
abstract
Recombinant antibody single-chain variable fragments (scFv) are difficult to purify homogeneously from a protein complex mixture. The most effective, specific and fastest method of purification is an affinity chromatography on Protein L (PpL) matrix. This protein is a multi-domain bacterial surface protein that is able to interact with conformational patterns on kappa light chains. It mainly recognizes amino acid residues located at the VL FR1 and some residues in the variable and constant (CL) domain. Not all kappa chains are recognized, however, and the lack of CL can reduce the interaction. From a scFv composed of IGKV10-94 according to IMGT®, it is possible, with several mutations, to transfer the motif from the IGKV12-46 naturally recognized by the PpL, and, with the single mutation T8P, to confer PpL recognition with a higher affinity. A second mutation S24R greatly improves the affinity, in particular by modifying the dissociation rate (kd). The equilibrium dissociation constant (KD) was measured at 7.2 10-11 M by surface plasmon resonance. It was possible to confer PpL recognition to all kappa chains. This protein interaction can be modulated according to the characteristics of scFv (e.g., stability) and their use with conjugated PpL. This work could be extrapolated to recombinant monoclonal antibodies, and offers an alternative for protein A purification and detection.
Abbreviations
| scFv | = | single-chain variable fragment |
| PpL | = | Peptostreptococcus Protein L |
| VL | = | light variable domain |
| IGKV | = | Immunoglobulin Kappa Variable |
| FR | = | Framework |
| CL | = | light constant domain |
| HRP | = | horseradish peroxidase |
| SPR | = | surface plasmon resonance |
| SDS–PAGE | = | sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
Introduction
Downstream process optimization is a crucial step in the recovery of highly purified recombinant proteins from flask cultures or fermentation broth. Many purification processes have been developed, of which most include an affinity chromatography step. One of the most popular, Ni-NTA agarose-gel affinity chromatography is widely used to isolate His tagged proteins from cleared cell lysates. However, these methods seldom yield pure products, and contaminants containing multiple histidine residues are often co-purified.Citation1,2
The purification of recombinant antibodies is greatly facilitated by the use of specific ligands of bacterial origin, such as Protein A from Staphylococcus aureus or Protein G from streptococci. These proteins are able to interact with specific structural motifs common to constant heavy chain domains of many antibody isotypes and Protein A is now considered the most popular ligand for affinity purification of therapeutic antibody molecules. An alternative to Proteins A and G is Protein L (PpL) from Peptostreptococcus magnus, which is able to bind to some kappa light chain variable domains without interfering with the antigen-binding site.Citation3,4 This characteristic has the great advantage of allowing purification not only of IgGs, but also antibody fragments, such as single-chain variable fragments (scFv), which are devoid of constant domains. However, PpL is only effective in binding certain subtypes of kappa light chains.Citation5 For example, it binds human VκI, VκIII and VκIV subtypes, but does not interact with VκII subtype. Binding of mouse immunoglobulin is restricted to those having VκI light chains. PpL is thus not a versatile antibody-binding protein tool, and many scFv fragments still require the use of a tag for purification, which has associated drawbacks. Nevertheless, PpL matrices have been developed for the capture and purification of antibody fragments using easy, fast, cost-effective and very specific protocols.Citation6-8 PpL enables pure and homogeneous antibody fragments to be obtained from very heterogeneous samples, even when these antibody molecules are present in limited (trace) amounts. In addition, PpL-affinity purification prevents the use of potentially immunogenic tags. PpL is now produced in recombinant form and marketed as a high affinity matrix with improved flow properties for purification, manufacturing and even industrial applications. In addition PpL conjugates have been engineered in order to facilitate detection of antibody fragments, either free or in complex with their antigen, allowing detection and titration in a wide range of assays.
Commercially available PpL is a 35 kDa protein with homogeneous domains that show an affinity for kappa light chains that is fully comparable with the wild-type PpL.Citation9-11 To elucidate PpL-binding, interaction studies between Fab 2A2 (Homo sapiens IGKV1-39*01 or VκI subtype) and PpL domain C* (PDB code 1HEZ) have been performed.Citation12 For the first time, the existence of 2 interaction interfaces in the protein was shown. The first interface is located on strands β1, β2 and the α helix. The second interface is between the α helix and strand β3. Thirteen residues of the antibody light chain are involved in the first VL-PpL interface. All are located in the framework region 1 (FR1) with the exception of 2 residues (K127 and E143). In the second VL-PpL interface, 15 residues on the antibody VL domain are involved. Ten of them are common with the first interface and are located mainly on the β strands A and B. The remainder being located on strands D and E. By using mutants to alter the first or second interfaces, it was shown that the interface 1 had a stronger affinity than interface 2. Conversely, Housden et al.Citation13 established that the KD of sites 1 and 2 were 37.5 nM and 4.6 nM, respectively, and demonstrated that the relative affinities of each site for κ –chains may depend upon the subclass and origin of the κ –chain. This dual interaction has also been shown with murine Fab (IMGT®: IGKV 10-96*01) of the monoclonal antibody anti-SAG1 4F11E12.Citation14
All of these studies were performed with whole immunoglobulin or Fab fragments naturally recognized by PpL. However, when the antibody is in the minimal antigen-binding format (scFv) and lacks constant domains, its ability to be recognized by PpL can be lost, as is the case for 4F11E12 antibody (unpublished data).Citation14 In addition, some naturally variable light chains are not recognized by PpL. To overcome this difficulty, Muzard et al.Citation15 suggested a versatile method to confer PpL binding activity on any antibody. This method consists of substituting the VL-kappa FR1 domain of the antibody with V-Kappa FR1 of a murine antibody (9C2; IGKV12-46*01) that naturally binds PpL with high affinity. This approach was validated on various VLs, including IGKV1-110*01, IGKV1-117*01, IGKV3-10*01.
The aim of this study was to confer PpL binding ability to any scFv or antibody fragment consisting of a murine variable light chain IGKV10-94*01, IGKV10-95*01 or IGKV10-96*01 (VκV subtype) that differs from all other murine or human VLs at one position: threonine 8 (IMGT®, the international ImMunoGeneTics information system®, www.imgt.org).Citation16,17 The FR1 of these 3 subfamilies share the same sequence, except at position 24 (Ser or Arg). This study should contribute to the identification of important residues on antibody VL domains involved in the interaction between scFv and PpL, and thus minimize the changes required to confer PpL binding activity.
Results
A scFv with VL IGKV10-94*01, referred to here as scFv 10-94, was selected because of its high-yield production in prokaryotic systems. After induction of bacteria transformed with pSW1-scFv10-94, periplasmic proteins were extracted and submitted to sodium dodecyl sulfate–PAGE (SDS-PAGE) and Western blot analysis (using an anti-flag antibody) to confirm the scFv production (data not shown). As scFv could neither be detected by PpL-HRP conjugate (ELISA) nor by PpL-agarose affinity chromatography (data not shown), we concluded that wild type scFv10-94 does not interact with PpL.
Mutations in the IGKV domain of scFv 10-94
The first strategy to transfer PpL recognition to scFv 10-94 used the technique described by Muzard et al.Citation15 We created scFv10-94m by substituting FR1 of IGKV10-94 with the FR1 of IGKV 12-46, which contains the PpL recognition site (A). SDS-PAGE and Western blot showed that this mutation does not affect the yield of production; however, scFv 10-94m did not exhibit significant interaction with PpL. The PpL-agarose column flow-through fraction represents the entire production, and the scFv 10-94m was hardly detected in the purified fraction (B). Muzard et al.Citation15 showed that E17Q mutation had a beneficial effect on the interaction with PpL, and therefore scFv 10-94mQ (mutation E17Q) was produced, purified and analyzed as previously described. But, when affinity chromatography using PpL-agarose was performed, a significant quantity of scFv was still present in the flow-through fraction (A and B). An additional mutation was therefore introduced into the FR3 (T90K) as found in scFv 12-46 (A). This restored PpL binding activity onto scFv 10-94 mQK and this allowed efficient purification from the periplasmic extract (B).
Figure 1. Design, production and purification of the scFv mutants and functional analysis of the interaction with the PpL by ELISA. (A) Mutations performed on the IGKV12-46 and IGKV10-94 light variable domain. (B) SDS-PAGE stained with Coomassie brilliant blue and Western blot revealed using the anti-flag monoclonal antibody MRC-OX74. Lane M : molecular mass standards (315, 250, 180, 140, 95, 72, 55, 42, 26 17, 10 and 4.6 kDa); lane 1: periplasmic fraction of induced bacteria loaded onto PpL-agarose; lane 2: PpL-agarose column flow-through fraction; lane 3: PpL-agarose column eluted fraction. (C) scFvs were directly immobilized on the plates. The calibrated scFv preparations were then detected using PpL–HRP conjugate.
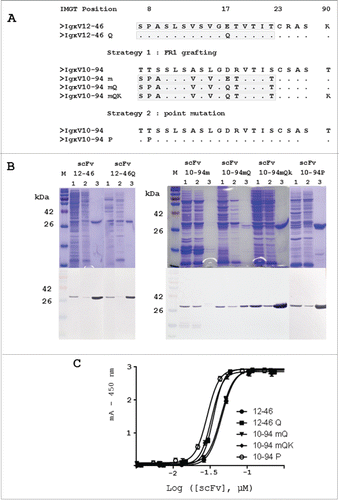
In the second approach, the single mutation T8P was made in FR1 of scFv 10-94 (A). Similarly to scFv 10-94 mQK, a large proportion of scFv10-94 P was purified from the bacterial periplasmic extract (B), but the elution of the scFv from the PpL-agarose column required a more acidic pH (2.0 and not 3.0).
The purification efficiency is less than 5% and 15% for the scFv 10-94m and scFv 10-94mQ respectively. For other scFv, it is of the order of 90% under the experimental conditions reported here. These results are in agreement with the purification yield: 0.5 to 1 mg/L of culture for the scFv 12-46 and scFv 12-46Q; 1 to 2 mg/L of culture for the scFv 10-94mQK and scFv 10-94P; 75 μg/L and 200 μg/L of culture for the scFv 10-94m and scFv10-94mQ respectively for a similar level of production to scFv 10-94 mutants.
PpL-binding activity was also investigated using a direct ELISA. Each PpL-purified scFv was directly fixed onto the plates in equimolar quantities and detection was carried out using the anti-flag antibody to calibrate the assay. Immobilized scFv was subsequently detected with PpL-HRP conjugate (C) and thus the mutants were compared with scFv 12-46 and scFv 12-46 Q.Citation15 The assay showed that PpL scFv interaction is variable. Three groups could be identified. The weakest interaction corresponded to scFv 12-46 and 10-94 mQ. When the 2 sequences were optimized with Q17 or with K90, the interaction became similar for both mutants scFv12-46 Q and scFv 10-94 mQK (A and C). The strongest interaction was obtained for the scFv mutant 10-94 P with only one mutation (C).
Optimization of the FR1 of IGKV10-94 P for the interaction with PpL
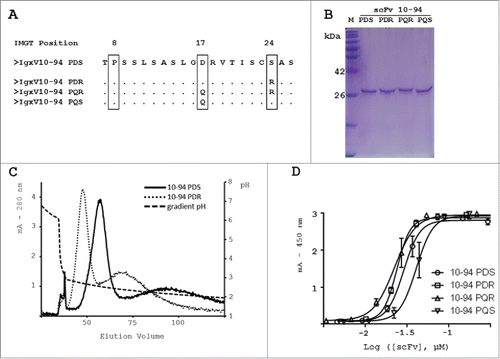
scFv 10-94 P interacted strongly with PpL, but this could be further optimized through double mutations : D17Q as in scFv 12-46, and in addition S24R as is the case in IGKV-95 / 96 FR1. Indeed, R24 plays a critical role in the interaction between Fab 4F11E12 (1YNT.pdb) and PpL.Citation14 Single point and double mutants were constructed (A). From the scFv 10-94 P, also designated as scFv 10-94 PDS, scFv 10-94 PDR, PQR and PQS were designed. After production and affinity purification using PpL-agarose, each mutant was obtained in pure and homogeneous yields (B). However, during the purification, and more specifically during the elution at pH 2.0, scFv variants behaved differently. scFv 10-94 PDR was eluted promptly while elution of scFv 10-94 PDS lasted longer, leading to dilution of the product. This was observed after purification on a CaptoL column onto which the same quantities of purified scFv 94-10 PDS and scFv 94-10 PDR were injected (C). The elution was carried out with a pH gradient (7 to 2). In both cases, at pH 3.0 a small fraction was eluted that corresponded to aggregates. Both mutants eluted with different retention times, and scFv 10-94 PDR was eluted first, being more sensitive to pH.
Figure 2. Design, production and purification of the scFv 10-94 P mutants and functional analysis of the interaction with the PpL by ELISA. (A) Mutations performed on the IGKV10-94 P(DS) light variable domain. (B) SDS-PAGE stained with Coomassie brilliant blue (C) PpL purified scFv 10-94 PDS and PDR were eluted with pH gradient from CaptoL column. (D) scFvs were directly immobilized on the plates. The calibrated scFv preparations were then detected using PpL–HRP conjugate.
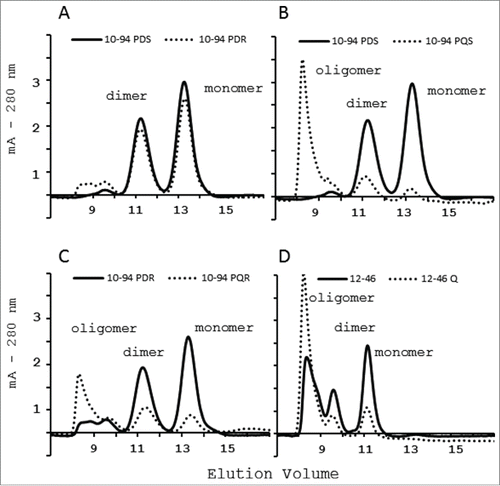
To better analyze the interaction with PpL, it was important to understand the effects caused by D17Q or S24R mutations on the mutants’ oligomeric profiles. scFv 10-94 PDS and scFv 10-94 PDR are mainly in monomeric form under these experimental conditions (A). Conversely, scFv 10-94 PQS and scFv 10-94 PQR have a strong tendency to form oligomers that are eluted from the size exclusion chromatography in the dead volume (B and C). These observations were confirmed by size exclusion chromatography analysis of scFv 12-46 and 12-46Q (D).
Figure 3. Size-exclusion chromatography on a calibrated SuperdexTM 75 10/300 GL column of the PpL-agarose purified scFv. Effect on the oligomeric profile of R24 mutation (A) or Q17 mutation (B, C and D).
The interaction of scFv mutants with PpL was also analyzed by ELISA. PpL-purified scFvs were immobilized onto the plate and then detected either using the anti-flag MRC-OX74 antibody for calibration or using PpL-HRP conjugate for titration (D). Some differences were observed depending on the scFv mutant. The Q17 mutation reduced the interaction with PpL while R24 (± Q17) apparently increased the interaction.
Affinity between PpL and scFv mutants
Because of the existence of several domains and sites of interaction on PpL, and because of the existence of oligomers in scFv, 2 subsequent experiments were performed.
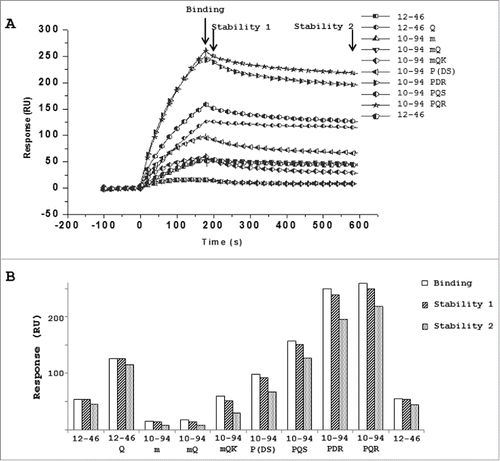
To compare scFv binding profiles, PpL-biotin was immobilized on a streptavidin sensor chip (SA) and purified scFv was injected at 18 nM onto a coated protein surface. The kinetic profiles of all scFvs are shown in A. From these sensorgrams, 3 RU values corresponding to the binding (175s), short-time (200s) and long-time stabilities (580s) were compared for all the scFvs on PpL (B). The m, mQ and mQK successive mutations on the scFv 10-94 showed progressively higher binding and stability values on PpL; however, the single P8 mutation (scFv 10-94P) yielded a better interaction that was strengthened by the mutation S24R (scFv 10-94PDR). Mutation Q17 led to a stronger apparent binding of scFv 12-46Q, scFv 10-94PQS and scFv 10-94PQR but this is likely due to presence of oligomeric structures of which a limited number of scFv are truly involved in the interaction with PpL ( and ).
Figure 4. Surface plasmon resonance analysis of the binding of PpL-purified scFv on immobilized PpL represented as sensorgrams (A) or histograms (B) based on 3 parameters: binding (180s), stability 1 (200s) and stability 2 (580s).
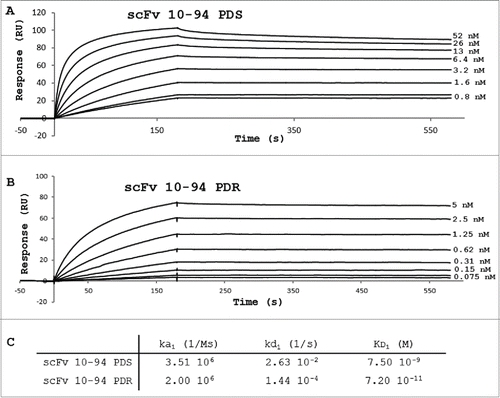
To determine the affinity and kinetic constants of scFv 10-94PDS and PDR, scFvs were covalently immobilized on a CM5 sensor chip and different concentrations of PpL were injected onto the coated-scFv. For both scFvs, the mixture of monomers and dimers observed could influence the non-stoichiometric kinetics. The bivalent model gave better global fitting of the sensorgrams, and a KD1 of 7.5 10-9 M and 7.2 10-11 M were obtained for the PDS and PDR versions, respectively (). The difference of affinity was essentially due to a difference of dissociation rate (a kd1 of 2 log lower for PDR) (C).
Figure 5. Sensorgrams showing the binding of PpL on immobilized scFv 10-94 PDS (A) or scFv 10-94 PDR (B) and Biacore analyses of kinetic parameters (C).
Discussion
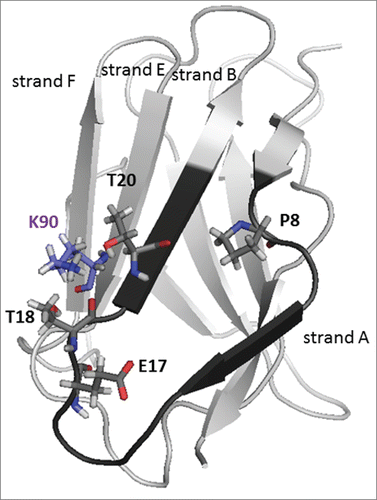
In order to confer PpL-binding activity to scFv 10-94, the first strategy developed here consisted in transferring the PpL recognition motif of scFv 12-46 as described by Muzard et al.Citation15 This strategy was unsuccessful and did not allow efficient purification of scFv 10-94m using PpL-affinity chromatography. The additional mutation E17Q showed low efficiency when used in the PpL recognition purification process. These results clearly indicated that critical residues that are essential for strong interaction with PpL are not all present in the IGKV 12-46 FR1. In addition, no structural analysis or 3-dimensional models of the interaction between PpL and IGKV12-46 have yet been reported. The only data available concern one PpL domain in complex with 3 distinct IGKV (PDB : 1HEZ, 1MHH and 1YNT) : Homo Sapiens IGKV1-39 (2A2Citation12), Mus musculus IGKV8-21 (19F9D6Citation18) and Mus musculus IGKV 10-96 (4F11E12Citation14). The alignment of their sequences with IGKV 12-46 shows a significant difference in position 18 with a threonine residue in place of a basic amino acid (A). A model of scFv 12-46 shows that a lysine (K90 in strand E) is located in close proximity to T18 instead of threonine as in the 3 other sequences (). We performed a T90K mutation and this enabled the efficient purification of scFv 10-94 mQK by affinity chromatography. scFv 10-94 mQK and scFv 12-46Q possess the same mQK pattern for interaction with PpL consisting of the FR1 of variable domain IGKV12-46 (position 7 - 22), of the mutation E17Q and the presence of a lysine in position 90. The study of the interaction with the PpL-HRP by ELISA confirms the results of the purifications with a higher affinity for the mutant scFv mQK (scFv 12-46 Q and scFv 10-94 mQK), than scFv 12-46 and scFv 10-94 mQ. The surface plasmon resonance (SPR) analysis showed that mutant scFv 10-94mQK exhibits higher binding and stability values than scFv 10-94mQ on the PpL immobilized. The presence of different proportion of oligomers in the purified fractions makes the comparison of scFv 12-46(Q) and scFv 10-94m(Q)K difficult by this technique. However, from the sensorgrams shown in A, the dissociation rate calculated for scFv 12-46(Q) is slower than for scFv 10-94m(Q)K (data not shown). This could be explained by the presence of the arginine 24 in the IGKV 12-46, which is the only difference in the FR1 of the IGKV 10-94 m(Q)K.
Figure 6. Alignment of the FR1 of the light variable domains. Alignment of 1HEZ, 1YNT and 1MHH with IGKV12-46 (A) and IGKV10-94 (B) according to http://multalin.toulouse.inra.fr/multalin/ (High consensus color: red; Low consensus color: blue; Neutral color: Black).

Figure 7. Model of the variable domain IGKV 12-46. In black, sequence m transferred according to Muzard et al.Citation15
The sequence alignment of VL-FR1 from the IGKV 10-94 and the Fab 4F11E12 (IGKV 10-96 with M4V mutation) shows only one difference situated at position 24 (serine or arginine) (B). The Fab fragment recognizes PpL via these 2 interfaces, but there is no detectable interaction with scFv. This could be explained partly by the existence of interactions between the PpL and the light constant domain via, in particular, the aspartic acid (PpL-D36), but also by a lower affinity of the VL-FR1 of Fab for the PpL. Thus, arginine 24 is not a key amino acid in the interaction with the PpL. The alignment VL-FR1 10-94 or 96 with sequences IGKV1-39 (2A2) and IGKV8-21 (19D9D6) shows a difference in position 8 with the presence of a proline instead of the threonine (B). This proline 8 is crucial because it interacts with many residues on PpL, especially with tyrosine 34 (interface 1) and tyrosine 32 (interface 2).Citation12 Furthermore, it probably enables a conformational modification of the separation between strands A and B that would favor the interaction with PpL. Therefore, scFv 10-94 P was produced and purified under the form of a concentrated and homogeneous product. Thus the simple transfer of T8P into the VL of scFv 10-94 resulted in a very strong interaction with the PpL, even better than with the multiple mutations mQK, as demonstrated by ELISA and SPR (C and 4).
Could the interaction be further improved? The previous results suggested 2 particularly interesting mutations: D17Q as the transfer E17Q for IGKV12-46 and S24R. Arginine is present in other VLs but, according to the structure of 1HEZ, especially participates in the interaction with both interfaces of the PpL. scFv 10-94P was therefore named more precisely as scFv 10-94PDS. The mutant scFv 10-94PDR was produced and obtained in pure form in a similar way to the PDS. The arginine did not cause any modification of the oligomeric profile. However, the behavior relative to PpL differs during the PpL-agarose affinity chromatography, the faster elution at pH 2.0 yielding more concentrated samples. This was verified during the elution of an identical quantity of scFv 10-94PDS or PDR fixed on a captoL column with a pH gradient (C). The scFv 10-94PDR seems to be more sensitive to pH and to show a better affinity for PpL, regardless of experimental methodology. Their affinities for PpL were determined by SPR with KD1 of 7.5 10-9 M and 7.2 10-11 M for PDS and PDR, respectively, with a much slower dissociation rate for the scFv 10-94PDR.
The second mutation is more questionable. Indeed, the results obtained by size exclusion chromatography show that the mutation VL-17Q favors an important multimerization of scFv 10-94 P and 12-46, the amide being more inclined to interact with residues on the other scFvs. Only the immobilization of mutants scFv Q really permits a comparison of their strength of interaction with PpL. This mutation should strengthen the interaction with the interface 1 of the PpL by interacting with PpL-Q16, as seen in IGKV 12-46 Q (). However, in IGKV 10-94 PQ(S or R), it may decrease the interaction when combined with S24 and shows no change in the strength of interaction when combined with R24. Thus, this mutation is of little interest in the case of IGKV10-94 P(S/R).
The comparison of VL IGKV12-46 with IGKV10-94 P shows that for most of the positions that differ, the amino acid residues are similar, that is to say small (A, S or T), hydrophobic (A, V, L) or negatively charged (D, E) (A). On the other hand, the amino acid residues of the positions 18 and 90 change from T-K to R-T. Being close in space, it would thus be important to have a basic amino acid that could interact with the α helix of PpL and would lock down the interaction between the ß sheets of PpL and of VL. The best interaction with PpL would be due to an arginine in position 18 that could interact more favorably with both interfaces of the PpL. According to Graille et al.Citation12, only the interface 2 of PpL interacts with amino acids in position 65, 72 and 74 (or S79, T88 and T90 in IMGT). Lysine 90 could interact only with the interface 2 of the PpL which could explain the lower global affinity. The interaction of the PpL with IGKV 12-46 merits study in a simplified system using a domain of the PpL.Citation12,13,18,19 Nevertheless our study underlines the importance of the lysine 90 when a threonine is present in position 18. The VL IGKV12-46 and IGKV10-94 P also show that the proline 8 is essential for a strong interaction with PpL. Moreover, similar results with the IGKV 10-94 P were obtained with the scFv of the 4F11E12 (unpublished data) and the scFv of the antibody 3J24.Citation20
Identification of the key residues on IGKV 12-46 and IGKV 10-94 P (S/R) should allow the optimization of the recognition of all kappa chains by commercially available PpL with only a minimum of modification. This should reduce the risk of affecting overall folding or conformation/accessibility of antigen-binding sites. However, even the grafting of the IGKV12-46 FR1 on the scFv 4C1 did not change its functionality.Citation6,15 The interaction site of the IGKV 12-46 has a good affinity for PpL, enabling the purification and detection of scFv. The site can be transferred to another scFv by preserving the characteristics of the interactions, in particular a fast elution at an acid pH of 3.0. As for the interaction site of IGKV 10-94 P, this possesses the best affinity (PDR) for commercial PpL and enables a rapid elution of the concentrated scFv. The PDS variant would have the advantage of yielding less concentrated purified scFv, which would be profitable for scFv that aggregate at low concentrations. A subnanomolar affinity is essential to optimize the purification efficiency to recover most of the antibody fragments and to develop an immunoassay with PpL.
Our work has led us to speculate as to whether the transfer of PpL recognition could be achieved on lambda chains not naturally recognized by PpL and characterized by the absence of amino acid 10 in the VL-FR1. Such a result would be useful to future research on antibodies.
Material and methods
Nomenclature
The variable domains are numbered according to the IMGT, www. IMGT.org.Citation21 ScFv used is named according to the gene of the light variable domain. The characteristics of scFv, molecular mass and coefficient of extinction molar, were defined with http://web.expasy.org/protparam/.
Recombinant scFv
The scFv 9C2Citation7 and 9C2-Q15 are named, for homogenization, scFv 12-46 and scFv 12-46 Q. The cassette corresponding to the sequence 7 - 22 FR1 is symbolized by m. The scFv consisting of the light variable domain IGKV10-94 is named scFv 10-94 (not published). scFvs result from the association of the heavy and light variable domains of an antibody via a (Gly4Ser)3 peptide link and from a peptide flag MRC-OX74 in C-terminal.Citation22,23
Transfer of the cassette m corresponding to the FR1 of domain IGKV12-46*01
The modification of the FR1 of the scFv 10-94 was realized by PCR overlaps. At first, the couples of primers oligo3bxxl / K7pLFor (CC ATG ATT ACG CCA AGC TTG CAT GC / AAC GCT AAC GCT CAG GCT TGC CGG AGA CTG GGT CT CTG) and K7pLRev / pswFor2xxl (AGC CTG AGC GTT AGC GTT GGT GAG ACC GTT ACC ATT ACC TGT / GGC GAT TAA GTT GGG TAA CGC CAG GG) enabled the respective amplification of the sequence encoding VH-VLFR1 and VL with the modification of the sequence of the FR1. Secondly, the overlap PCR was performed with the external primers oligo3bxxl and pswFor2xxl. The amplified and modified gene was inserted into the plasmid pSW1 by double PstI /XhoI digestion. The absence of mutations was verified by sequencing (GATC Biotech). The gene so modified codes the scFv 10-94m.
Site-directed mutagenesis
The protocol of QuikChange® Site-Directed Mutagenesis Kit (Stratagene, 200518) was followed. Plasmids containing the genes of the scFv 94-10m or scFv 94-10 were first amplified by PCR with the pfu polymerase by means of complementary primers allowing introduction of the various mutations (Promega).
From the plasmid pSW1 scFv 94-10m, the following primers were used - to introduce the mutation Q: mE17QRev (GCGTTAGCGT TGGTCAGACC GTTACCATTA DC) / E17QFor (GGTAATGGTA ACGGTCTGAC CAACGCTAAC GC); - to introduce the mutation K: T90KRev (GGCACCGATT ATAGCCTGAA AATTTCTAAT CTGGAACCGG) / T90KFor (CCGGTTCCAG ATTAGAAATT TTCAGGCTAT AATCGGTGCC). From the plasmid pSW1 scFv 94-10, the mutations in position 8, 17 and 24 can be performed in an independent manner. Primers are - to introduce the mutation T8P: T8PRev (CA GATGACCCAG ACCCCGTCTA GCCTGAGCGC) / T8PFor (GCGCTCAGGC TAGACGGGGT CTGGGTCATC TG); - to introduce the mutation D17Q: D17Qrev (GCCTGAGCGC AAGCCTGGGT CAGCGTGTTA CCATTAGC) / D17QFor (GCTAATGGTA ACACGCTGAC CCAGGCTTGC GCTCAGGC); - to introduce the mutation S24R: S24Rrev (GTTACCATTA GCTGTCGCGC AAGCCAGGG) / S24Rfor (CCCTGGCTTG CGCGACAGCT AATGGTAAC). Nucleotides allowing the introduction of the mutations are underlined. The program consisted of 14 cycles, each of 3 stages: 30s of denaturation in 94°C, 1 min of hybridization in 50 and 8 min of elongation. These cycles were preceded by denaturation at 94°C for 2 minutes and completed by elongation for 10 minutes.
After digestion by the DpnI restriction enzyme (Promega), bacteria were transformed with the non-digested neo-formed plasmids. Afterwards the plasmid was extracted from a transformed bacteria and verified by sequencing.
Protein expression and purification
The constructed vectors were cloned into Escherichia coli HB2151 (K12, ara, D(lac-pro), thi/F0 proA+B+, laclq lacZDM15) for protein expression. scFv expression was induced with isopropyl b-D-1-thiogalactopyranoside (IPTG, 0.1 mM). Periplasmic extracts were collected after mild osmotic shock, as reported in Aubrey et al.Citation7, and were then extensively dialyzed against phosphate-buffered saline (PBS) at pH 7.4 and clarified by centrifuging (10,000g, 4°C, 20 min). Preparations containing the scFv protein were stored at –20°C until use.
scFv proteins were purified by loading periplasmic preparations onto a column of Pierce® PpL Agarose (Life Technologies, 20512). The column was washed with 20 ml of PBS (pH 7.4), and the recombinant proteins were eluted in 1 ml fractions with glycine (0.1 M, pH 3.0 or 2.0). Fractions containing the recombinant proteins were selected at A280 nm, pooled, dialyzed against PBS (pH 7.4), and centrifuged (10,000g, 4°C, 10 min).
Protein preparations were analyzed by SDS–PAGE on homogeneous 12% gel under reducing conditions and underwent Western blotting using the anti-Flag monoclonal antibody MRC-OX74. The immunocomplexes formed were then detected using a phosphatase alkaline conjugate goat anti-mouse antibody and the 5-bromo-4-chloro-3-indolyl-phosphatase /nitroblue tetrazolium liquid substrate system (Sigma–Aldrich).
The purified scFv preparations were resolved by size-exclusion FPLC on a Superdex 75 10/300 GL column (molecular mass range 3,000–70,000) (GE Healthcare Life Sciences, 17-5174-01) with an Akta purifier. The column was loaded with 200 pmol of scFv. Proteins were eluted with PBS at a rate of 0.4 ml/min, and detected with a UV detector at 280 nm.
Binding to PpL-agarose of purified scFv
The HiScreen Capto L column (GE Healthcare Life Sciences, 17-5478-14) was loaded with 200 pmol of PpL-agarose purified scFv 10-94 PDS or PDR at a rate of 0.4 ml/min. After washing with PBS, scFvs were eluted with a gradient of 0% glycine (0.1 M, pH 6.0) to 100% of glycine-HCl (0.1 M, pH 2.0) on 155 ml in 0.4 ml/min and detected with a UV detector at 280 nm.
Binding to PpL by ELISA
The 96-well plates were coated with successive dilutions of the PpL-purified scFv preparations at 2 μM (100 μL), incubated for 16 h at 4°C and then were saturated with 3% BSA for 2 h at 37°C. Plates were then incubated for 1 h with the anti-Flag monoclonal antibody MRC-OX74, followed by incubation with an HRP-conjugated goat anti-mouse IgG antibody (Fc specific, Sigma–Aldrich) or with 100 μL of 1.25 μg/ml PpL–HRP conjugate (Life Technologies, 32420). 100 μL of TMB Substrate solution (Sigma–Aldrich, T0440) was then added to the wells. The reaction was stopped with 50 μL of H2SO4 (1 M). Absorbance was read at 450 nm in an ELISA plate reader. The wells were washed 3 times with PBS containing 0.05% Tween 20 (Sigma–Aldrich) between the intermediate steps. The background signal was determined by coating the wells directly with the blocking solution. All assays were conducted in triplicate.
SPR analysis (PpL binding)
SPR analysis were performed at 25°C on a BIA3000 apparatus (GE Healthcare, Uppsala, Sweden) at 30 μL/min in HBS-EP buffer (0.01M HEPES pH7.4, 0.15 M NaCl, 3 mM EDTA and 0.005% polysorbate P20). Biotinylated PpL (Pierce Biotechnology) was immobilized (500 RU) on an SA sensor chip (GE Healthcare). PpL-purified scFv at 18nM were injected onto immobilized PpL for 180s. After a dissociation step of 400s, PpL was regenerated by a pulse of glycine-HCl pH2.2. To be independent from the fit quality, we compared the binding responses in RU of the different scFvs at 3 times: 180s, 200s and 580s, called binding, stability 1 and stability 2, respectively. The stability of the coated surface was checked by injection of 9C2 at the beginning and the end of the series of experiments.
Measurement kinetics were performed on T200 apparatus (GE healthcare) to measure affinity. scFv 10-94 PDR and scFv 10-94 PDS (500-600 RU) were covalently immobilized on a CM5S sensorchip by the amine coupling method according to the manufacturer's instructions. Different concentrations of PpL given in were injected at 50 μL/min for 180s and after a dissociation of 400s, regenerated with glycine-HCI pH1.7. The sensorgrams were analyzed with a bivalent global-fitting model using the T200 Evaluation software 2.0.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work has been funded with support from the French Higher Education and Research Ministry under the program “Investissements d’avenir” Grant Agreement: LabEx MAbImprove ANR-10-LABX-53–01.
References
- Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, et al. Protein production and purification. Nat Methods 2008; 5:135–46; PMID:18235434; http://dx.doi.org/10.1038/nmeth.f.202
- Das D, Allen TM, Suresh MR. Comparative evaluation of two purification methods of anti-CD19-c-myc-His6-Cys scFv. Protein Expr Purif 2005; 39:199–208; PMID:15642471; http://dx.doi.org/10.1016/j.pep.2004.10.007
- Bjorck L. Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J Immunol 1988; 140:1194–7; PMID:3125250
- Myhre EB, Erntell M. A non-immune interaction between the light chain of human immunoglobulin and a surface component of a Peptococcus magnus strain. Mol Immunol 1985; 22:879–85; PMID:3930951; http://dx.doi.org/10.1016/0161-5890(85)90073-2
- Nilson BH, Solomon A, Bjorck L, Akerstrom B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J Biol Chem 1992; 267:2234–9; PMID:1733930
- di Tommaso A, Juste MO, Martin-Eauclaire MF, Dimier-Poisson I, Billiald P, Aubrey N. Diabody mixture providing full protection against experimental scorpion envenoming with crude Androctonus australis venom. J Biol Chem 2012; 287:14149–56; PMID:22375011; http://dx.doi.org/10.1074/jbc.M112.348912
- Aubrey N, Devaux C, Sizaret PY, Rochat H, Goyffon M, Billiald P. Design and evaluation of a diabody to improve protection against a potent scorpion neurotoxin. Cell Mol Life Sci 2003; 60:617–28; PMID:12737321; http://dx.doi.org/10.1007/s000180300053
- Royce J. An industrial platform solution for antibody fragment purification. BioProcess International 2015; 13:6
- Kastern W, Sjobring U, Bjorck L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J Biol Chem 1992; 267:12820–5; PMID:1618782
- Bottomley SP, Beckingham JA, Murphy JP, Atkinson M, Atkinson T, Hinton RJ, Gore MG. Cloning, expression and purification of Ppl-1, a kappa-chain binding protein, based upon protein L from Peptostreptococcus magnus. Bioseparation 1995; 5:359–67; PMID:8767928
- Nilson BH, Logdberg L, Kastern W, Bjorck L, Akerstrom B. Purification of antibodies using protein L-binding framework structures in the light chain variable domain. J Immunol Methods 1993; 164:33–40; PMID:8360508; http://dx.doi.org/10.1016/0022-1759(93)90273-A
- Graille M, Stura EA, Housden NG, Beckingham JA, Bottomley SP, Beale D, Taussig MJ, Sutton BJ, Gore MG, Charbonnier JB. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure 2001; 9:679–87; PMID:11587642; http://dx.doi.org/10.1016/S0969-2126(01)00630-X
- Housden NG, Harrison S, Housden HR, Thomas KA, Beckingham JA, Roberts SE, Bottomley SP, Graille M, Stura E, Gore MG. Observation and characterization of the interaction between a single immunoglobulin binding domain of protein L and two equivalents of human kappa light chains. J Biol Chem 2004; 279:9370–8; PMID:14668335; http://dx.doi.org/10.1074/jbc.M312938200
- Graille M, Stura EA, Bossus M, Muller BH, Letourneur O, Battail-Poirot N, Sibai G, Gauthier M, Rolland D, Le Du MH, et al. Crystal structure of the complex between the monomeric form of Toxoplasma gondii surface antigen 1 (SAG1) and a monoclonal antibody that mimics the human immune response. J Mol Biol 2005; 354:447–58; PMID:16242717; http://dx.doi.org/10.1016/j.jmb.2005.09.028
- Muzard J, Adi-Bessalem S, Juste M, Laraba-Djebari F, Aubrey N, Billiald P. Grafting of protein L-binding activity onto recombinant antibody fragments. Anal Biochem 2009; 388:331–8; PMID:19268418; http://dx.doi.org/10.1016/j.ab.2009.02.035
- Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res 2009; 37:D1006–12; PMID:18978023; http://dx.doi.org/10.1093/nar/gkn838
- Martin AC. Accessing the Kabat antibody sequence database by computer. Proteins 1996; 25:130–3; PMID:8727325; http://dx.doi.org/10.1002/(SICI)1097-0134(199605)25:1%3c130::AID-PROT11%3e3.3.CO;2-Y
- Graille M, Harrison S, Crump MP, Findlow SC, Housden NG, Muller BH, Battail-Poirot N, Sibai G, Sutton BJ, Taussig MJ, et al. Evidence for plasticity and structural mimicry at the immunoglobulin light chain-protein L interface. J Biol Chem 2002; 277:47500–6; PMID:12221088; http://dx.doi.org/10.1074/jbc.M206105200
- Beckingham JA, Bottomley SP, Hinton R, Sutton BJ, Gore MG. Interactions between a single immunoglobulin-binding domain of protein L from Peptostreptococcus magnus and a human kappa light chain. Biochem J 1999; 340 (Pt 1):193–9; PMID:10229674; http://dx.doi.org/10.1042/bj3400193
- Zahid M, Loyau S, Bouabdelli M, Aubrey N, Jandrot-Perrus M, Billiald P. Design and reshaping of an scFv directed against human platelet glycoprotein VI with diagnostic potential. Anal Biochem 2011; 417:274–82; PMID:21771576; http://dx.doi.org/10.1016/j.ab.2011.06.036
- Lefranc MP. Unique database numbering system for immunogenetic analysis. Immunol Today 1997; 18:509; PMID:9386342; http://dx.doi.org/10.1016/S0167-5699(97)01163-8
- Mousli M, Devaux C, Rochat H, Goyffon M, Billiald P. A recombinant single-chain antibody fragment that neutralizes toxin II from the venom of the scorpion Androctonus australis hector. FEBS Lett 1999; 442:183–8; PMID:9928998; http://dx.doi.org/10.1016/S0014-5793(98)01647-0
- Devaux C, Moreau E, Goyffon M, Rochat H, Billiald P. Construction and functional evaluation of a single-chain antibody fragment that neutralizes toxin AahI from the venom of the scorpion Androctonus australis hector. Eur J Biochem 2001; 268:694–702; PMID:11168408; http://dx.doi.org/10.1046/j.1432-1327.2001.01923.x