
Abstract
Histone deacetylases (HDACs) are modification enzymes that regulate a plethora of biological processes. HDAC1, a crucial epigenetic modifier, is deregulated in cancer and subjected to a variety of post-translational modifications. Here, we describe the generation of a new monoclonal antibody that specifically recognizes a novel highly dynamic prophase phosphorylation of serine 406-HDAC1, providing a powerful tool for detecting early mitotic cells.
Histone deacetylases (HDACs) catalyze the removal of acetyl moieties from ϵ-NH3 groups of lysines of target substrates, including both histone and non-histone proteins.Citation1,2 In humans, Class I HDACs (HDAC1-3 and HDAC8) are ubiquitously expressedCitation3 and regulate cell cycle progression, cellular proliferation and differentiation during development. In particular, knockout of HDAC1 in mice causes dramatic impairment in cellular proliferation, and HDAC1-deficient mice die at embryonic day 9.5 of development.Citation4 Different human cancer cell lines depleted of HDAC1 are strongly perturbed in their cell cycle, and undergo an increase of apoptosis and loss of mitotic cells.Citation5 HDAC1 and HDAC2 bind to the promoters of the cell cycle regulators p21WAF/CIP1 and p57Kip2 to modulate their expression and regulate G1/S transition.Citation6,7 More obscure is the role of HDAC1 at the G2/M transition. Inhibition of HDAC enzymatic activities by trichostatin A (TSA) or depletion of HDAC1/HDAC2 affect the G2/MCitation5 progression, but deeper knowledge about the molecular mechanisms is still missing.
HDAC1 is modified by a plethora of post-translational modifications (PTMs) (reviewed in ref. 8). For example, casein kinase II (CKII) phosphorylation of HDAC1 stabilizes HDAC1 interaction with binding partners in multiprotein complexes, such as RbAP48, Sin3a and MTA-2.Citation9 To contribute in deciphering the PTM code of HDAC1 during cell cycle progression, we recently identified a new mitotic, Aurora kinase-dependent phosphorylation of serine 406-HDAC1 (unpublished data). Our goal in this study was to generate a highly specific monoclonal antibody that recognizes solely this modified form of HDAC1. Since this phosphorylation is highly dynamic and it is restricted to a specific temporal window from mitotic prophase to metaphase, this antibody is a valuable read-out for early mitotic cells.
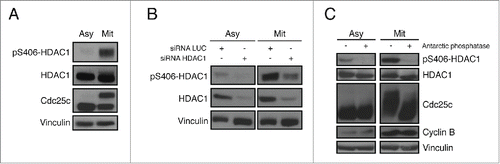
The HDAC1 phosphopeptide 397-Acetyl-DEDDPDKRIpSISSSDKRIA-[C] was used as the immunogen. In vitro purified HDAC1 was subjected to an in vitro kinase assays as described in Materials and Methods. The product of the reaction was analyzed by TiO2-enriched mass spectrometry. The singly Aurora kinase-dependent phosphorylated peptide RISICSSDK from HDAC1 was identified from both MS2 () and MS3 () spectra. The most dominant peak in the MS2 spectrum corresponding to the neutral loss of one phosphoric acid from the peptide molecular ion was selected for MS3. To assess the validity of our antibody, HeLa cells were first synchronized in mitosis by nocodazole treatment, and, upon western blot analysis, a clear signal appeared at the expected molecular weight only in mitotic cells. As anticipated, since in mitosis phosphorylated and hyperphosphorylated isoforms of HDAC1 are present,Citation10-12 the pS406-HDAC1 antibody (clone BT-15) recognizes both modified bands (). Upon depletion of endogenous HDAC1 by RNAi, the signal with the BT-15 antibody also decreased in the interfered samples, as did the signal of total HDAC1 both in asynchronous and in mitosis (). To further assess the specificity of our antibody for this phosphorylated form of HDAC1, cellular extracts of asynchronous and mitotic HeLa cells were treated with Antarctic phosphatase, which dephosphorylated total HDAC1 and consequently induced the complete loss of the BT-15 signal ().
Figure 1. MS Spectrum and Full Annotation of HDAC1 phospho-S(406). TiO2-enriched mass spectrometry of HDAC1. In vitro purified HDAC1 was previously subjected to an in vitro kinase assays as described in Materials and Methods. (A) MS2 and (B) MS3 spectra.
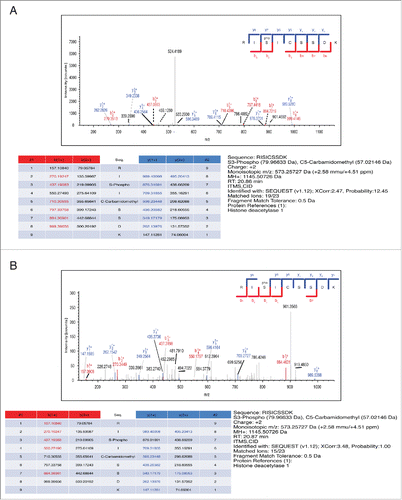
Figure 2. Characterization of the pS406-HDAC1 monoclonal antibody BT-15. (A) Mitotic synchronization of HeLa cells by nocodazole treatment. Samples were analyzed by protein gel blot with the indicated antibodies. Cdc25c is used as mitotic marker, vinculin as loading control. (B) RNA interference of HDAC1 in asynchronous and mitotic HeLa cells. Samples were analyzed by western blot using the indicated antibodies. Vinculin is used as loading control. (C) Antarctic phosphatase assay on asynchronous and mitotic HeLa cells. Samples were analyzed by protein gel blot with the indicated antibodies. Cdc25c was used as positive control for Antarctic phosphatase, Cyclin B as mitotic marker, Vinculin as loading control.
Confocal immunofluorescence analysis showed the behavior of the subpopulation of pS406-HDAC1 in all the different phases of mitotic progression. HeLa cells were plated on poly-D-lysine-coated coverslips and stained with BT-15 antibody, total HDAC1 antibody and DAPI for the DNA (). Surprisingly, we observed that the pattern of phosphorylation of HDAC1 on serine 406 was not constant during the different phases of mitosis: the peak of phosphorylated HDAC1 occurs in prophase, then it decreases in prometaphase. Mitotic MEF cells knockout for HDAC1Citation7 and stably expressing human HDAC1 wild type, S406A or S406E mutants were tested by protein gel blot with the BT-15 antibody. As expected, HDAC1 wild type was recognized, but not the un-phosphorylatable S406A mutant. On the contrary, the S406E was recognized by the BT-15 antibody with the same efficiency as the wild type (). This is consistent with the notion that the glutamate mimics the structure and charge of a phosphorylated serine, and is thus recognized by the BT-15 antibody as an appropriate epitope. We then evaluated the BT-15 antibody's efficiency and specificity in immunoprecipitating only the mitotic phosphorylated HDAC1. As shown in , when incubating both asynchronous and mitotic HeLa cellular extracts with different amounts of the antibody, HDAC1 protein was immunoprecipitated only in mitotic samples, where HDAC1 is heavily phosphorylated on serine 406 in comparison to asynchronous samples.
Figure 3. Specificity and potential applications of the pS406-HDAC1 monoclonal antibody BT-15. (A) Immunofluorescence images of HeLa cells synchronized by double thymidine block and fixed with 4% paraformaldehyde after 9.5 hours. Cover slips were stained with the indicated antibodies and DAPI and analyzed at confocal microscopy. Subphases of mitosis were identified by the condensation status of the chromatin. Asterisks indicate the cell of interest in every field. Scale bar corresponds to 5 µm. (B) Mitotic synchronization of immortalized HDAC1−/− MEF stably expressing HDAC1 wt, phospho null HDAC1 S406A or phospho mimetic HDAC1 S406E mutants. Samples were analyzed by western blot with the indicated antibodies. GAPDH is used as loading control. (C) Immunoprecipitations of pS406-HDAC1 in asynchronous and mitotic HeLa cells. The indicated quantities of BT-15 supernatant and 0.5 mg of protein lysates were used. Asterisks indicate the pS406-HDAC1 form, arrowhead the basal HDAC1. (D) Immunoprecipitations of HDAC2 in asynchronous and mitotic HeLa cells. IP against HDAC2 was performed and analyzed by protein gel blot with total HDAC2 antibody (right panel, to show that the IP properly worked) and the BT15 antibody (left panel). Asterisks indicate the IgG, arrowhead the HDAC2 protein. HA-tag was used as internal control of the IP.
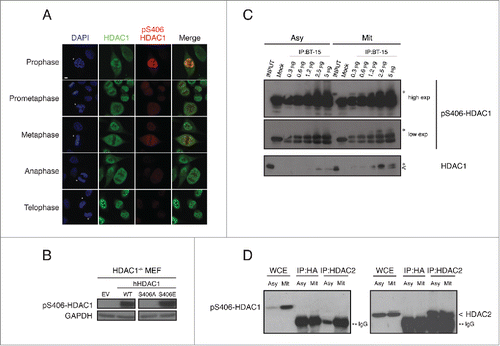
Since S421/423 of HDAC1 are constitutively phosphorylated by casein kinase II (CKII) to regulate its activity,Citation9 we wanted to rule-out a potential cross-recognition of the BT-15 antibody with the CKII phosphorylation sites. Upon synchronization, cellular extracts were collected at the indicated time points and tested with the antibody specific for the CKII phosphorylation (pS421/423 HDAC1) and the BT-15 antibody. Supplementary Figure 1 represents the time-course trend of the 2 phosphorylation events during cell cycle: HDAC1 is constitutively phosphorylated by CKII at serine 421/423, whereas BT-15 antibody specifically recognizes the mitotic phosphorylation of HDAC1. Because of similarities in the protein sequence of HDAC1 and HDAC2, we wanted to exclude the possibility of cross-recognition of the BT-15 antibody with HDAC2. Upon immunoprecipitating both mitotic and asynchronous cellular extracts with HDAC2, we could not detect a signal with the BT-15 antibody, indicating that it is specific for HDAC1 and does not cross-react with HDAC2 ().
From data described here, we conclude that the BT-15 monoclonal antibody is specific for pS406-HDAC1 and represents a powerful tool to detect and study prophase, including the prophase phosphorylation of HDAC1 in cells. Prophase, a critical phase in which cells prepare for mitosis, marks the onset of events fundamental for cell division such as the nuclear envelope breakdown, spindle microtubules attachment to chromosomes and spindle assembly checkpoint (SAC) activation.Citation13,14 Since treatment with spindle poisons that activate the SAC arrest cells in prophase, the BT-15 antibody could be a helpful tool to detect them more easily. Importantly, in prophase cohesins are removed from the arms of condensing chromosomes,Citation15 an essential event for the separation of sister chromatids. In conclusion, aside from marking HDAC1-related mitotic-specific events, this antibody may serve as a crucial research tool to specifically identify mammalian cells in prophase.
Materials and Methods
In vitro protein purification and Aurora kinase assay
Recombinant GST-fusion proteins were produced in BL-21 E.coli strain and purified with Glutathione® beads (Sigma). Proteins were eluted using reduced glutathione and the GST tag was removed through a proteolytic cleavage with prescission protease (produced as recombinant protein at our Campus facilities). After SDS-PAGE and staining with Coomassie brilliant blue, quantification of purified recombinant protein was performed by comparison of the band with bands corresponding to known amounts of bovine serum albumin protein.
Aurora kinase assays were performed as previously described.Citation16 Briefly, 1 µg of purified substrate (HDAC1, HDAC3 or histone H3) were incubated with 50 ng of recombinant-purified Aurora A/TPX2 or Aurora B/INCENP kinase in the presence of 5 µCi of [32P]-labeled ATP at 30°C for 1 hour and analyzed by SDS-PAGE followed by autoradiography. Histone H3 was used as a positive control for Aurora kinases activity;Citation17 HDAC3 was the negative control because it is not phosphorylated in mitosis and does not have a putative consensus site for Aurora-mediated phosphorylation.
Mass spectrometry: In-Gel Digestion, Phosphopeptide Enrichment and LCMS/MS
Wild type HDAC1 was phosphorylated in vitro by Aurora kinases as described above. The mixture of proteins was divided into 2 portions. Half was separated by one-dimensional SDS-polyacrylamide gel electrophoresis, 10% gel, 1 mm thickness. The gel was stained with SimplyBlue SafeStain. Bands of interest were cut and trypsinized. The in-solution portion of the protein mixture was also trypsinized. Peptides resulting from both in-gel and in-solution digestion were enriched for phosphopeptides using a modified procedure previously described.Citation18 1 M glycolic acid was used as loading buffer and the self-packed TiO2 tips were conditioned with 10 μL of loading buffer. Phosphopeptide eluant volume was reduced (speedvac) to 1 μL and acidified by adding 1% trifluoroacetic acid (TFA) to reach the final volume of 7 μL for LC-MS loading. The TiO2-unbound peptides were desalted, lyophilized and re-suspended in 0.1% TFA, then injected into the LC-MS. Nano-LC-MS analysis was carried out (Agilent 1100) on-line with a Fourier transformed-LTQ mass spectrometer (Thermo Electron, San Jose, CA). Peptides were loaded, purified and concentrated on a C18 precolumn (Applied Biosystems), and separation performed on an in-house C18 analytical column (10.5 cm x100 μm ID fused-silica emitter, New Objective, Inc.., Woburn, MA USA). The LC linear gradient was as follows: 100% solvent A (5% acetonitrile, 0.1% formic acid) to 20% solvent B (acetonitrile, 0.1% formic acid) over 20 min and from 20% to 80% solvent B in 5 min at a constant flow rate of 0.3 μL/min. Data acquisition mode was set as follow: for protein identification, data acquisition mode was set to obtain one MS scan across m/z 400 to 2 000, followed by 5 MS/MS (MS2) scans of the 5 most intense ions of each MS scan. MS spectra were limited to one scan per precursor ion followed by 180 sec. of exclusion. For phosphorylation site determination, data acquisition mode was set to obtain one MS scan followed by 3 MS2 scans of the most intense ions in each MS scan. MS2 spectra were limited to one scan per precursor ion followed by 180 seconds of exclusion. Data-dependent settings were chosen to trigger an MS3 scan when a neutral loss of 98, 49 or 32.7 Da was detected for the 3 most intense fragment ions. Former target ions selected for MS2 were dynamically excluded for 180 sec. Database search was performed using MASCOT (www.matrixscience.com) and Sequest in Proteome Discoverer 1.1 (1.1.0.263 Thermo Fisher Scientific Inc..) with the following search parameters: database: NCBInr, taxonomy: human; enzyme: trypsin; missing cleavages: 2; fixed modification: carbamidomethyl cysteine; variable modification: oxidation methionine, phosphorylation serine, threonine or tyrosine. Parent tolerance set to 10 ppm, MS2 tolerance: 0.5 Da. All Mascot and Sequest phosphorylation spectra were manually validated.
Immunization of the animals
Peptide pS406-HDAC1 (produced in vitro by Cambridge Research Biochemicals (Billingham, UK), conjugated to the carrier protein keyhole limpet hemocyanin through the terminal cysteine residue was used for immunization. Four SJL mice were immunized intraperitoneal (IP) with 25 μg of antigen every 2 weeks. The first immunization was performed with complete Freund's adjuvant, and this was followed by 3 injections in incomplete Freund's adjuvant. Ten days after the fourth immunization, the animals were tail bled, and the antibody response was measured by ELISA using both the unphosphorylated (DEDDPDKRISICSSDKRIA) and phosphorylated (DEDDPDKRI(pS)ICSSDKRIA) peptides as baits.
Generation of BT-15 stable clone
The mouse selected for generation of monoclonal antibodies was boosted by both IP and intravenous administration of antigen in saline. Five days later the spleen was harvested and used for hybridoma production. Splenocytes were fused to SP2/0-Ag14 myeloma cells using polyethyleneglycol 1500 (Roche) as previously described.Citation19 Fused cells were initially seeded in 96-well tissue culture plates, and clones that tested positive for the production of anti-pS406-HDAC1 antibodies were subsequently subcloned by limiting dilution. One of them, clone BT-15, was then subjected to a variety of tests to assess its specificity and optimal working conditions in different techniques as described in legends.
Cell cultures, plasmids and site directed-mutagenesis
HeLa cells were maintained in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (Lonza), 2 mM L-glutamine (Lonza) and antibiotics (250 μg/ml penicillin and 25 μg/ml streptomycin). MEFs HDAC1−/− were maintained in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (Lonza), 2 mM L-glutamine (Lonza), antibiotics (250 μg/ml penicillin and 25 μg/ml streptomycin) and non-essential amino acids. All cells were cultured in a humidified 37°C incubator with 5% CO2.
For expression in cells, the pBabe-hygro (empty or encoding HDAC1) plasmids were used. Point mutant HDAC1 proteins were generated by PCR site-directed mutagenesis of pBabe-hygro-HDAC1 wild type using primers carrying the desired mutations (Supplementary Table 1).
Cell synchronization
Cells were synchronized at the G1/S boundary by double thymidine block: cells were treated with 2 mM thymidine for 16 hours washed and released in fresh medium for 9 hours, blocked again in 2mM thymidine containing media and collected 16 hours later. G1 populations were obtained from mitotic cells released from nocodazole block for 5 hours in fresh medium. If modifications were introduced, they are indicated in the corresponding experiment. To synchronize cells in mitosis, 330 nM nocodazole was added for 16 hours and mitotic rounded-up cells were harvested by shaking them off. The G2-enriched population is the one that remains attached to the plate after nocodazole treatment and mitotic shake-off.
Drug treatments, retroviral infection and RNA interference
Nocodazole (Sigma, #M1404) and thymidine (Sigma, #T1895), were used at the concentrations and time indicated in each experiment.
Packaging Phoenix cells were transfected with retroviral constructs using calcium phosphate protocol. The medium containing viral particles was collected and added to the target cells for 6 hours in the presence of 5 μg/ml polybrene. The day after, cells were subjected to another cycle of infection.
siRNA oligos for RNA interference were purchased from MGW Operon. RNA interference was performed according to the oligofectamine protocol (Invitrogen®). The following siRNA were used:
Luciferase (Luc): 5′-CGUACGCGGAAUACUUCGATT-3′
HDAC1: 5′-CUAAUGAGCUUCCAUACAATT-3′
In vitro phosphatase assays
Cells were lysed in JLB buffer (50 mM Tris pH 8, 150 mM NaCl, 10% glycerol, 0.5% Triton X-100, PMSF, aprotinin and leupeptin) and indicated units of Antarctic phosphatase (New England Biolabs) were added to 50 µg of protein lysates. The reaction was carried on at 30°C for 1 hour.
Immunoprecipitation and protein analysis
For Western blot analysis cells were lysed in denaturing SDS lysis buffer [one volume of Buffer I (5% SDS, 150 mM Tris-HCl pH 6.8, 30% glycerol) + 3 volumes of Buffer II (25 mM Tris-HCl pH 8.3, 50 mM NaCl, 0.5% NP-40, 0.5% deoxycholate, 0.1% SDS) + aprotinin and leupeptin, sodium fluoride, sodium pyrophosphate and sodium orthovanadate].
For immunoprecipitation, non-denaturing E1A lysis buffer (50 mM HEPES pH 7.5, 250 mM NaCl, 0.1% NP-40, PMSF, aprotinin and leupeptin, sodium fluoride, sodium pyrophosphate and sodium orthovanadate) was used.
The following antibodies were used. For western blot: HDAC1 mouse, clone 10E2 (produced in-house by Christian Seiser), HDAC1 rabbit (Abcam, #ab7028), HDAC3 rabbit (Abcam, #ab7030), vinculin mouse (Santa Cruz), phospho-serine 10 histone H3 rabbit (Upstate, #06–570), Cdc25c rabbit (Santa Cruz, #sc327), GAPDH mouse (Abcam), phospho-serine 406 HDAC1 mouse, clone BT-15 (final concentration 3.6 µg/ml, produced at the IFOM-IEO-Campus and described in this report), phospho-serines 421/423-HDAC1 rabbit (Millipore, #07–1575). For immunoprecipitation: Flag-M2 Agarose® (Sigma, #A2220).
Immunofluorescence
For indirect immunofluorescence analysis, cells were plated on coverslips coated with poly-D-lysine and fixed with paraformaldehyde 4%. The following primary antibodies were used: HDAC1 (Abcam), phospho-serine 406 HDAC1 (final concentration 11 µg/ml, clone BT-15, produced at the IFOM-IEO-Campus). The following secondary antibodies were used: Cy3-conjugated goat anti-mouse (Jackson Laboratories), FITC-conjugated goat anti-rabbit. DAPI was used to stain DNA. Images were taken with an Olympus BX61 upright widefield microscope equipped with a Photometrics CoolSNAP EZ camera and analyzed with Metamorph software and with a confocal laser microscopy Leica TCS SP2.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Serge et al Supplementary Data
Download PDF (169 KB)Acknowledgments
We are very grateful to all lab members. We thank Claudia Miccolo for precious technical help. We thank all the support facilities at IEO and IFOM, in particular Andrea di Fonzo for the mass spectrometric analysis, Marisa Aliprandi for help in antibody production, Amanda Oldani and Sara Barozzi for technical support with imaging techniques. Most importantly, we thank Prof. Andrea Musacchio for his crucial help. This work was supported by the Associazione Italiana per la Ricerca sul Cancro (IG5732 and IG12075 to S.C.) and the Italian Ministry of Health (to S.C.). C.V.S. was supported by a Fondazione Umberto Veronesi fellowship. St.Sa. was supported by fellowships from the Italian Foundation for Cancer Research. C.S. was supported by the Austrian Science fund (FWF P25807)
References
- Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007; 26:5310-8; PMID:17694074; http://dx.doi.org/10.1038/sj.onc.1210599
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008; 9:206-18; PMID:18292778; http://dx.doi.org/10.1038/nrm2346
- Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett 2009; 277:8-21; PMID:18824292; http://dx.doi.org/10.1016/j.canlet.2008.08.016
- Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. Embo J 2002; 21:2672-81; PMID:12032080; http://dx.doi.org/10.1093/emboj/21.11.2672
- Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser C, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol 2007; 27:4784-95; PMID:17470557; http://dx.doi.org/10.1128/MCB.00494-07
- Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev 2010; 24:455-69; PMID:20194438; http://dx.doi.org/10.1101/gad.552310
- Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G, Lagger S, et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol 2010; 30:1171-81; PMID:20028735; http://dx.doi.org/10.1128/MCB.01500-09
- Segre CV, Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol 2011; 2011:690848; PMID:21197454; http://dx.doi.org/10.1155/2011/690848
- Pflum MK, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem 2001; 276:47733-41; PMID:11602581; http://dx.doi.org/10.1074/jbc.M105590200
- Cai R, Kwon P, Yan-Neale Y, Sambuccetti L, Fischer D, Cohen D. Mammalian histone deacetylase 1 protein is posttranslationally modified by phosphorylation. Biochem Biophys Res Commun 2001; 283:445-53; PMID:11327722; http://dx.doi.org/10.1006/bbrc.2001.4786
- Khan DH, He S, Yu J, Winter S, Cao W, Seiser C, Davie JR. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J Biol Chem 2013; 288:16518-28; PMID:23612983; http://dx.doi.org/10.1074/jbc.M112.440446
- He S, Khan DH, Winter S, Seiser C, Davie JR. Dynamic distribution of HDAC1 and HDAC2 during mitosis: association with F-actin. J Cell Physiol 2013; 228:1525-35; PMID:23280436; http://dx.doi.org/10.1002/jcp.24311
- Guttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 2009; 10:178-91; PMID:19234477; http://dx.doi.org/10.1038/nrm2641
- Stukenberg PT, Burke DJ. Connecting the microtubule attachment status of each kinetochore to cell cycle arrest through the spindle assembly checkpoint. Chromosoma 2015; http://dx.doi.org/10.1007/s00412-015-0515-z
- Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000; 103:399-410; PMID:11081627; http://dx.doi.org/10.1016/S0092-8674(00)00132-X
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 2010; 190:73-87; PMID:20624901; http://dx.doi.org/10.1083/jcb.201001036
- Zeitlin SG, Shelby RD, Sullivan KF. CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J Cell Biol 2001; 155:1147-57; PMID:11756469; http://dx.doi.org/10.1083/jcb.200108125
- Thingholm TE, Jorgensen TJ, Jensen ON, Larsen MR. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nature protocols 2006; 1:1929-35; PMID:17487178; http://dx.doi.org/10.1038/nprot.2006.185
- Harlow EaL D. Antibodies: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1988.