fc was apparent for P1 samples.
Figure 1. SDS-PAGE of the two Fc1 produced in NS0 cell. P1 = pool 1 from IEX purification, P2 = pool 2 from IEX purification, R = reduced, NR = non-reduced.
Intact mass of IEX fractions of Fc1
In order to determine the difference between the two pools or fractions collected from IEX of Fc1, mass spectrometry was performed on each pool after N-deglycosylation and subsequent reduction. In the deconvoluted mass spectrum of the IEX P1 fraction in supplemental information (Figure S1A), the major peak has the molecular weight (Mw) of 26,105 Dalton (Da). There is also a minor peak in this spectrum that agrees with the predicted average Mw of the protein sequence with C-terminal lysine truncation. C-terminal lysine truncation is a common modification of recombinant antibodies produced in mammalian cell lines such as CHO, NS0, and SP2/0.Citation15 The major peak is approximately 979 Da greater than the minor peak at Mw 25,126 Da. In the deconvoluted mass spectrum of IEX P2 fraction (Figure S1B), the major peak has a mass of 25,125 Da. This Mw agrees with the predicted average Mw of the protein sequence with C-terminal lysine truncation. The increase in Mw of approximately 979 Da was identified to be the result of O-glycosylation.
Tryptic peptide map of IEX P1 of Fc1
Through reduction, alkylation, trypsin digestion, and LC-MS/MS of the IEX P1 fraction, the site of O-glycosylation was determined to be threonine located at position 3 (i.e., Thr3). ETD fragmentation of the N-terminal tryptic peptide was used to determine the amino acid with O-glycosylation. PMi Byonic™ software (Protein Metrics) was used to match the O-glycosylated peptide, and determine the site of O-glycosylation (Figure S2A). In the fragmentation data, the C2 ion was only observed without glycosylation, and the C3 ion was only observed with the O-glycosylation (Figure S2B). Based on accurate mass of common glycans, Byonic assigned the glycosylation as HexNAc(1)Hex(1)NeuGc(2) with a Mw of 979 Da. The parent peptide, C2, and C3 ions were all identified with less than 1 ppm error. The O-glycosylation has been reported located mostly in the hinge regionCitation16 or possibly on the Fc region.Citation17 Hereafter, the IEX P1 and P2 fractions for Fc1 produced in NS0 cells are referred to as O-glycosylated Fc1 and non-O-glycosylated Fc1, respectively.
Conformational stability of O-Glycosylated and non-O-Glycosylated Fc1
The O-glycosylated Fc1 and non-O-glycosylated Fc1, as well as the Fc1 fragment produced from endoproteinase Lys-C enzymatic cleavage of the parent IgG1 protein,Citation12 were each tested with DSC to compare their conformational stability and unfolding transition(s). Fc1 from each source shows two endotherms (peaks) at each pH condition, with the lower-T endotherm corresponding to unfolding of the CH2 domain and the higher-T endotherm resulting from unfolding of the CH3 domainCitation18,19,20 (see Figure S3 in supplemental information for DSC thermograms). At each pH, the peak positions of the endotherms are the same for the three different Fc1 samples, within statistical uncertainty, although the unfolding enthalpies appear to be somewhat different. Based on run-to-run variability and the data-averaging protocol for data acquisition in the DSC, all peak positions (temperature values) that are within 0.3°C were considered effectively indistinguishable. The heat capacity of the native state for the non-O-glycosylated Fc1 is lower than that for the O-glycosylated Fc1 and the Fc1 fragment, especially at pH 4.0. It was not apparent that the heat capacity differences between scans of the native baselines were of consequence in subsequent analyses, as the thermograms were not fully reversible, and therefore were not amenable to determinations of temperature-dependent unfolding equilibrium.
Isothermal aggregation of O-Glycosylated vs. non-O-Glycosylated Fc1
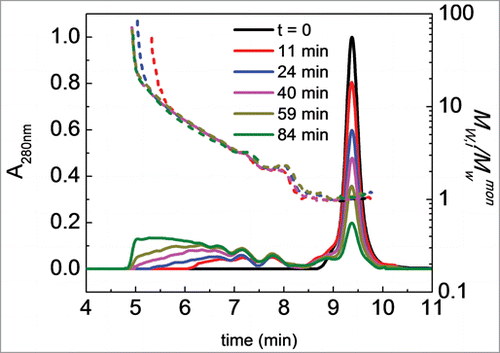
Isothermal aggregation kinetics were measured at selected temperatures for the O-glycosylated Fc1, non-O-glycosylated Fc1, and the Fc1 cleaved from the IgG1 at a protein concentration of 1 mg/ml at pH 4.0, 5.0, and 6.0 to compare the aggregation rates of the three Fc1 sample types. The incubation temperatures (60°C at pH 4.0, 70°C at pH 5.0, and 75°C at pH 6.0) were chosen so as to lie above the CH2 unfolding transition at a given pH, while also lying as far as possible below the CH3 unfolding peak temperature (Tm).Citation12 Monomer concentration was quantified from analytical size exclusion chromatography (SEC), described below. Example chromatograms from analytical SEC are shown in for illustrative time points during aggregation of the O-glycosylated Fc1 at pH 4.0, 60°C. For the given mobile phase conditions, the monomer elutes at approximately 9.6 min. Dimers and trimers that formed during aggregation are partially resolvable in SEC, with increasing amounts of high molecular-weight species at longer times. Soluble aggregates larger than trimer (HMW aggregates) co-eluted near 5.8 min. Dimers are clearly present at early incubation times. At pH 5.0 (70°C), relatively low concentrations of small oligomers (dimers and trimers) are detectable in SEC at early times (data not shown). Aggregates at pH 4.0 remained soluble throughout isothermal aggregation. Aggregates formed at pH 5.0 and 6.0 within the given incubation time range (about 70% monomer loss) were soluble and no centrifugation was required. Visible haze formation and precipitation occurred after aggregation had proceeded past approximately 90% monomer loss at pH 5.0 (70°C) and pH 6.0 (75°C) (data not shown). The Mw of the species that eluted at each 1-s slice of a given peak was determined with inline MALS. An example of Mw versus retention time overlaid with the SEC chromatograms is also shown in .
Figure 2. Illustrative SEC chromatograms and corresponding Mw,i/Mwmon profiles (short dashed lines, i represents each one-second “slice” of column eluate) of Fc1 at pH 4.0, 60°C. Different colors indicate different incubation times (see also panel legends). Initial protein concentration is 1 mg/ml. Colors in online version only.
Monomer fraction (m) was determined from SEC chromatograms at each incubation time. m is defined as the ratio of the monomer concentration in each sample to the monomer concentration in the initial, unheated sample (C0). Monomer loss vs. incubation time for the three different Fc1 sample types at pH 4.0, 60°C; pH 5.0, 70°C; and pH 6.0, 75°C is shown in . The observed rate coefficient for monomer loss (kobs) and half life (t50) were determined by fitting the experimental monomer loss data to a simple rate expression, as in Equation Equation1(1)
(1) .Citation21,22,23
(1)
(1) where m is defined as the monomer concentration at a given incubation time, divided by C0, making the units for kobs equal to 1/s. The scaling exponent (α) was also regressed, but no mechanistic interpretation was based on the resulting value or fitted curves. The fits were used only to more precisely interpolate half-lives and fit kobs values, along with their associated statistical uncertainties. In the results below, α = 2 was used, and conventional units for a second-order rate coefficient (M−1 s−1) are recovered by dividing kobs from Equation Equation1
(1)
(1) by the initial protein concentration.Citation24
Figure 3. Monomer fraction as a function of incubation time at (A) pH 4.0, 60°C; (B) pH 5.0, 70°C; and (C) pH 6.0, 75°C. In all panels: non-O-glycosylated Fc1 produced from NS0 cells (red, right-facing triangles), Fc1 cleaved from IgG1 (black squares), and O-glycosylated Fc1 produced from NS0 cells (blue, downward-facing triangles). Color in online version only.
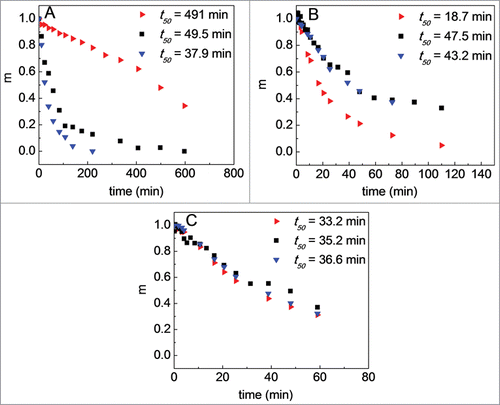
The monomer-loss profiles of the O-glycosylated Fc1 and the Fc1 cleaved from the IgG1 overlap, and the half-lives are similar within statistical uncertainty at each incubation condition. The half-life for the non-O-glycosylated Fc1 is about 10-fold longer than that of the Fc1 cleaved from the IgG1 at pH 4.0, 60°C but about 3-fold shorter than that of the Fc1 cleaved from the IgG1 at pH 5.0, 70°C. The monomer loss profiles of the three Fc1 sample types effectively superimpose at pH 6.0, 75°C. As such, the O-glycosylated Fc1 produced from NS0 cell lines best mimics the Fc1 produced from the cleavage of the IgG1.Citation12 The remainder of the results focus on the O-glycosylated Fc1, and hereafter is referred to simply as Fc1 for brevity.
Net charge on Fc1 from electrophoretic mobility
shows the theoretical charge or valence (zcalc) on the Fc1 as a function of pH, which was calculated from the number of titratable amino acids located in the Fc1 and theoretical pKa values of each titratable amino acid under an assumption that each amino acid was isolated in solution.Citation25 The experimental net charge or valence (z*) was determined from values of electrophoretic mobility (μ)obtained from capillary zone electrophoresis (CE) (Equation Equation2(2)
(2) ) and diffusion coefficient values (D) from sedimentation velocity experiments.Citation26
(2)
(2) where e denotes the elementary charge (1.6 × 10−19 coulombs), and kB is Boltzmann's constant (1.38 × 10−16gcm2s−2K−1). z* is a combination of the actual charged amino acid side chains, any charged covalent structural modifications (e.g., sialic acid, phosphorylation), and any territorially associated counter-ions that are localized to regions near highly charged patches on a protein (i.e., not the same as the ions that make up the Debye-Hückel “counter-ion cloud”).Citation27 z* is always considered to be less than the actual charge (zDHH) on a protein due to Debye-Hückel counter-ion shielding, the coupling of ionic flows, and the deformation of the electric field in the vicinity of the protein.Citation27 zDHH is calculated from z* by Equation Equation3
(3)
(3) .
(3)
(3) where κ is the inverse Debye length ([=] cm−1),Citation28 a = Rs + Ri is the sum of the Stokes radii for the protein and its counterion ([=] cm) and f(κa) is Henry's function (unitless).Citation29 Calculation of κ and Henry's function was performed with the ZUtilities software program,Citation27 that can be freely downloaded from ftp://ftp.eos.sr.unh.edu/pub/bitc/ZUtilities via the ‘Electrophoresis utilities’ link at the top of the webpage. The results for z* and zDHH are listed in , in comparison with zcalc. The charge is positive at pH 4.0, indicating electrostatic repulsions between folded Fc1 molecules. However, the charge is close to zero at both pH 5.0 and 6.0, which suggests the net electrostatic repulsions between Fc1 monomers are minimal. Statistical uncertainties on z* are 95% confidence intervals based on propagating uncertainties in measured μ and D values.
Table 1. Calculated, net, and corrected charges, second virial coefficients for the Fc1 at pH 4.0, 5.0, and 6.0 (30 mM sodium citrate), reported with standard deviation
Protein-protein interactions based on G22 from laser scattering
Boltzmann-averaged monomer-monomer interactions were determined versus pH at room temperature by examining static light scattering at protein concentrations of the Fc1 ranging from 1 to 10 mg/ml. Ionic strength values for these solutions were 35, 70, and 112 mM for pH 4.0, 5.0, and 6.0 solutions (with sodium citrate fixed at a concentration of 30 mM), respectively, which were calculated using pKa values of citrate of 3.013, 4.76, and 6.40.Citation30 An illustrative plot of Rex/K as a function of monomer concentration is shown in the supplemental information (Fig. S4). The light scattering (LS) results for each of the solution conditions examined here were analyzed using Equation Equation4(4)
(4) ,Citation31
(4)
(4) where c is the concentration ([=] mass/vol) of protein. Rex is the excess Rayleigh ratio measured from SLS with a toluene standard, and K is an optical constant once a value of the refractive index increment (dn/dc) is set (see Materials and Methods). The protein-protein Kirkwood Buff (KB) integral (G22) is a more broadly applicable measure of net interactions obtainable from SLS than the traditional osmotic second virial coefficient (B22), especially at higher concentrations or in the presence of strong attractions and repulsions.Citation31,32
Conceptually, G22 is similar to the more familiar osmotic second virial coefficient for quantitatively measuring protein-protein interactions, except that it carries the opposite sign to B22, and, in the limit of low protein concentration, it has double the magnitude of B22.Citation31 A scaled version of G22 is defined as g2* ≡ - G22/2B22HS-1, with B22HS defined as the steric-only or “hard sphere (HS)” value of the second osmotic virial coefficient. The effective hard sphere or steric radius is 3.1 nm for the Fc1, as obtained from sedimentation velocity experiments.Citation26 Illustrative fits can be found in supplemental information (Fig. S4).
The resulting g2* values at pH 4.0, 5.0, and 6.0 are listed in . Based on the definition above, g2* has a value of zero for a purely HS solute, or for a system in which attractive and repulsive non-steric forces effectively balance each other. The absolute values of g2* for Fc1 at each pH / buffer condition are close to zero, but with a positive sign at pH 4.0 and a negative sign at pH 5.0 and 6.0. This indicates that the non-steric protein-protein interactions at pH 4.0 are slightly net repulsive, and vice versa at pH 5.0 and 6.0. Interestingly, the values of g2* for Fc1 are much closer to zero and much weaker functions of pH than those typically reported for IgG1 examples in the literature.Citation33,34,35 This may have implications for arguments that have been proposed regarding the importance of large differences in net charge of the Fc and Fab regions of IgG proteins when one considers aggregation and clustering at much higher protein concentrations than were considered here.
Temperature dependence of aggregation rates and mechanism(s)
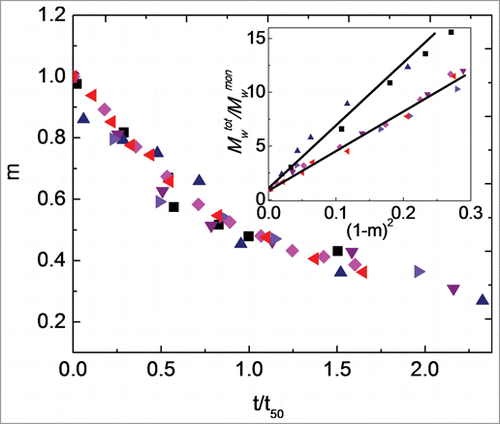
Isothermal aggregation was monitored for Fc1 over a broad temperature range, from 30°C to 77°C, as a function of pH to investigate the effect of temperature on aggregation rates and mechanism(s). Scaling the incubation times by the time required for 50% monomer loss (t50) at a given temperature allows direct comparison of kinetics that occur on greatly different time scales.Citation23,36 At the highest (coldest) temperatures, the rates of monomer loss were too fast (slow) to allow t50 to be accurately quantified with the techniques here, and those temperatures were therefore not included in the results shown next. shows m as a function of t/t50 at pH 4.0 for an initial protein concentration of 1 mg/ml at T = 40°C, 45°C, 50°C, 55°C, 60°C, and 65°C. t50 values reported in the caption of decrease greatly with increasing temperature, especially from 45°C to 50°C (∼102 fold). However, monomer loss profiles over the first two half-lives superimpose across the different incubation temperatures.
Figure 4. Main panel: Monomer fraction as a function of aggregation time for pH 4.0 at incubation temperatures of 40°C (black squares; t50 = 117 days), 45°C (blue upward triangles; t50 = 10.7 days), 50°C (purple downward triangles; t50 = 126 min), 55°C (right-facing triangles; t50 = 100 min), 60°C (pink diamonds; t50 = 50.3 min), and 65°C (left-facing triangles; t50 = 18.0 min). Inset: Average degree of polymerization, Mwtot/Mwmon, versus squared extent of reaction, (1-m)2, for incubation temperatures as in main figure. The solid lines are only guides to the eyes. Color in online version only.
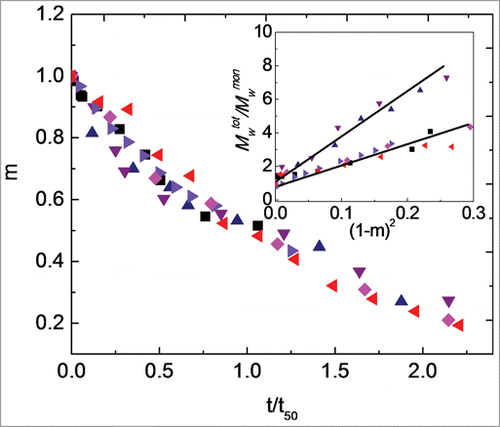
On the basis of theoretical considerations and mass balance analysis of aggregation mechanisms, a potentially useful way to examine the mechanism of aggregate growth is by scaling the total molecular weight, including monomers and soluble aggregates (Mwtot), by the molecular weight of the monomer (Mwmon) and plotting Mwtot/Mwmon as a function of (1-m)2, as shown in inset.Citation36 A linear dependence of Mwtot/Mwmon vs. (1-m)2 is observed within the first half-life at all incubation temperatures in . From a mechanistic perspective, a linear dependence of Mwtot/Mwmon versus (1-m)2 indicates aggregates grow predominantly by monomer addition (chain polymerization).Citation36,37 The inset shows that the sizes of aggregates formed at 40°C are similar to those for aggregates formed at the temperatures of 55°C, 60°C, and 65°C within the first half-life for monomer loss. The sizes of aggregates are similar at a given amount of monomer loss for 45°C to 50°C, but decrease as temperature increases from 50°C to 55°C, especially at longer times (smaller m values). Qualitatively, the results in and visual inspection of SEC chromatograms () suggest that aggregation at pH 4.0 for most, if not all, temperatures share some common features during the initial stages of aggregate growth (i.e., over the first one to two half-lives). Aggregates originally form as dimers, and then grow to trimers and somewhat larger oligomers by adding monomers.Citation36,37
shows m as a function of t/t50 at pH 5.0 for a protein concentration of 1 mg/ml at T = 50°C, 55°C, 60°C, 65°C, 70°C, and 72°C. t50 values are reported in the caption. The monomer-loss results follow a common decay profile with respect to t/t50 for all the temperatures within the first two half-lives. The inset to is analogous to that for ; aggregate Mw is linear vs. (1-m)2 over the first half life at all temperatures. The data in the inset correspond to only conditions before any insoluble aggregates were observed. The aggregates formed at temperatures of 50°C and 55°C are somewhat larger than those formed at temperatures of 60°C, 65°C, 70°C, and 72°C, for a given extent of monomer loss. Similar to the behavior at pH 4.0, the aggregate growth within the first two half-lives at pH 5.0 at all the temperatures shares common features for the growth mechanism. In general, the aggregates formed at pH 4.0 are smaller than those at pH 5.0, consistent with what has been reported qualitatively for other IgG1 and non-antibody systems. Citation35,38,Citation39
Figure 5. Main panel: Monomer fraction as a function of aggregation time for pH 5.0 at incubation temperatures of 50°C (black squares; t50 = 59.2 days), 55°C (blue upward triangles; t50 = 21.1 days), 60°C (purple downward triangles; t50 = 130 min), 65°C (right-facing triangles; t50 = 40.9 min), 70°C (pink diamonds; t50 = 28.1 min), and 72°C (red left-facing triangles; t50 = 18.2 min). Inset: Average degree of polymerization, Mwtot/Mwmon, vs. squared extent of reaction, (1−m)2, for incubation temperatures as in main panel. The solid lines are only guides to the eyes. Color in online version only.
Similar monomer loss and Mwtot/Mwmon versus (1-m)2 profiles are only available for pH 6.0 at temperatures of 70°C, 72°C, 75°C, and 77°C (Fig. S5 in supplemental information). The monomer loss rates are extremely slow for pH 6.0 at temperatures lower than 70°C, and only initial rates (from less than 10 percent monomer loss) were calculated from the incubation of Fc1 samples at those lower temperatures. As a result, Mw scaling with (1-m)2, and t50 values are not reported for those conditions.
Discussion
Conformational stability and aggregation rates of the two IEX pools of NS0-expressed Fc1 were first compared to identify which pool is comparable to the Fc1 fragment cleaved from the parent IgG1. P1 and P2 from IEX were identified to be different in terms of O-linked glycosylation as shown via mass spectral analysis (Fig. S1). The presence of sialic acids on the O-linked glycans of the O-glycosylated Fc1 introduces additional negative charges, which results in its earlier elution on cation-exchange chromatography.Citation40,41,42 The thermal unfolding curves and isothermal aggregation kinetics of the Fc1 from these 3 different sources were compared (Fig. S3 and ) to determine which Fc1 pool represents the Fc1 fragment corresponding to the parent IgG1 molecule.Citation12 Two unfolding transitions in DSC were observed at pH 4.0, 5.0 and 6.0, similar to what has been observed for Fc1 molecules by other researchers.Citation43,44,45,46
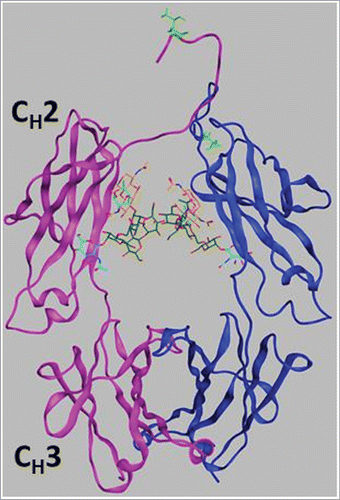
As noted in the Results, the peak positions of the endotherms are effectively the same in all cases, and the area under a given peak was also similar for each of the three Fc1 samples. The thermal unfolding curve of the O-glycosylated Fc1 superimposes slightly better with that of the Fc1 fragment produced from enzymatic cleavage, especially with regard to the heat capacity of the native state baseline in the vicinity of 25°C. Previous workCitation45 has shown the important stabilizing role of N-glycosylation at the conserved Asn-297 in the CH2 domain of human Fc1. Compared to the large effect of N-glycosylation on the conformational stability of the domains of Fc1, the O-glycans identified at Thr3 at the N-terminus of the Fc1 do not appear to have a significant effect on the conformational stability of the CH2 domain or CH3 domain. The structural model of the Fc1 (from homology modeling of an Fc1 crystal structure in the Protein Data Bank (PDB), ID: 3AVE)Citation47 in shows that the position of O-glycosylation is distinct from the canonical N-glycosylation sites, and occurs in a regime that is relatively unstructured. It is therefore perhaps unsurprising that O-glycosylation at Thr3 does not greatly affect the unfolding thermogram of the CH2 domain.
Figure 6. Ribbon diagram of the Fc1 structure from homology modeling of an Fc crystal structure in the PDB (ID: 3AVE)Citation47 by adding hinge region sequence (THTCPPCP) to the N-terminus. One chain including the CH2 and CH3 domains is colored in magenta and the other chain is in blue, with Asn297-linked N-glycans shown as light blue, orange and green sticks. The side chain of the O-glycosylation site, Thr3, is shown as light green sticks.
Although the effect of the O-glycosylation on the conformational stability of the Fc1 appears to be minimal, its effects on the observed aggregation kinetics or rate coefficient (kobs) is pronounced and pH-dependent, as shown in . The non-O-glycosylated Fc1 should have more positive charge than the O-glycosylated Fc1 without the additional negative charge(s) from the O-glycosylation, so it is perhaps unsurprising that the observed aggregation rate for the non-O-glycosylated Fc1 is lower than that for the O-glycosylated Fc1 at pH 4.0.Citation40,41,42 As pH increases, the net charge on Fc1 approaches zero, and presumably the differences in O-glycosylation due to charge-charge interactions, would still be present because the non-O-glycosylated Fc1 would be missing the negative charge(s) due to the glycans. By that reasoning, the aggregation rates for the non-O-glycosylated Fc1 should be lower than that for the O-glycosylated Fc1 at pH 5. However, the opposite result is observed. One hypothesis to account for this apparent discrepancy is that citrate ion bindingCitation48 at pH 5 and 6 can compensate for the change in charge by binding to the glycans, mitigating the net charge of the O-glycosylated Fc1, compared to the Fc1 that is missing those O-glycans. However, a detailed characterization of all of the different glycosylated species was beyond the scope of this work. The major purpose of comparison for the present work was to identify that the biologically produced O-glycosylated Fc1 was comparable to the Fc1 fragment cleaved from the parent IgG1. As a result, only the O-glycosylated Fc1 was used in what is considered further below.
Protein net charge and Kirkwood Buff integrals were quantified as a function of pH to provide insights in understanding aggregation mechanism(s) of the Fc1. The second virial coefficient or the protein-protein KB integral quantifies the net interactions between two proteins in solution, and reflects an spatial average of both attractions and repulsions.Citation49,50 Attractive interactions include hydrophobic and van der Waals forces, and electrostatic attractions between oppositely charged side chains. Non-steric repulsive interactions are primarily those between charged side chains with the same charge sign. Both attractive and repulsive electrostatic interactions will typically decrease in magnitude upon addition of simple, non-adsorbing electrolytes as a result of counterion screening and the effects of a Debye double-layer.Citation51 Hydrophobic and van der Waals interactions are relatively short ranged (∼1 nm or less), and deducing their contributions requires one to perform a systematic determination of B22 or G22 as a function of ionic strength.Citation35,52 A detailed test of ionic strength dependence was not performed here, but is the subject of ongoing work for the Fc1.
If electrostatic repulsions dominate over all types of attractions, g2* will lie above 1, and vice versa. As pH increases, g2* (last column in ) changes sign from positive to negative but with absolute values close to zero, within statistical uncertainty. Because a constant buffer concentration (30 mM) was maintained, the higher pH values also have higher net ionic strength due to charge titration with increasing pH. At pH 5 and pH 6, the net charge is essentially zero, and significant non-electrostatic attractions (e.g., van der Waals attractions and hydrophobic interactions) between proteins presumably result in the negative sign of g2*. Those attractions were presumably also present at pH 4.0, but were offset to a greater degree by electrostatic repulsions. This is qualitatively similar to what has been observed for antibodies and other proteins,Citation12,33,35,53 but, as noted above, the magnitude of the charges and g2* values, and the effects of pH are much less pronounced for Fc1 compared to examples for IgG proteins.Citation33,34,35 Therefore, altering pH as a means to stabilize Fc1 with respect to aggregation is a strategy that appears to rely almost exclusively on altered conformational stability. This is in contrast to what has been found for IgG systems.Citation34,48,54 It also suggests that the Fab domains of IgG proteins carry most of the net charge in vitro, which is not what would be anticipated from theoretical calculations and amino acid sequence.Citation33
The effects of temperature on aggregation rates and mechanism(s) of the Fc1 were investigated by monitoring isothermal aggregation of the Fc1 over a broad temperature range, from 30°C to 77°C. The observed aggregation rate coefficient for monomer loss was determined by fitting experimental monomer loss data to Equation Equation1(1)
(1) . As noted above, aggregation over the first half life of monomer loss resulted generally in the formation of dimers, trimers, and oligomers. shows the measured kobs values for monomer loss as a function of temperature. They are plotted as ln kobs vs. inverse temperature to highlight regions of Arrhenius and non-Arrhenius behavior. All kobs values are orders of magnitude below conservative estimates of diffusion limits (3.3e6 mL·mg−1·min−1) (). The aggregation rates span more than 5 orders of magnitude, and from temperatures well above the Tm for CH2, down to roughly room temperature. For a given temperature, aggregation rates are higher at lower pH, which has been observed for IgG proteins and the human Fc1 and Fc2.Citation38,45 This behavior is consistent with the lower conformational stability of both CH2 and CH3 domains as pH decreases, in that this loss of stability causes the net concentration of partially unfolded proteins to be higher at lower pH (for a given temperature).
Figure 7. Observed aggregation rates (kobs) as a function of temperature for Fc1, 1 mg/ml at pH 4.0 (filled squares), pH 5.0 (filled upward triangles), and pH 6.0 (filled left-side triangles) on the left-hand axis; the right-hand axis is labeled with the number of days for the monomer loss of 1%, which is calculated from kobs labeled on the left-hand axis by the equation, t1% = 0.01/kobs. Two regimes are observed to exhibit Arrhenius behavior with clear breakpoints around unfolding temperature of CH2 domain, which are marked with solid arrows (53 °C for pH 4.0, 65°C for pH 5.0, and 70°C for pH 6.0). At lower temperatures, there is divergence upward from the first Arrhenius regime. The short dash lines are only guides to the eyes. The similar data set for the mAb1 (open left-side triangles) and mAb2 (open upward triangles) are extracted from of Drenski et al.Citation55 and re-plotted here for comparison (breakpoints around 67°C, marked by the short dash-dot arrow). The short dash-dot lines are also only guides to the eyes.
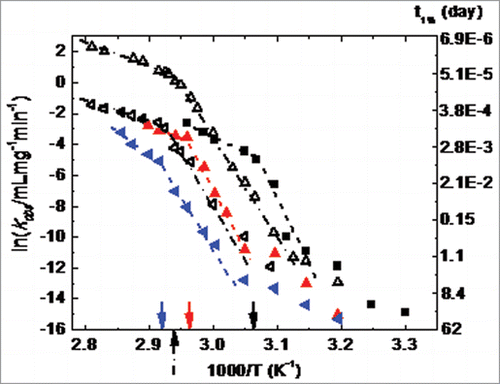
A striking feature of is that the temperature profile (ln kobs vs. 1/T) at each pH shows a distinct “breakpoint” that separates two qualitatively different regimes. The temperatures at the breakpoints are the same as the Tm for the CH2 domain from DSC (Fig. S3) at a given pH. Above (below) the CH2 Tm, the profiles are Arrhenius (non-Arrhenius) with a relatively low (high) effective activation energy. A similar qualitative pattern has also been observed for two mAbs,Citation55 where the breakpoint corresponds to the Tm of CH2 for two different IgG1 (data shown as overlays in , with data extracted from of Drenski et al.Citation55
Preliminary results for selected conditions above and below the breakpoint temperature show that kobs is independent of initial protein concentration at the high temperature conditions; in contrast, kobs depends on initial protein concentration at lower-temperature conditions. This type of qualitative change in aggregation kinetics is consistent with model predictions and aggregation mechanisms that involve a combination of unfolding, refolding, and net-irreversible aggregation of unfolded or partially unfolded proteins.Citation22,56
In such cases, the high-temperature Arrhenius regime corresponds to aggregation with unfolding as the rate-limiting step, and the activation energy corresponds to that for unfolding. Significantly below the Tm, unfolding is no longer rate-limiting and instead unfolding/refolding is fast compared to aggregation events. Unfolded or partially unfolded monomers are still required for aggregation, but now folding-unfolding pre-equilibrates. As a result, the effective activation energy is a combination of the enthalpy of unfolding (ΔHun) and any intrinsic activation barriers to forming stable aggregates from unfolded monomers.Citation21 At the breakpoint for ln kobs vs. 1/T , one expects the activation energy be lower (higher) at temperatures above (below) the breakpoint. This follows because ΔHun near Tm is generally much larger than the activation energy for unfolding. Finally, based on general principles of protein unfolding thermodynamics, ΔHun usually decreases greatly with decreasing temperature, and therefore diagrams such as are anticipated to show upward curvature for the non-Arrhenius portion of the diagram.Citation57
Returning to , and with the above arguments in mind, the breakpoint occurring at the Tm of the CH2 domain argues that CH2 unfolding is required for aggregation. That is qualitatively in keeping with prior results at much lower pH conditions for Fc1 fragments.Citation45 The other features outlined above are also observed in . For example, the effective activation energy is reduced at temperatures above the CH2 Tm compared to immediately below it; and the low-temperature regime shows non-Arrhenius behavior with upward curvature. Interestingly, the data from Drenski et alCitation55 lie within roughly the same range of kobs values as the data reported here. However, that is possibly coincidental because Drenski et alCitation55 did not report the solution conditions, although the protein concentrations were similar (less than ten-fold different) for those measurements and the results reported here. Based on these examples and the above theoretical arguments, it is anticipated that similar kinetic behavior will be found for Fc-based constructs other than IgG examples.
Finally, it is not currently possible to quantitatively predict, a priori, the curvature in the non-Arrhenius regimes for or analogous conditions for other proteinsCitation34,57,58,59 without accurate determination of quantities such as the heat capacity of unfolding for the regime or domain that needs to unfold to aggregate. Unfortunately, unfolding in DSC is often not reversible and one can only qualitatively or semi-quantitatively estimate this key quantity.Citation57,59 Therefore, predicting low-temperature stability or shelf life from high temperature conditions remains a significant and long-standing challenge for any system that displays behaviors such as those illustrated in . This highlights challenges that are anticipated to hold for any laboratory that seeks to develop Fc-based constructs for biotechnology applications that require long-term product stability.
In conclusion, we compared conformational stability and aggregation kinetics of a human Fc1 for the O-glycosylated Fc1, non-O-glycosylated Fc1, and the Fc1 fragment cleaved from the parent IgG1. O-glycosylation did not affect conformational stability of the Fc1, but significantly altered aggregation kinetics, in a pH-dependent manner. Comparison with the other sources of Fc1 indicated that O-glycosylated Fc1 was representative of the Fc1 within the parent anti-CD40 IgG1,Citation12 and thus was used for a systematic characterization of pH- and temperature-dependent aggregation kinetics and protein-protein interactions.
Aggregation involved unfolding of the CH2 domain and formation of low- to intermediate-Mw soluble oligomers. The size of the oligomers depended on pH, with pH values farther below the pI resulting in smaller aggregates, on average. The aggregation mechanism was temperature dependent in all cases, and showed two regimes. For temperatures at or above the Tm of the CH2 domain, aggregation rates were Arrhenius and consistent with unfolding-limited mechanisms such as suggested previously for other proteins.Citation56,Citation59 At lower temperatures, aggregation rates were non-Arrhenius and consistent with aggregation occurring slowly after partial Fc1 unfolding. The conditions include perhaps the broadest range of aggregation rates (over 5 orders of magnitude) and temperatures (30°C to well above Tm values) that have been reported to date for an Fc1, and provide a context for stability assessment, prediction, and engineering of Fc regions for researchers focused on Fc-based constructs.
Materials and Methods
Production of Fc1
The Fc1 gene was inserted into a pcDNA3.1(−) vector and transiently transfected into NS0 cells. Fc1 protein was expressed in batch suspension culture in 4 L flasks. 24 L of biomass was purified via protein A affinity chromatography, followed by ion-exchange chromatography. Purified Fc1 was confirmed as mono-dispersed natural homodimer (99%) in solution using analytical ultracentrifugation and gel electrophoresis ().
SDS-PAGE
NuPAGE 4–12% Bis-Tris gels with 1.0 mm ×10 well (Life Technologies, Catalog no NP0321BOX) were used for separation in fresh 1x MES SDS running buffer prepared from 20× MES SDS running buffer (Life Technologies, Catalog no NP0002). The reduced sample was boiled at 95°C for 5 min with NuPAGE sample reducing agent (Life Technologies, Catalog no NP0004). Protein ladders (Life Technologies, Catalog no LC5925) and samples were electrophoresed at a constant voltage of 200 (Volts) for 35 min, and stained via Coomassie blue using standard methods.
Mass spectrometry
Deglycosylation was performed using Glycerol-Free PNGase F (New England BioLabs, Catalog no P0705S). Subsequent reduction was performed using Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) (Thermoscientific, Catalog no 20490). The deglycosylated, reduced samples were separated using a Poroshell 300SB-C3 LC column, 1.0 × 30 mm, 5 µm (Agilent Technologies Inc., Catalog no 822600–909). Mobile Phase A was 99% water, 1% acetonitrile, 0.1% formic acid. Mobile Phase B was 70% n-propanol, 20% acetonitrile, 10% water, 0.1% formic acid. Intact samples were analyzed using an Agilent Time-of-Flight (TOF) 6210 mass spectrometer.
Prior to trypsin digestion, samples were reduced using DL-dithiothreitol (DTT) solution (Sigma, Catalog no 646563–10X.5ML) and alkylated using iodoacetamide (IAM) (Sigma, Catalog no A3221–10VL). Samples were then digested with trypsin from porcine pancreas (Sigma, Catalog non T6567–5 × 20UG) for 16 hours at 37°C. The peptides are separated using a C18 Hypersil™ GOLD HPLC column, 5 µm: 100L X 1.0mm ID size (Thermoscientific, Catalog no 25005–101030). Mobile Phase A was 99% water, 1% acetonitrile, 0.1% formic acid. Mobile Phase B was 95% acetonitrile, 5% water, 0.1% formic acid. The peptides were analyzed using a Thermo Orbitrap Fusion mass spectrometer. Electron Transfer Dissociation (ETD) fragmentation was used to fragment the O-glycosylated peptide, while retaining the glycan on the modified residue.
Sample preparation and storage
Samples used in each experiment were buffer exchanged at 4 – 10°C to desired buffer and pH conditions via Zeba desalting columns (Thermoscientific), after which the protein concentration and solution pH were verified separately. Buffers at pH 4.0, 5.0, and 6.0 with 30 mM sodium citrate were prepared by dissolving citric acid monohydrate, sodium dibasic citrate or sodium tribasic citrate (Sigma) in distilled and deionized water from a Milli-Q filtration system (Millipore) with a Quantum EX cartridge, and titrating to the target pH with 5 M sodium hydroxide (Sigma) solution. Samples were diluted gravimetrically to desired protein concentrations for each experiment described below. For all measurements, samples (aggregated and monomeric) were used within a week of preparation and stored at 2 – 8°C between measurements.
Differential scanning calorimetry
Thermal unfolding and aggregation of 1 mg/ml solutions of the initially monomeric Fc1 fragment, O-glycosylated Fc1, and non-O-glycosylated Fc1 in 30 mM sodium citrate buffer were monitored from 20°C to 90°C at a commonly used scan rate of 60°C/hr via an automated capillary DSC (MicroCal). Alternative scan rates were tested but the interpretations and conclusions presented in this report were independent of the scan rates tested (6 to 300°C/hr). Two scans with the corresponding buffer were performed to establish instrument thermal history and to obtain the instrument baseline for each sample, with the average of these scans subtracted from the subsequent protein thermogram to obtain the apparent heat capacity. Normalized scans were then analyzed with Origin 7.0. Pre-transition baselines were subtracted from each resulting heat capacity thermogram, to give the resulting excess heat capacity (Cp,ex) as a function of temperature.
Formation of protein aggregates
For aggregate preparation, monomeric protein at a given pH and initial protein solution concentration of 1 mg/ml was pipetted into HPLC vials (Waters, Catalog no 186000273DV) and hermetically sealed with HPLC screw caps (Waters, Catalog no 186000274). Aggregated protein samples were then prepared by incubating the solutions for predetermined times and temperatures in either a controlled temperature water bath (to within 0.2°C) or an incubator (to within 0.5°C). At each temperature and solution condition, incubation times were selected to ensure sufficient monomer loss to obtain aggregation rates based on the change in monomer concentration vs. time, measured by SEC.
Aggregated samples were quenched in an ice-water bath for at least 10 minutes after a given incubation time at elevated temperature. Ice-quenching was empirically observed to arrest aggregation, based on a lack of changes in chromatographic, light scattering, and spectroscopic results on time scales of weeks or longer.Citation33 All samples with a turbid appearance were centrifuged at 10,000 g for a minimum of 7 minutes, and the supernatant was collected for subsequent experimental analysis.
Analytical size exclusion chromatography
An Agilent Technologies 1200 Series with a photodiode array (PDA) detector, model G1365B and a TSK-GEL G3000SWxl SEC (Tosoh) were used to quantify monomer concentration of aggregated protein samples. 100 μL aliquots of protein samples were loaded into individual HPLC vials (Waters, Catalog no 186000384DV) and sealed with HPLC pre-slit screw caps (Waters, Catalog no 186000384DV). The vials were placed into the separation module autosampler, maintained at 4 ± 3°C. The column was held at room temperature (∼20 –22°C). The mobile phase was isocratic and was 0.5% (v/v) phosphoric acid and 100 mM sodium chloride in distilled deionized water, adjusted to pH 3.5 with 5 M sodium hydroxide.Citation33 Mobile phase flow rate was maintained at 1 mL/min. The injection volume for standards and samples was selected to assure nominally 100 μg of protein in each injection. Monomer concentrations were calculated based on absorbance at 280 nm using the Agilent ChemStation software, with separately prepared external standards.
The monomer concentrations for the Fc1 sample at each incubation condition were plotted and regressed as a function of incubation time. The observed rate coefficient for monomer loss (kobs) was determined by fitting the experimental monomer loss data to Equation Equation1(1)
(1) .Citation21,22,23
Analytical ultracentrifugation
The monomeric protein sample was diluted to 0.4 mg/ml in the corresponding buffer. Sedimentation velocity analysis, which is one of analytical ultracentrifugation methods,Citation60 was done to determine the tracer diffusion coefficient of the monomeric protein sample in an optima XL-I (Beckman Coulter) at 20°C using an An60Ti four-hole rotor running at 40,000 rpm. The sedimentation process was monitored by ultraviolet absorbance at 280 nm with a scan interval of 1sec, using corresponding dilution buffer as the reference buffer. The variation in the concentration distribution in the ultracentrifuge cell with time was collected using XL-I operating software and was analyzed using the continuous c(S) distribution model in the SEDFIT software (version 12.1c) to give the distribution of sedimentation coefficient through numerical solutions of the Lamm equationCitation61 and the tracer diffusion coefficient was calculated from sedimentation coefficient with the Sednterp software program,Citation62 which is freely available online (http://bitcwiki.sr.unh.edu/index.php/Main_Page). The hydrodynamic radius was calculated from the tracer diffusion coefficient using the Stokes-Einstein equation.
Capillary zone electrophoresis
Monomeric human Fc1 samples for CE were prepared as described above (cf., Sample Preparation and Storage), at selected pH values and a protein concentration of 1 mg/ml. The net charge measurement was performed using a ProteomeLab™ PA800 apparatus (Beckman Coulter) equipped with an ultraviolet (UV) absorbance detector with a working wavelength of 214 nm. Temperature was held at 20°C. An eCap amine capillary with an inner diameter of 50 μm (Beckman Coulter, Catalog no477431) was used, with a total capillary length of 60 cm (length to the detector = 50 cm). The capillary was rinsed with 0.1 N NaOH, amine regeneration solution (Beckman Coulter, Catalog no477433) and running buffer before each sample injection. Three voltages (10 kV, 14 kV, and 18 kV) were applied for each sample condition. 0.005% dimethylformamide (Sigma) in corresponding buffer was used as an electroosmotic flow (EOF) marker that was injected along with each sample. The electrophoretic mobility (µ) was obtained by subtracting the slope of a linear relationship between the EOF velocity (νEOF) and the electric field from the slope of a linear line for the sample velocity (νsample) vs. the electric field.Citation27,63 The observed charge, z*, was calculated from Equation Equation2(2)
(2) using µ from CE, and the tracer diffusion coefficient (D) from sedimentation velocity experiments via Beckman Coulter ProteomeLab™ XL-I.Citation26 The observed charge, z*, is always considered less than the net charge on a protein, zDHH, due to counter-ion shielding, the coupling of ionic flows and deformation of the electric field in the vicinity of the protein. The conversion between z* and zDHH can be calculated using Equation Equation3
(3)
(3) .
Protein-protein interactions from static light scattering
Monomeric protein samples were prepared, as described above, at protein concentrations ranging from 1 to 10 mg/ml. SLS measurements were performed using a Wyatt DAWN HELEOS II with a micro-cuvette accessory (Wyatt Technology). The instrument is equipped with a 120 mW solid state laser that operates at 658 nm. The weight averaged molecular weight (Mw) and the protein-protein Kirkwood Buff (KB) integral (G22) were obtained using Equation Equation4(4)
(4) ,Citation32,34 with additional details provided elsewhere. A dn/dc value of 0.187 cm3/g was used for all solution conditions.
Size exclusion chromatography with in-line multi-angle light scattering
The HELEOS II MALS detector was used in flow-cell mode along with an Optilab rEX refractive index detector (Wyatt Technology) and an Agilent Technologies 1200 Series HPLC (described above). Light scattering was measured at 16 angles between 32° and 147° and averaged over each one-second “slice” of column eluate with the in-line MALS detectors. Signals from the detectors were imported into ASTRA 5.3.4 software for data processing using standard methods.Citation32,34,38
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Material.pdf
Download PDF (139.5 KB)Acknowledgments
The authors thank Dr. John Miglietta, Dr. Irina Rybina, and Dr. Ertan Eryilmaz for helping design the Fc DNA construct, purify the Fc protein, and re-build the structure of the Fc from the published X-ray crystal structure of Fc with similar sequences in PDB, respectively.
Funding
Boehringer Ingelheim Pharmaceuticals Inc. is gratefully acknowledged for financial support.
References
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs 2015; 7:9-14; PMID:25529996; http://dx.doi.org/10.4161/19420862.2015.989042
- Aggarwal S. What's fueling the biotech engine - 2012 to 2013. Nat Biotechnol 2014; 32:32-9; PMID:24406926; http://dx.doi.org/10.1038/nbt.2794
- Carter P. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 2011; 317:1261-9; PMID:21371474; http://dx.doi.org/10.1016/j.yexcr.2011.02.013
- Kontermann R, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015; 0:1-10
- Beck A, Reichert J. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. mAbs 2011; 3:415-6; PMID:21785279; http://dx.doi.org/10.4161/mabs.3.5.17334
- Rath T, Baker K, Dumont J, Peters R, Jiang H, Qiao S, Lencer W, Pierce G, Blumberg R. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol 2015; 35:235-54; PMID:24156398; http://dx.doi.org/10.3109/07388551.2013.834293
- Levin D, Golding B, Strome S, Sauna Z. Fc fusion as a platform technology: potential for modulating immunogenicity. Trends Biochem Sci 2015; 33:27-34
- Kontermann R. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs 2009; 23:93-109; PMID:19489651; http://dx.doi.org/10.2165/00063030-200923020-00003
- Braun A, Kwee L, Labow M, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon α (IFN-α) in normal and transgenic mice. Pharm Res 1997; 14:1472-8; PMID:9358564; http://dx.doi.org/10.1023/A:1012193326789
- Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev 2002; 1:457-62
- Ratanji K, Derrick J, Dearman R, Kimber I. Immunogenicity of therapeutic proteins: Influence of aggregation. J Immunotoxicol 2014; 11:99-109; PMID:23919460; http://dx.doi.org/10.3109/1547691X.2013.821564
- Wu H, Kroe-Barrett R, Singh S, Robinson A, Roberts C. Competing aggregation pathways for monoclonal antibodies. FEBS Lett 2014; 588:936-41; PMID:24530501; http://dx.doi.org/10.1016/j.febslet.2014.01.051
- Weiss IV W, Young T, Roberts C. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci 2009; 98:1246-77; PMID:18683878; http://dx.doi.org/10.1002/jps.21521
- Kumar S, Wang X, Singh S. Identification and impact of aggregation-prone regions in proteins and therapeutic monoclonal antibodies. In: Aggregation of therapeutic proteins. Hoboken, NJ: John Wiley & Sons, Inc; 2011; 103-15
- Zhong X, Wright J. Biological insights into therapeutic protein modifications throughout trafficking and their biopharmaceutical applications. Int J Cell Biol 2013; 2013: Article ID 273086; 1-15; PMID:23690780; http://dx.doi.org/10.1155/2013/273086
- Kim H, Yamaguchi Y, Masuda K, Matsunaga C, Yamamoto K, Irimura T, Takahashi N, Kato K, Arata Y. O-glycosylation in hinge region of mouse immunoglobulin G2b. J Biol Chem 1994; 269:12345-50; PMID:7512967
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 97:2426-47; PMID:17828757; http://dx.doi.org/10.1002/jps.21180
- Demarest S, Rogers J, Hansen G. Optimization of the antibody CH3 domain by residue frequency analysis of IgG sequences. J Mol Biol 2004; 335:41-8; PMID:14659738; http://dx.doi.org/10.1016/j.jmb.2003.10.040
- Souillac P. Biophysical characterization of insoluble aggregates of a multi-domain protein: an insight into the role of the various domains. J Pharm Sci 2005; 94:2069-83; PMID:16052560; http://dx.doi.org/10.1002/jps.20423
- Ionescu R, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci 2008; 97:1414-26; PMID:17721938; http://dx.doi.org/10.1002/jps.21104
- Roberts C. Non-native protein aggregation kinetics. Biotechnol Bioeng 2007; 98:9237-8; http://dx.doi.org/10.1002/bit.21627
- Roberts C. Kinetics of irreversible protein aggregation: analysis of extended Lumry-Eyring models and implications for predicting protein shelf life. J Phys Chem B 2003; 107:1194-207; http://dx.doi.org/10.1021/jp026827s
- Andrews J, Roberts C. A Lumry-Eyring nucleation polymerization model of protein aggregation kinetics: 1. aggregation with pre-equilibrated unfolding. J Phys Chem B 2007; 111:7897-913; PMID:17571872; http://dx.doi.org/10.1021/jp070212j
- Andrews J, Roberts C. Non-native aggregation of α-Chymotrypsinogen occurs through nucleation and growth with competing nucleus sizes and negative activation energies. Biochemistry (Mosc) 2007; 46:7558-71; http://dx.doi.org/10.1021/bi700296f
- Voet D, Voet JG. Biochemistry (3rd ed). Hoboken, NJ: John Wiley and Sons, Inc; 2004
- Brown P, Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J 2006; 90:4651-61; PMID:16565040; http://dx.doi.org/10.1529/biophysj.106.081372
- Chase S, Laue T. The determination of protein valence by capillary electrophoresis. PACE Setter 2008; 12:1-5
- Edsall J, Wyman J. Biophysical Chemistry: Volume 1 (1st ed.). San Diego, CA: Academic Press; 1958; 282-95
- Henry D. The cataphoresis of suspended particles. Proc Roy Soc A 1931; 133:106-40; http://dx.doi.org/10.1098/rspa.1931.0133
- Kertes A, King C. Extraction chemistry of fermentation product carboxylic-acids. Biotechnol Bioeng 1986; 28:269-82; PMID:18555324; http://dx.doi.org/10.1002/bit.260280217
- Blanco M, Sahin E, Li Y, Roberts C. Reexamining protein-protein and protein-solvent interactions from Kirkwood-Buff analysis of light scattering in multi-component solutions. J Chem Phys 2011; 134:225103-12; PMID:21682538; http://dx.doi.org/10.1063/1.3596726
- Blanco M, Perevozchikova T, Martorana V, Manno M, Roberts C. Protein-protein interactions in dilute to concentrated solutions: α-chymotrypsinogen in acidic conditions. J Phys Chem B 2014; 118:5817-31; PMID:24810917; http://dx.doi.org/10.1021/jp412301h
- Brummitt R, Nesta D, Chang L, Chase S, Laue T, Roberts C. Nonnative aggregation of an IgG1 antibody in acidic conditions: Part 1. unfolding, colloidal interactions, formation of high-molecular-weight aggregates. J Pharm Sci 2011; 100:2087-103; PMID:21213308; http://dx.doi.org/10.1002/jps.22448
- Kim N, Remmele R Jr., Liu D, Razinkov V, Fernandez E, Roberts C. Aggregation of anti-streptavidin immunoglobulin gamma-1 involves Fab unfolding and competing growth pathways mediated by pH and salt concentration. Biophys Chem 2013; 172:26-36; PMID:23334430; http://dx.doi.org/10.1016/j.bpc.2012.12.004
- Sahin E, Grillo A, Perkins M, Roberts C. Comparative effects of pH and ionic strength on protein-protein interactions, unfolding, and aggregation for IgG1 antibodies. J Pharm Sci 2010; 99:4830-48; PMID:20821389; http://dx.doi.org/10.1002/jps.22198
- Li Y, Roberts C. A Lumry-Eyring nucleated-polymerization model of protein aggregation kinetics 2. Competing growth via condensation and chain polymerization. J Phys Chem B 2009; 113:7020-32; PMID:19368365; http://dx.doi.org/10.1021/jp8083088
- Li Y, Weiss IV W, Roberts C. Characterization of high-molecular-weight nonnative aggregates and aggregation kinetics by size exclusion chromatography with inline multi-angle laser light scattering. J Pharm Sci 2009; 98:3997-4016; PMID:19283773; http://dx.doi.org/10.1002/jps.21726
- Brummitt R, Nesta D, Chang L, Kroetsch A, Roberts C. Nonnative aggregation of an IgG1 antibody in acidic conditions, Part 2: nucleation and growth kinetics with competing growth mechanisms. J Pharm Sci 2011; 100:2104-19; PMID:21213307; http://dx.doi.org/10.1002/jps.22447
- Li Y, Ogunnaike B, Roberts C. Multi-variate approach to global protein aggregation behavior and kinetics: Effects of pH, NaCl, and temperature for α-chymotrypsinogen A. J Pharm Sci 2010; 99:645-62; PMID:19653264; http://dx.doi.org/10.1002/jps.22159
- Santora L, Krull I, Grant K. Characterization of recombinant human monoclonal tissue necrosis factor-α antibody using cation-exchange HPLC and capillary isoelectric focusing. Anal Biochem 1999; 275:98-108; PMID:10542114; http://dx.doi.org/10.1006/abio.1999.4275
- Santora L, Stanley K, Krull I, Grant K. Characterization of maleuric acid derivatives on transgenic human monoclonal antibody due to post-secretional modifications in goat milk. Biomed Chromatogr 2006; 20:843-56; PMID:16425344; http://dx.doi.org/10.1002/bmc.603
- Lyubarskaya Y, Houde D, Woodard J, Murphy D, Mhatre R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Anal Biochem 2006; 348:24-39; PMID:16289440; http://dx.doi.org/10.1016/j.ab.2005.10.003
- Garber E, Demarest S. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun 2007; 355:751-7; PMID:17321501; http://dx.doi.org/10.1016/j.bbrc.2007.02.042
- McConnell A, Spasojevich V, Macomber J, Krapf I, Chen A, Sheffer J, Berkebile A, Horlick R, Neben S, King D, et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng Des Sel 2013; 26:151-63; PMID:23173178; http://dx.doi.org/10.1093/protein/gzs090
- Latypov R, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J Biol Chem 2012; 287:1381-96; PMID:22084250; http://dx.doi.org/10.1074/jbc.M111.297697
- Vermeer A, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J 2000; 78:394-404; PMID:10620303; http://dx.doi.org/10.1016/S0006-3495(00)76602-1
- Ahmed A, Giddens J, Pincetic A, Lomino J, Ravetch J, Wang L, Bjorkman P. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol 2014; 426:3166-79; PMID:25036289; http://dx.doi.org/10.1016/j.jmb.2014.07.006
- Barnett G, Razinkov V, Kerwin B, Laue T, Woodka A, Butler P, Perevozchikova T, Roberts C. Specific-ion effects on the aggregation mechanisms and protein-protein interactions for anti-streptavidin immunoglobulin gamma-1. J Phys Chem B 2015; 119:5793-804; PMID:25885209; http://dx.doi.org/10.1021/acs.jpcb.5b01881
- Ben-Naim A. Statistical thermodynamics for chemists and biochemists (1st ed.). New York, NY: Plenum Press; 1992
- McQuarrie D. Statistical mechanics (2nd ed.). Sausalito, CA: University Science Books; 2000
- Israelachvili J. Intermolecular & surface forces (2nd ed.). London, UK: Academic Press; 1992
- Roberts D, Keeling R, Tracka M, van der Walle C, Uddin S, Warwicker J, Curtis R. The role of electrostatics in protein-protein interactions of a monoclonal antibody. Mol Pharm 2014; 11:2475-89; PMID:24892385; http://dx.doi.org/10.1021/mp5002334
- Tessier P, Sandler S, Lenhoff A. Direct measurement of protein osmotic second virial coefficients by cross-interaction chromatography. Protein Sci 2004; 13:1379-90; PMID:15075404; http://dx.doi.org/10.1110/ps.03419204
- Arosio P, Jaquet B, Wu H, Morbidelli M. On the role of salt type and concentration on the stability behavior of a monoclonal antibody solution. Biophys Chem 2012; 168–169:19-27; PMID:22750560; http://dx.doi.org/10.1016/j.bpc.2012.05.004
- Drenski M, Brader M, Alston R, Reed W. Monitoring protein aggregation kinetics with simultaneous multiple sample light scattering. Anal Biochem 2013; 437:185-97; PMID:23481914; http://dx.doi.org/10.1016/j.ab.2013.02.014
- Roberts C, Darrington R. Irreversible aggregation of recombinant bovine granulocyte-colony stimulating factor (bG-CSF) and implications for predicting protein shelf life. J Pharm Sci 2003; 92:1095-111; PMID:12712430; http://dx.doi.org/10.1002/jps.10377
- Wang W, Roberts C. Non-Arrhenius protein aggregation. AAPS J 2013; 15:840-51; PMID:23615748; http://dx.doi.org/10.1208/s12248-013-9485-3
- Kayser V, Chennamsetty N, Voynov V, Helk B, Forrer K, Trout BL. Evaluation of a Non-Arrhenius model for therapeutic monoclonal antibody aggregation. J Pharm Sci 2011; 100:2526-42; PMID:21268027; http://dx.doi.org/10.1002/jps.22493
- Saluja A, Sadineni V, Mungikar A, Nashine V, Kroetsch A, Dahlheim C, Rao VM. Significance of unfolding thermodynamics for predicting aggregation kinetics: A case study on high concentration solutions of a multi-domain protein. Pharm Res 2014; 31:1575-87; PMID:24398696; http://dx.doi.org/10.1007/s11095-013-1263-5
- Cole J, Lary J, Moody T, Laue T. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 2008; 84:143-79; PMID:17964931; http://dx.doi.org/10.1016/S0091-679X(07)84006-4
- Schuck P, Perugini M, Gonzales N, Howlett G, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 2002; 82:1096-111; PMID:11806949; http://dx.doi.org/10.1016/S0006-3495(02)75469-6
- Harding S, Rowe A, Horton J. Analytical ultracentrifugation in biochemistry and polymer science (1st ed.). London, UK: Royal Society of Chemistry; 1992
- Adamson N, Reynolds E. Rules relating electrophoresis mobility, charge, and molecular size of peptides and proteins. J Chromatogr B 1997; 699:133-47; http://dx.doi.org/10.1016/S0378-4347(97)00202-8