
Abstract
An alternative method to traditional 2-dimensional gel electrophoresis (2D-PAGE) and its application in characterizing the inherent charge heterogeneity of chromatographically isolated monoclonal antibody heavy and light chains is described. This method, referred to as ChromiCE, utilizes analytical size-exclusion chromatography (SEC), performed under reducing and denaturing conditions, followed by imaged capillary isoelectric focusing (icIEF) of the chromatographically separated heavy and light chains. Under conditions suitable for the subsequent icIEF analysis, the absolute and relative SEC elution volumes of the heavy and light chains were found to be highly pH dependent, a phenomenon that can be exploited in optimizing chromatographic separation. Compared to 2D-PAGE, the ChromiCE method substantially decreases the time and labor needed to complete the analysis, improves reproducibility, and provides fully quantitative assessment of charge heterogeneity. The ChromiCE methodology was applied to a set of diverse monoclonal antibodies to demonstrate suitability for quantitative charge variant analysis of heavy and light chains. A typical application of ChromiCE in extended characterization and stability studies of a purified antibody is shown.
Abbreviations
| 2D-PAGE | = | two-dimensional polyacrylamide gel electrophoresis |
| cIEF | = | capillary isoelectric focusing |
| FWHH | = | full width half height |
| HC | = | heavy chain |
| icIEF | = | imaged capillary isoelectric focusing |
| IEF | = | isoelectric focusing |
| IEX | = | ion exchange chromatography |
| LC | = | light chain |
| RD-SEC | = | reduced and denatured size exclusion chromatography |
| SEC | = | size exclusion chromatography |
Introduction
With over 30 approved products worldwide and hundreds of on-going clinical trials, therapeutic antibody development is a well-established and vital segment of the pharmaceutical industry.Citation1 Moreover, advances in antibody engineering have resulted in novel targeted therapies and antibody formats such as antibody-drug conjugates and bispecific antibodies, which hold tremendous therapeutic potential.Citation2,3 Compared to non-proteinaceous drugs, therapeutic antibodies are several orders of magnitude larger and significantly more complex.Citation4 This complexity arises, in part, because therapeutic antibodies are manufactured in biological systems (e.g., Chinese hamster ovary cells), leading to differences in post-translational modifications (e.g., amino or carboxy terminal processing, glycosylation).Citation5,6 Additionally, various chemical alterations (e.g., deamidation, oxidation, fragmentation) may occur during purification and storage.Citation7-9 The combined effect of these modifications, which occur during both up-stream and down-stream processes, is micro-heterogeneity in the physicochemical properties of the proteins that requires advanced analytical technologies to properly characterize.
While micro-heterogeneity is a generally accepted property of both natural and recombinant proteins, methods to assess, control, and, if possible, reduce the occurrence in biotherapeutics intended for clinical use are needed.Citation10 A suite of orthogonal and complementary assays are typically employed to characterize the molecular properties of therapeutic antibodies such as size, glycosylation, and isoelectric point.8 These quality attributes support drug safety and efficacy profiles, which are ultimately related to clinical performance.Citation11 Charge heterogeneity of a purified protein is manifest in the presence of minor populations with slightly different isoelectric points (either acidic or basic relative to the main species). These charge variants may stem from several distinct pathways, including deamidation, glycation, and differences in glycosylation.Citation12 Although evidence suggesting isolated charge variants have comparable safety and efficacy is beginning to emerge, the results cannot necessarily be extended to all molecules, a priori, and is likely to be highly dependent on the nature, location, and extent of the modifications.Citation13 Furthermore, charge heterogeneity must be monitored during development and production to ensure product consistency, and over the course of normal shelf-life to monitor stability.Citation14 Obtaining detailed quantitative analysis of the charge variant profile is an early requisite step toward uncovering the various pathways by which a therapeutic antibody acquires charge heterogeneity. This information has the potential to better inform structure/function/stability relationships, and to clarify any effect of charge heterogeneity on clinical safety and efficacy and shelf-life.
Charge variant analysis of intact antibodies may be achieved using isoelectric focusing techniques (IEF) or ion exchange chromatography (IEX). Gel and capillary-IEF methods use a pH gradient and an electric field to separate charge variants based on the intrinsic charge of the molecule.Citation14-16 Although readily implemented without the need for sophisticated equipment, flatbed and vertical gel-IEF have been largely supplanted by capillary-based IEF (cIEF) methods because the latter provides rapid, quantitative characterization with greater resolution. Chromatographic methods are also relatively straightforward to implement and the different combinations of ion exchange resins, solution conditions, and elution gradients facilitate method optimization for each individual antibody.Citation17 In IEX, charge variants are resolved by disrupting the interactions between the stationary phase and the charged groups on the molecular surface; therefore, it is believed to reveal the more biologically relevant charge variant profile and alterations thereof.Citation18 While both IEF and IEX methods provide a convenient fingerprint of monoclonal antibody charge heterogeneity, they do not provide specific information related to the molecular events giving rise to each species. Incorporating IEX separation up-stream of mass spectral analysis has been shown to provide greater detail regarding the molecular origin of charge variants.Citation19 Intact mass of resolved peaks can reveal differential glycan forms, but additional processing and data analysis (e.g., tryptic peptide mapping) is required to identify the location and nature of specific chemical modifications (e.g., residue specific deamidation).Citation20,21 Thus, a rapid and simple separation method with greater information content would be highly beneficial.
The typical charge variant profile of an intact monoclonal antibody is composed of a single main species and between 3 to 10 acidic and basic charge variants. Often the IEX chromatograms and IEF electropherograms are complex, with poorly resolved peaks, a situation that is exacerbated in accelerated degradation studies. Thus, it is frequently desirable to examine the charge heterogeneity of the heavy chain (HC) and light chain (LC) separately. The conventional approach is to employ 2-dimensional polyacrylamide gel electrophoresis (2D-PAGE), in which the monoclonal antibody is chemically denatured and reduced, then separated according to isoelectric point in the first dimension and subsequently resolved according to molecular weight in the second dimension. Although widely regarded as the “gold-standard” in gel-based separations, fundamentally the power of 2D-PAGE is in separation of proteins from a complex mixture rather than discrete analysis of purified proteins. Several innovations have improved sensitivity and the ability to identify low abundance proteins; however, streaking or warping in either dimension is frequently observed, making it difficult to achieve highly consistent results with 2D-PAGE. An additional drawback with the use of 2D-PAGE in therapeutic antibody development is that preparation and manipulation of the IEF strips and polyacrylamide gels is not amenable to automation, which limits overall throughput. Lastly, and most significantly, rigorous analysis of the relative charge variant percentages and pI values is not readily accomplished, inhibiting routine quantitative lot-to-lot comparisons.
Given the importance of charge heterogeneity in therapeutic antibody development, we sought an alternative to 2D-PAGE for the charge variant analysis of antibody heavy and light chains. By taking a non-standard approach to analytical size-exclusion chromatography (SEC), sufficient amounts of reduced and denatured heavy and light chains were separated to subsequently perform capillary IEF with whole column imaging, more commonly known as imaged capillary IEF (icIEF).Citation22 This novel combination of 2 relatively simple methods, referred to as ChromiCE, has the flexibility to allow for antibody-specific optimization, increases throughput while reducing time and labor, and provides quantitative charge variant data on the individual heavy and light chains. These attributes make ChromiCE an excellent characterization assay for in-process monitoring of charge heterogeneity and detection of changes in product quality during routine storage. The utility and general applicability of this approach is demonstrated by comparing the capillary isoelectric fingerprint of heavy and light chains from an antibody with the corresponding 2D-PAGE image and subsequently detecting changes in the charge variant profile of this antibody under accelerated stability conditions.
Results
Reduced and denatured SEC
Canonical high performance SEC is typically performed with flow rates of ∼1 mL/min; significant changes in the flow rate may dramatically alter the resolution and will affect the duration of SEC. The van Deemter equation predicts that optimal resolution (minimal peak broadening) is achieved at a linear velocity that allows the most uniform mass transfer while minimizing longitudinal diffusion.Citation23 Generally, optimum column efficiency is achieved with lower flow rates; higher flow rates reduce run time, but at the expense of chromatographic resolution. Initially, a flow rate of 0.25 mL/min was used to counteract the increase in backpressure induced by the high viscosity of 8 M urea in the mobile phase. When the flow rate was increased in an attempt to screen conditions more rapidly by shortening the SEC runs, the peaks did not appear to be adversely affected by the increased flow rate. Indeed, upon measuring the full width at half height (FWHH), we determined that increasing the flow rate from 0.25 to 0.425 mL/min reduced the FWHH of the HC peak from 1.73 to 0.91 (). Column and system back pressure limitations, however, prevented flow rates greater than 0.425 mL/min. Subsequently, all optimization and semi-preparative separations were performed at this flow rate.
Figure 1. RD-SEC elution profiles as a function of flow rate (A & B) and ionic strength (C & D). Five hundred milligrams of reduced and denatured mAb-1 was injected into 2 Shodex 802.5 columns pre-equilibrated with 8 M urea, 2 mM DTT, and 10 mM HEPES, pH 7.5 and eluted with a flow rate of (A) 0.25 mL/min or (B) 0.425 mL/min. The effect of ionic strength was examined under similar conditions except the buffer was (C) 10 mM and (D) 35 mM sodium acetate, pH 4.5.
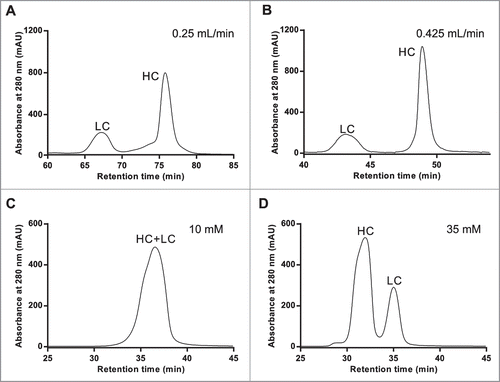
In the mAb-1 elution profile shown in , the heavy and light chains appear to elute in reverse order (i.e., the LC elutes before the HC). This indicated non-specific interactions between the protein and the stationary phase, were present under these conditions. Non-specific interactions between antibodies and SEC resins under aqueous conditions are well-documented and believed to be dominated by electrostatic interactions.Citation24 This phenomenon is typically abrogated with high ionic strength mobile phase, an approach not well-suited for subsequent isoelectric focusing.Citation25 To alter any potential electrostatic interactions, the chromatography was performed in mildly acidic mobile phase (pH 4.5) so that both the heavy and light chains would have a net positive charge. Under these conditions, the elution profile of mAb-1 () was markedly different than the elution profile observed under near neutral pH (); however, the heavy and light chain peaks were not well resolved. This suggested the non-specific interactions were significantly weaker, but not completely eliminated. Silica-based separation matrices are known to have residual anionic character, which can be shielded with minimal conductivity mobile phase. We hypothesized that marginally increasing the conductivity would make optimization of reduced and denatured SEC (RD-SEC) a more straightforward process and not adversely affect the subsequent isoelectric focusing. Rather than introduce an additional variable by including sodium chloride, the concentration of buffer salts was increased. When reduced and denatured mAb-1 (100 μg) was separated with a mobile phase containing 35 mM buffer salt, the resolution between heavy and light chain peaks was greatly improved (). This beneficial effect was observed for several different monoclonal antibodies (data not shown); however, increasing the buffer strength above 35 mM may adversely affect icIEF performance and is not recommended.
The striking difference observed between the RD-SEC elution profiles of mAb-1 performed under neutral and acidic conditions suggested chromatographic separation of antibody heavy and light chains could be fine-tuned. Therefore, the effect of mobile phase pH using 35 mM buffer salt concentration and 0.425 mL/minute flow rate on RD-SEC elution profile was investigated more broadly. When mAb-1 was separated under slightly acidic conditions, the HC and LC appeared to be well-separated, but with a highly distorted baseline (). The separation between HC and LC progressively decreased as the mobile phase pH increased (), eventually resulting in overlapped peaks at pH 9.0. The optimized elution was achieved with a mobile phase pH of 7.0, which is approximately one pH unit below the pI values for both the HC and LC (HC pI: 8.0; LC pI: 7.8). Differences in elution profiles were not solely dependent on pH, indicating the chemical nature of the buffer and the relative difference between the pKa and pH may also influence the chromatographic behavior.
Figure 2. Demonstration of antibody specific optimization of RD-SEC by screening different mobile phase pH's. One hundred milligrams of reduced and denatured mAb-1 was injected into 2 Shodex 802.5 columns pre-equilibrated with 8 M urea, 2 mM DTT, and (A) 10 mM MES, pH 6.0, (B) 10 mM MOPS, pH 7.0, (C) 10 mM MOPS, pH 8.0, and (D) 10 mM HEPES, pH 9.0.
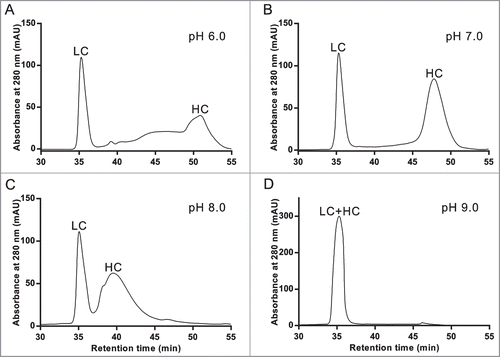
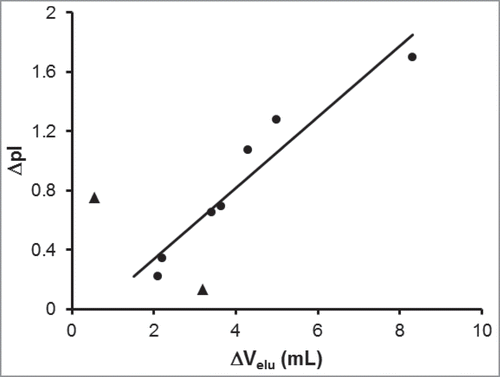
The behavior observed in the mAb-1 chromatograms is representative of a general phenomenon found for a panel of 9 antibodies comprising IgG1 and IgG4 isotypes with isoelectric points ranging from 6.0 to 8.7 that were separated by RD-SEC on silica-based analytical size-exclusion columns. Optimal separation was generally achieved with a mobile phase buffer approximately one pH unit below the lowest pI value of the 2 chains. This provides a simple way to determine the initial conditions from which to perform RD-SEC optimization of antibody heavy and light chain separation. Interestingly, the magnitude of the difference in elution volume was correlated with the difference in pI values of the heavy and light chains (). For antibodies with large differences in heavy and light chain isoelectric points, greater resolution of the 2 peaks was obtained. If the isoelectric points were more similar, the peaks eluted more closely and with greater variability, as indicated by the 2 outliers (triangles).
Figure 3. Correlation between the difference in heavy and light chain elution volumes with difference in isoelectric points. The chromatographic separation of 9 different monoclonal antibodies under reducing and denaturing conditions was optimized to determine the difference in elution volume between the heavy and light chains (ΔVelu). These values are plotted against the difference in theoretical pI values between the heavy and light chains. The linear correlation line (r2 = 0.92) was determine from 7 mAbs (circles); 2 mAbs (triangles) were excluded from the regression.
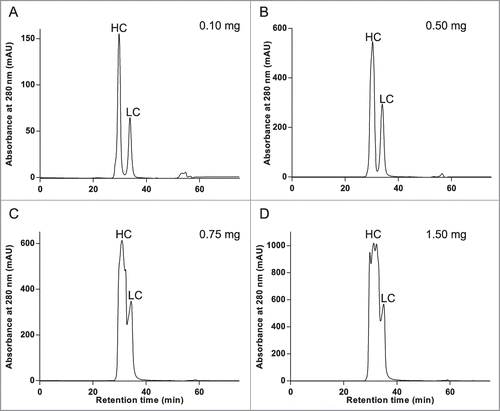
Analytical chromatography generally operates on a scale that rarely affords material for follow-on analysis, with the exception of extremely sensitive methodologies such as mass spectral analysis. Therefore, it was necessary to determine the maximum load amount that maintained adequate resolution of the heavy and light chains under RD-SEC conditions optimized on the analytical scale. An antibody (mAb-2) with more standard SEC elution properties was chosen for this analysis because non-standard elution profiles (i.e., large difference in heavy and light chain elution volumes, such as with mAb-1) can in fact be exploited to load extremely high amounts of material. Thus, mAb-2 represents a more realistic test case to probe the limits of resolution with respect to load amount under the ChromiCE conditions. As shown in , sufficient resolution was obtained with a load amount of 500 μg, whereas higher load amounts resulted in distorted peak shapes and decreased resolution between HC and LC. Although identification of the HC and LC RD-SEC peaks was intuitive based on the differential UV absorbance at 280 nm (in general, HC and LC have 8–9 and 2–3 tryptophan residues, respectively), the peak identities were confirmed experimentally with SDS-PAGE () and mass spectrometry (Fig. S2) prior to proceeding to icIEF. An LC-MS method is arguably the most robust method; however, polyacrylamide gel analysis is likely the most convenient. The high concentration of urea may interfere with other methods, for example the upper limit of urea concentration for microfluidic capillary electrophoresis analysis is 6 M. The identities of the proteins that composed the pooled fractions collected from the apex of the 2 peaks in the RD-SEC chromatogram of mAb-2 are HC and LC, respectively.
Figure 4. Chromatographic separation of reduced and denatured heavy and light chains in sufficient amounts for icIEF is possible with RD-SEC. Increasing amounts, from 0.1 milligrams to 1.5 milligrams, of reduced and denatured mAb-2 was separated under previously optimized conditions (8 M urea, 1 mM TCEP, and 35 mM sodium citrate, pH 6.0; 0.425 mL/min flow rate).
Charge variant analysis
Having demonstrated good control over separation of heavy and light chains using RD-SEC, we next investigated the conditions best suited for subsequent icIEF. Initially, HC and LC RD-SEC fractions were diluted into icIEF buffer prepared without urea, thus reducing the final urea concentration to ∼4 molar. This focusing condition was found to be suitable for most antibodies; however, the HC electropherograms from a few mAbs were extremely noisy with inconsistent absorbance between replicate injections, which is indicative of poor solubility. Therefore, the icIEF buffer was prepared with 8 M urea; this condition was found to improve HC solubility and provide run-to-run consistency. The increase in viscosity can be offset with increased focusing time and can be optimized for each molecule. The charge variant profiles of RD-SEC separated HC and LC of mAb-2 are shown in , respectively. For comparison, the corresponding 2D-PAGE is shown in .
Figure 5. Imaged capillary isoelectric focusing (icIEF) of chromatographically separated reduced and denatured mAb-2 heavy and light chains. Pooled RD-SEC fractions containing either (A) heavy or (B) light chain protein were focused for 8 minutes on an iCE280 Analyzer. For comparison, the corresponding 2D-PAGE is shown in C.
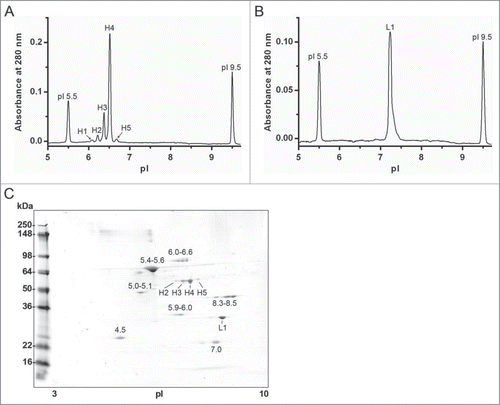
In the electropherogram shown in , the dominant peak (H4), corresponding to a pI of ∼6.5, is consistent with the theoretical pI of the heavy chain after C-terminal lysine processing with a theoretical pI of 6.8.Citation26 The identity of the species composing the minor basic peak (H5, pI 6.7) is likely the heavy chain with C-terminal lysine and the minor acidic peaks (H1–H3) may represent HC with one or more deamidated asparagine residues.Citation26 A single peak was observed for the separated LC with a pI value of 7.2, consistent with the theoretical value of 6.9 determined from the primary sequence. The quantitative icIEF results are summarized in . Direct identity testing of these peaks remains a technical challenge; however, the tentative identities are supported by LC/MS/MS tryptic peptide mapping of reduced and alkylated mAb-2.
Table 1. Summary of isoelectric focusing of mAb-2 heavy chain by imaged capillary isoelectric focusing
Forced degradation studies
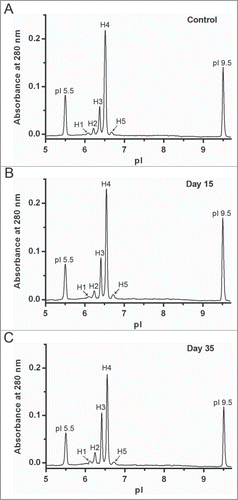
Quantitative analysis is required to clearly detect degradation products formed during accelerated and real-time stability studies. Here, mAb-2 was incubated at 45°C to create conditions favorable for deamidation, then analyzed with the ChromiCE method to demonstrate its ability to detect and quantify changes in charge variant profiles relative to control samples. Significant changes in the charge distribution profile of reduced and denatured mAb-2 HC were observed after icIEF between the control and samples incubated for both 15 and 35 days (). At the 35 day time point, the percent peak area of peak H4 (main species) decreased by ∼20% with a concomitant increase in total acidic species percentages distributed over peaks H1-H3 (). This is consistent with increased deamidation of 2 Asn-Gly dipeptide sequences in the heavy chain; no novel charge variants were identified.Citation27 The peak area percentage of the minor basic charge variant (peak H5) was somewhat variable, but overall largely unchanged. The mAb-2 LC electropherogram generated after 35 d of heat stress was highly similar to the control, containing a single peak with a pI value of ∼7.2 (data not shown).
Figure 6. Assessment of accelerated stability studies using ChromiCE. mAb-2 was incubated at 45°C and processed for ChromiCE to provide quantitative charge variant analysis at specific time points.
Discussion
Analytical characterization of physicochemical properties is essential to quality assessment of biotherapeutics and the detection of changes that may occur during manufacturing and storage. Therapeutic monoclonal antibodies have long been characterized with 2D-PAGE to qualitatively assess charge heterogeneity of the heavy and light chains. Here, we have presented a rapid, straightforward approach to quantify the charge variants of isolated heavy and light chains. The methodology utilizes automated platforms (SEC and icIEF) to minimize time and labor; results are readily obtained in a single day. The SEC segment, performed under reducing and denaturing conditions, can be optimized by screening mobile phase pH for optimal chromatographic resolution. The icIEF electropherograms, also obtained under reducing and denaturing conditions to prevent protein aggregation at the isoelectric point, provide quantitative analysis (pI and peak area percentage) of the charge variants. This method, referred to as ChromiCE, has been used to characterize over 10 different monoclonal antibodies, including both IgG1 and IgG4 isotypes, with isoelectric points ranging from 6.0 to 8.7, demonstrating the general applicability of the method during therapeutic antibody development.
Chromatographic separation of heavy and light chains from reduced monoclonal antibodies has been demonstrated previously. Strong organic acids can effectively dissociate the heavy and light chains of reduced antibodies and allow chromatographic separation of the chains by size-exclusion, reverse-phase, or cation-exchange columns.Citation28,29 Unfortunately, these methods also required the use of trifluoroacetic acid/acetonitrile or sodium chloride gradients for proper elution, both of which are incompatible with capillary isoelectric focusing. Furthermore, the antibodies are often modified (alkylated) prior to chromatography. By incubating the antibodies in the presence of a strong reductant (TCEP) and denaturant (∼6 M guanidinium), we were able to achieve complete reduction and denaturation without the use of elevated temperatures or modifications, which could alter the pI profile. The SEC step also performs the essential buffer exchange of guanidinium for urea, thereby maintaining the proteins in a denatured state and transferring them into a solvent that is compatible with icIEF.
Baseline resolution of globular proteins differing in molecular weight by a factor of 2 is readily achieved with analytical SEC columns when appropriate load amounts (typically less than 100 μg total) are injected. The need to inject several fold greater quantities of antibody in order to recover sufficient material for subsequent icIEF analysis was immediately recognized. A primary concern was the effect these elevated load amounts would have on column performance and lifetime. Therefore, we examined the utility of several commercially available lab-scale columns. We determined that resins with average particle size greater than ∼10 μm, packed in standard columns, were not capable of adequately resolving the heavy and light chain peaks under reduced and denatured conditions (data not shown). Conversely, the smaller average particle size of UPLC resins provided excellent resolution; however, the column dimensions available at the time were not sufficient to handle several hundred microgram protein loads. Based on our results, standard Shodex or Tosoh SEC columns (8 mm by 300 mm) appear to be the most appropriate commercially available columns for the ChromiCE method. We routinely injected 500 μg of reduced and denatured mAb and obtained near baseline resolution between heavy and light chains. No significant impact of high load amounts on column performance was observed if a simple cleaning procedure with high purity water is performed regularly.
In principle, SEC separates proteins based on their hydrodynamic volume, assuming there are no interactions between the separation matrix and the eluting proteins. After applying the ChromiCE method to several different monoclonal antibodies, the elution volumes for the population of heavy and light chains analyzed were found to be inconsistent, suggesting some of the denatured proteins were interacting with the SEC resin. Deviation from ideal chromatographic elution behavior in low ionic strength mobile phases has been previously shown to be primarily caused by electrostatic effects.Citation30,31 Altering the electrostatic profile of the heavy and light chains by changing the pH of the mobile phase induced antibody-specific effects in the associated elution profiles and was found to be a useful parameter for empirical optimization of SEC conditions on an analytical (100 μg) scale. In some cases the optimal resolution, measured solely by peak-to-peak separation, was found with the LC eluting ahead of the HC. This unexpected elution profile may be explained, in part, by the previous observation that urea denatured proteins exhibit different levels of compactness and may contain extensive secondary structure.Citation32 Optimizing the separation conditions for RD-SEC is crucial because the amount of material that can be loaded while maintaining adequate resolution is generally correlated with peak separation; load amounts approaching one milligram are possible for antibodies with well-resolved heavy and light chain peaks.
Although not as striking as the effects of mobile phase pH, the chemical nature of the buffer also appears to influence the elution properties of reduced and denatured antibodies (data not shown). Buffers with higher molar conductivity (sodium phosphate and sodium acetate) resulted in narrower peaks than those with lower conductivity (i.e., HEPES or MOPS). We suspect this is likely an effect of charge density on ionic screening; however, keeping the overall ionic strength low in order to accelerate cIEF analysis is preferred. More subtle differences in elution profiles were observed for mobiles phases with similar pH, but different buffer species, indicating the chemical nature of the buffer and the relative difference between the pKa and pH may also influence the chromatographic behavior. Incorporating a dialysis step or desalting column into the procedure is feasible in cases when this is not possible. Although not tested within the context of ChromiCE, non-ionic detergents such as Brij-35 or NP-40 have been used to decrease nonspecific interactions with the gel filtration matrix,Citation33 and may impart better chromatographic behavior while remaining amenable to icIEF. In addition to properties of the mobile phase, flow rate also affects the elution behavior of analytes. In RD-SEC, higher flow rates generated narrower peaks. Elevated flow rates, along with the high viscosity of the mobile phase, markedly increases system back pressure, which may cause the denatured proteins to adopt a more uniform and compact size distribution, thereby improving peak shape. Kept within reasonable limits, higher flow rates have the added advantage of reduced run time.
The essentially seamless transition from chromatographic separation of monoclonal antibody heavy and light chains to analytical characterization of the respective protein charge profiles is a clear advantage of the ChromiCE method over standard 2D-PAGE. This reduces sample processing and incubation times substantially. Moreover, the overall chemical treatment is less harsh, and therefore the potential for artifacts is reduced. However, it is the ability to perform capillary isoelectric focusing, with its superior resolution, and the quantitative analysis of the resulting electropherograms that sets ChromiCE apart. This advantage enables a more direct comparison of charge variant profiles (isoelectric points and percent peak areas) within the context of antibody characterization and will likely make the early detection of degradation products easier and more reliable. ChromiCE can also be used to generate less complex electropherograms of antibodies that have undergone primary amine conjugation (e.g., biotin, ruthenium, or antibody-drug conjugates), which may provide a more thorough understanding of the conjugation process and greater quality assurance between batches of labeled reagents. Similarly, there is potential application in the comparison of innovator and biosimilar molecules to reveal differences that may be masked by the complexity of isoelectric profile of the intact molecule.Citation34 It remains to be seen if the ChromiCE method can be incorporated upstream of a mass spectrometry method; however, the potential wealth of information that may result is quite intriguing.
Although no method is without potential limitations, we have found the ChromiCE method well-suited to characterize reduced and denatured monoclonal antibodies. Optimization was typically achieved in less than a week and a complete lot-to-lot antibody comparison can be performed in a single day. The use of sensitive, quantitative technologies like the icIEF increases the robustness of the ChromiCE method compared to 2D-PAGE. We were able to detect and quantify changes in the charge variant profile of a monoclonal antibody subjected to heat-induced deamidation using ChromiCE. While it does consume significantly more protein, the approach is less laborious, making it more cost effective in the long run. We anticipate the ChromiCE approach may be developed more comprehensively for use in regulated environments,Citation22 as well as for applications beyond monoclonal antibodies, and it may be particularly beneficial for the analysis of hetero-oligomeric proteins that behave aberrantly on traditional 2D-PAGE.
Materials and Methods
Materials
All monoclonal antibodies (Chinese hamster ovary cell-derived; IgG1 and IgG4) used in this study were produced at Regeneron Pharmaceuticals, Inc. in the Protein Manufacturing and Process Development (Tarrytown, NY) or Industrial Operations (Rensselaer, NY) units. Chemical denaturants and buffer components were purchased from Sigma-Aldrich or VWR and were the highest grade available. Shodex columns and Bond-Breaker TCEP (tris(2-carboxyethyl)phosphine) solution were purchased from ThermoFisher Scientific. Capillary electrophoresis components and reagents were obtained from ProteinSimple (formerly Convergent Biosciences) with pI markers from GenWay Bio and Pharmalytes from GE Healthcare.
Size exclusion chromatography
Concentrated (50–200 g/L) mAb stock solutions were diluted into 6 M guanidine hydrochloride and 10 mM TCEP containing the same chemical buffer species (concentration and pH) as the subsequent SEC mobile phase, then incubated at ambient temperature for 30 minutes to achieve complete reduction and denaturation. Chromatography was performed on an Agilent Technologies HP Series 1100 system equipped with an Agilent Technologies 1260 Infinity micro-scale fraction collector. Two Shodex KW-802.5 columns (each 8 × 300 mm, 5 μm) connected in tandem were used for the separation with a 0.425 mL/min flow rate (achieved near max pressure rating for the columns). The SEC mobile phase included the sodium salt of the buffer (e.g., citrate, MOPS, or phosphate depending on the target pH), 8 M urea, and 1 mM TCEP. During optimization, 100 µg of protein was injected; the load amount was increased to 400–800 µg for semi-preparative separations. Typically, 3 and 2 fractions (∼150 µL each) from the apex of the HC and LC peaks, respectively, were pooled for further analysis.
Imaged capillary isoelectric focusing
Charge heterogeneity of chromatographically separated heavy and light chains was quantitatively assessed using an iCE280 Analyzer (ProteinSimple). Note: the ChromiCE method was not evaluated on a traditional cIEF platform (i.e., Beckman). Heavy and light chain samples were prepared at ∼0.2 and 0.1 mg/mL, respectively, in 8 M urea with 1% (w/v, final) methyl cellulose, 4% 3–10 Pharmalytes, and pI markers (icIEF buffer). The samples were maintained at room temperature in the autosampler until injected into the fluorocarbon-coated capillary (100 μm ID × 50 mm) and focused for 7–10 minutes at 3,000 V. Electropherograms were generated with CCD detection of the UVλ=280 nm absorbance across the entire capillary. Typically, samples were prepared in triplicate and injected once (n = 3 ).
Two-dimensional polyacrylamide gel electrophoresis
Antibody samples (3 µg) were adsorbed onto pH 3–10 immobilized pH gradient (IPG) strips (Invitrogen) during overnight incubation in IEF sample buffer (8 M urea, 2 M thiourea, 70 mM dithiothreitol (DTT), 3.0% (w/v) CHAPS, 2% (v/v) Triton X-100, 10% (v/v) glycerol, 2D-PAGE markers (Bio-Rad), and 13 µM bromophenol blue). The samples were then separated by IEF in the first dimension. The strips were then equilibrated in 2% (w/v) LDS sample buffer (Invitrogen) containing 50 mM DTT followed by 2% LDS sample buffer and 2.5% (w/v) iodoacetamide. In the second dimension, the proteins were separated according to molecular weight on 4–20% polyacrylamide gels (Invitrogen) and visualized by Coomassie blue staining.
Accelerated degradation study
A monoclonal antibody (mAb-2) was incubated at a temperature (45°C) previously shown to promote deamidation. Samples were taken at multiple time points and analyzed with the ChromiCE method previously optimized for mAb-2.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Material.pdf
Download PDF (239.3 KB)Acknowledgments
The authors gratefully acknowledge Biao Shen of Regeneron Pharmaceuticals' Analytical Chemistry group for providing extensive mass spectrometry support. We also thank Erica Pyles and Thomas Daly for their review of the manuscript and all of those in the Protein Biochemistry group for their patience and encouragement.
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413-5; PMID:22531442; http://dx.doi.org/10.4161/mabs.19931
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2003; 2:52-62; PMID:12509759; http://dx.doi.org/10.1038/nrd984
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol 2010; 10:301-16; PMID:20414204; http://dx.doi.org/10.1038/nri2761
- Declerck P. Biologicals and biosimilars: a review of the science and its implications. Generics Biosimilars Initiative J 2012; 1:13-6; http://dx.doi.org/10.5639/gabij.2012.0101.005
- Harris RJ. Processing of C-terminal lysine and arginine residues of proteins isolated from mammalian cell culture. J Chromatogr A 1995; 705:129-34; PMID:7620566; http://dx.doi.org/10.1016/0021-9673(94)01255-D
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 2011; 3:568-76; PMID:22123061; http://dx.doi.org/10.4161/mabs.3.6.17922
- Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B 2008; 870:55-62; http://dx.doi.org/10.1016/j.jchromb.2008.05.045
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426-47; PMID:17828757; http://dx.doi.org/10.1002/jps.21180
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs 2011; 3:253-63; PMID:21487244; http://dx.doi.org/10.4161/mabs.3.3.15608
- Rosati S, van den Bremer ET, Schuurman J, Parren PW, Kamerling JP, Heck AJ. In-depth qualitative and quantitative analysis of composite glycosylation profiles and other micro-heterogeneity on intact monoclonal antibodies by high-resolution native mass spectrometry using a modified Orbitrap. MAbs 2013; 5:917-24; PMID:23995615; http://dx.doi.org/10.4161/mabs.26282
- Goetze AM, Schenauer MR, Flynn GC. Assessing monoclonal antibody product quality attribute criticality through clinical studies. MAbs 2010; 2:500-7; PMID:20671426; http://dx.doi.org/10.4161/mabs.2.5.12897
- Kinoshita M, Nakatsuji Y, Suzuki S, Hayakawa T, Kakehi K. Quality assurance of monoclonal antibody pharmaceuticals based on their charge variants using microchip isoelectric focusing method. J Chromatogr A 2013; 1309:76-83; PMID:23958695; http://dx.doi.org/10.1016/j.chroma.2013.08.021
- Khawli L, Goswami S, Hutchinson R, Kwong Z, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs 2010; 2:613-24; PMID:20818176; http://dx.doi.org/10.4161/mabs.2.6.13333
- Flatman S, Alam I, Gerard J, Mussa N. Process analytics for purification of monoclonal antibodies. J Chromatogr B 2007; 848:79-87; http://dx.doi.org/10.1016/j.jchromb.2006.11.018
- Michels DA, Salas-Solano O, Felten C. Imaged capillary isoelectric focusing for charge-variant analysis of biopharmaceuticals. Bio Process Int 2011; 9:48-54
- Thurston CF, Henley LF. Isoelectric focusing under denaturing conditions. In: Walker JM, editor. New Protein Techniques. New York: Springer; 1988. p. 257-67.
- Fekete S, Beck A, Veuthey JL, Guillarme D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J Pharm Biomed Anal 2015; 113:43-55
- Rea JC, Moreno GT, Lou Y, Farnan D. Validation of a pH gradient-based ion-exchange chromatography method for high-resolution monoclonal antibody charge variant separations. J Pharm Biomed Anal 2011; 54:317-23; PMID:20884149; http://dx.doi.org/10.1016/j.jpba.2010.08.030
- Alvarez M, Tremintin G, Wang J, Eng M, Kao Y-H, Jeong J, Ling VT, Borisov OV. On-line characterization of monoclonal antibody variants by liquid chromatography–mass spectrometry operating in a two-dimensional format. Anal Biochem 2011; 419:17-25; PMID:21867674; http://dx.doi.org/10.1016/j.ab.2011.07.033
- Balaguer E, Demelbauer U, Pelzing M, Sanz-Nebot V, Barbosa J, Neusüß C. Glycoform characterization of erythropoietin combining glycan and intact protein analysis by capillary electrophoresis–electrospray–time-of-flight mass spectrometry. Electrophoresis 2006; 27:2638-50; PMID:16817164; http://dx.doi.org/10.1002/elps.200600075
- Dick LW, Mahon D, Qiu D, Cheng K-C. Peptide mapping of therapeutic monoclonal antibodies: Improvements for increased speed and fewer artifacts. J Chromatogr B 2009; 877:230-6; http://dx.doi.org/10.1016/j.jchromb.2008.12.009
- Mao Q, Pawliszyn J. Capillary isoelectric focusing with whole column imaging detection for analysis of proteins and peptides. J Biochem Biophys Methods 1999; 39:93-110; PMID:10344503; http://dx.doi.org/10.1016/S0165-022X(99)00006-8
- Gritti F, Guiochon G. The van Deemter equation: Assumptions, limits, and adjustment to modern high performance liquid chromatography. J Chromatogr A 2013; 1302:1-13; PMID:23838304; http://dx.doi.org/10.1016/j.chroma.2013.06.032
- Arakawa T, Ejima D, Li T, Philo JS. The critical role of mobile phase composition in size exclusion chromatography of protein pharmaceuticals. J Pharm Sci 2010; 99:1674-92; PMID:19894271
- Mao Q, Pawliszyn J. Effect of salt concentration on separation patterns in static capillary isoelectric focusing with imaging detection. J Chromatogr B, Biomed Sci Appl 1999; 729:355-9; PMID:10410962; http://dx.doi.org/10.1016/S0378-4347(99)00129-2
- Du Y, Walsh A, Ehrick R, Xu W, May K, Liu H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs 2012; 4:578-85; PMID:22820257; http://dx.doi.org/10.4161/mabs.21328
- Tyler-Cross R, Schirch V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J Biol Chem 1991; 266:22549-56; PMID:1939272
- GoTo Y, Calciano LJ, Fink AL. Acid-induced folding of proteins. Proc Natl Acad Sci 1990; 87:573-7; http://dx.doi.org/10.1073/pnas.87.2.573
- Liu H, Gaza-Bulseco G, Chumsae C. Analysis of reduced monoclonal antibodies using size exclusion chromatography coupled with mass spectrometry. J Am Soc Mass Spectrom 2009; 20:2258-64; PMID:19786356; http://dx.doi.org/10.1016/j.jasms.2009.08.015
- Kopaciewicz W, Regnier F. Nonideal size-exclusion chromatography of proteins: effects of pH at low ionic strength. Anal Biochem 1982; 126:8-16; PMID:7181119; http://dx.doi.org/10.1016/0003-2697(82)90102-6
- Golovchenko NP, Kataeva IA, Akimenko VK. Analysis of pH-dependent protein interactions with gel filtration medium. J Chromatogr 1992; 591:121-8; http://dx.doi.org/10.1016/0021-9673(92)80229-N
- Nick Pace C, Huyghues-Despointes BM, Fu H, Takano K, Scholtz JM, Grimsley GR. Urea denatured state ensembles contain extensive secondary structure that is increased in hydrophobic proteins. Protein Sci 2010; 19:929-43; PMID:20198681; http://dx.doi.org/10.1002/pro.370
- Fuhrer JP. Rapid analysis of membrane glycopeptides by gel permeation high-performance liquid chromatography. J Chromatogr 1990; 528:65-77; PMID:2384566; http://dx.doi.org/10.1016/S0378-4347(00)82363-4
- Cao J, Sun W, Gong F, Liu W. Charge profiling and stability testing of biosimilar by capillary isoelectric focusing. Electrophoresis 2014; 35:1461-8; PMID:24610636; http://dx.doi.org/10.1002/elps.201300471