
ABSTRACT
Frameshifts lead to complete alteration of the intended amino acid sequences, and therefore may affect the biological activities of protein therapeutics and pose potential immunogenicity risks. We report here the identification and characterization of a novel -1 frameshift variant in a recombinant IgG1 therapeutic monoclonal antibody (mAb) produced in Chinese hamster ovary cells during the cell line selection studies. The variant was initially observed as an atypical post-monomer fragment peak in size exclusion chromatography. Characterization of the fragment peak using intact and reduced liquid chromatography-mass spectrometry (LC-MS) analyses determined that the fragment consisted of a normal light chain disulfide-linked to an aberrant 26 kDa fragment that could not be assigned to any HC fragment even after considering common modifications. Further analysis using LC-MS/MS peptide mapping revealed that the aberrant fragment contained the expected HC amino acid sequence (1-232) followed by a 20-mer novel sequence corresponding to expression of heavy chain DNA sequence in the -1 reading frame. Examination of the DNA sequence around the frameshift initiation site revealed that a mononucleotide repeat GGGGGG located in the IgG1 HC constant region was most likely the structural root cause of the frameshift. Rapid identification of the frameshift allowed us to avoid use of a problematic cell line containing the frameshift as the production cell line. The frameshift reported here may be observed in other mAb products and the hypothesis-driven analytical approaches employed here may be valuable for rapid identification and characterization of frameshift variants in other recombinant proteins.
Abbreviations
| CEX | = | cation exchange chromatography |
| CHO | = | Chinese hamster ovary |
| CID | = | collision induced dissociation |
| Fab | = | fragment antigen-binding region of an antibody |
| Fc | = | fragment crystallizable region of an antibody |
| GS | = | Glutamine synthetase |
| HC | = | heavy chain |
| HexNAc | = | N-acetylhexosamine |
| HPLC | = | high pressure liquid chromatography |
| IgG | = | Immunoglobulin |
| LC−MS | = | liquid chromatography with online mass spectrometry detection |
| mAb | = | monoclonal antibody |
| MS | = | mass spectrometry |
| MS/MS | = | tandem mass spectrometry |
| NeuAc | = | N-acetylneuraminic acid |
| PFS | = | programmed frameshift |
| SEC | = | size exclusion chromatography. |
Introduction
Most recombinant monoclonal antibody (mAb) therapeutics are currently manufactured in mammalian expression systems, particularly Chinese hamster ovary (CHO) cell lines.Citation1,2 The process can be briefly outlined as follows. Genes coding for the light chain and heavy chain (HC) of the designed mAb molecule are inserted into expression plasmid(s), and the resulting plasmid(s) are used to transfect expression host cells such as CHO. The transfected cells are selected and optionally amplified in the presence of selection agents, e.g., methotrexate for a dihydrofolate reductase-based CHO expression system or methionine sulfoximine for a glutamine synthetase (GS)-based CHO expression system. Clonally-derived cell lines are then screened to identify those cell lines with optimal productivity, genetic stability and product quality and the “top” cell line is subsequently selected to generate a cell bank for producing the mAb in large scale bioreactors. Throughout the process starting from construction of the gene plasmid, transfection, selection, amplification, and during the cell growth in bioreactors, errors can occur during DNA replication, transcription, or translation, leading to unintended alteration of amino acid sequences. In recent years, numerous examples of such expression errors in production of recombinant proteins at various stages of the manufacturing process have been reported.Citation3-18 These reported examples are predominately single amino acid substitution caused by either DNA mutationsCitation3-11 or misincorporation during translation,Citation12-16 which are commonly referred as sequence variants. Expression errors leading to alteration of longer segments of amino acid sequence have also been reported.Citation8,17,18 In a special case of single base-pair mutation, the IgG light chain stop codon TAA was mutated to GAA (Glu), leading to an extension of the light chain by 18 amino acid sequence from the non-coding region of the light chain DNA construct.Citation8 In addition, rare cases of rearrangement leading to cross-over of the light chain and HC sequencesCitation17 or extension of the HC with expression of the non-coding region of the light chain DNA construct in both in-frame and out-of-frame fashions have been reported.Citation18 The product variants caused by the expression errors can potentially affect the safety and efficacy of the protein therapeutics, and therefore should be identified and characterized and ideally eliminated from the manufacturing process during early stages of development.Citation19 If these variants cannot be eliminated from the manufacturing process, they must be monitored when assessing comparability upon process changes and potentially tested as part of the batch release to ensure consistency of product quality.Citation19,20
Here, we report identification and characterization of a new type of product variant caused by frameshift in a humanized IgG1 monoclonal antibody (Mab-1) produced in CHO cells. Frameshift refers to expression of protein sequence out of the normal reading frame. Since frameshifts lead to complete alteration of the intended amino acid sequences and often premature termination of the protein expression, they may affect the biological activities and pose potential immunogenicity risks of protein therapeutics. Frameshift variants are more likely to be detected by chromatography analytic methods, such as size exclusion chromatography (SEC), than single amino acid substitution sequence variant due to the large changes to the protein sequence that result from the frameshift, but they might be ignored if they are incorrectly assumed to be common, known fragments (e.g., Fab) eluting in the same region of the chromatography. Therefore, it is critical to carefully examine the chromatography and characterize any ambiguous peaks with orthogonal techniques such as mass spectrometry (MS) during critical stages of development such as cell line selection.
The frameshift variant described here was initially observed as a post-monomer fragment peak in SEC that appeared to be an atypical fragment after careful examination of the chromatograph. It was then identified as a novel frameshift variant by MS techniques using a fraction enriched from the side-stream of a cation exchange chromatography (CEX) purification process step. Identification of the frameshift variant poses some unique analytical challenges. Edman sequencing and MS/MS de novo sequencing are the typical techniques for determining the sequences of unknown peptides in peptide maps, and they have been used to successfully identify novel peptide sequences, e.g., the aberrant sequences caused by TAA (Stop codon) to GAA (Glu) mutationCitation8 or DNA rearrangement,Citation18 respectively. However, these techniques may not always be feasible or sufficient: Edman sequencing will not work if there are other peptides co-eluting with the peak of interest, while MS/MS de novo sequencing requires presence of complete fragmentation ion series (e.g., b or y ions) in the MS/MS spectrum and it can be difficult to deduce sequence of a large peptide de novo. On the other hand, an automatic database search was not readily available to identify the frameshift either because the frameshifted sequence was not in the normal protein sequence database. Although we can translate the DNA sequence in all 3 reading frames in silico and use the translated sequences for the database search,Citation18 this would still not allow determination of the sequence containing the frameshift initiation site (i.e., the sequence joining the normal and frameshifted amino acid sequences). To overcome these challenges, we used a hypothesis-driven analytical approach as detailed in the following sections to effectively identify the frameshift sequence and the frameshift initiation site. Rapid identification of the frameshift variant allowed us to avoid choosing a problematic cell line containing the frameshift as the production cell line. The frameshift described here may be observed in other mAb products because the frameshift took place in the HC constant region of IgGs. The hypothesis-driven analytical approaches employed here may be useful for effective identification of frameshift variants in other recombinant proteins.
Results
Discovery of the aberrant IgG fragment
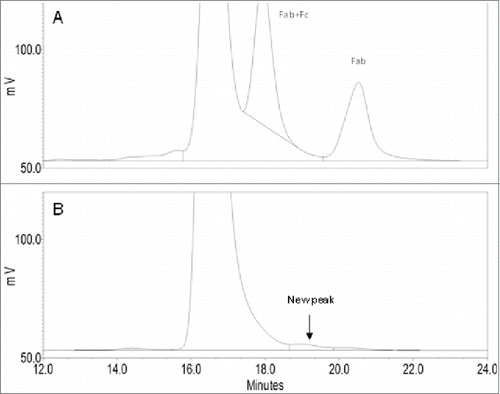
To identify a production cell line with the best product quality, Mab-1 samples were purified from the cell culture broth of the “top 4” clonally-derived cell lines (i.e., cell lines A,B,C,D) by protein-A affinity chromatography and were analyzed using SEC and other analytical methods. A unique small shoulder peak on the backside of the monomer main peak (i.e., the main peak) was observed in product expressed in cell line B, but not in the other 3 cell lines (). The small shoulder peak accounted for about 1% of the total protein peak area in the chromatography. Since cell line B was being considered at the time as the leading cell line candidate based on its more robust growth profile in cell culture and its high productivity, it was decided to identify the unknown species to better understand whether it may affect product safety, efficacy or quality. The knowledge would enable us to either include or exclude cell line B as a choice for the production cell line. It is well known that IgG1 molecules can undergo cleavages in the hinge region via various mechanisms to produce Fab and the complementary IgG1 fragment (i.e., IgG1 missing a Fab, also referred as Fab+Fc fragment).Citation21 Although the Fab and Fab+Fc fragments also eluted in the post-monomer region on the SEC, careful comparison of the unknown peak in cell line B with the Fab and Fab+Fc peaks produced in a stressed Mab-1 sample showed that it did not co-elute with either Fab or Fab+Fc peak on the chromatography (), indicating that it was likely a different species. Although only a small quantity of the unknown fragment directly collected from the SEC was available for the preliminary LC-MS analysis, we found that large quantity of the unknown species could be isolated more conveniently using a larger scale purification CEX chromatography. Analysis of the CEX fractions by the analytical SEC showed that the unknown species was mostly enriched in a front side fraction (namely F4) of the CEX chromatography using the protein-A purified Mab-1 sample from cell line B as the starting material. SEC analysis of the final F4 fraction sample confirmed that it contained ∼85% of the unknown fragment, 13% of the Mab-1 monomer (i.e., the intact IgG molecule) and 2% of the Fab fragment. The collected F4 fraction sample was used for all the subsequent LC-MS analyses as described in the remaining sections of this report.
Figure 1. Zoomed-in SEC chromatograms of protein A captured samples from the top 4 cell lines A, B, C, D of Mab-1.
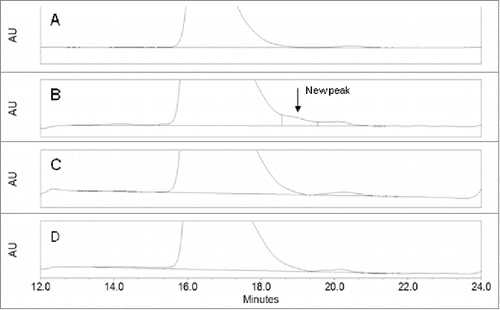
Figure 2. Comparison of zoomed-in SEC chromatograms of the unknown peak in cell line B (B) with Fab and Fab+Fc peaks produced in a stressed sample (pH 9.0 at 40oC for 8 weeks) (A).
Characterization of the aberrant fragment by top-down LC-MS analysis
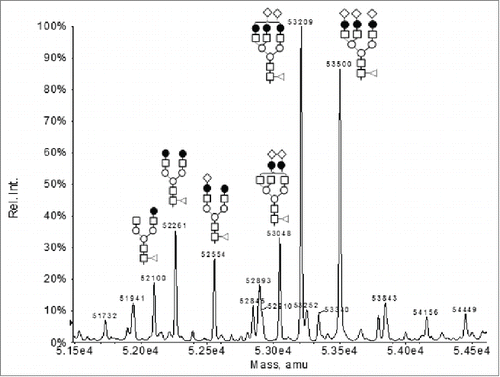
To determine the light chain and HC masses and modifications, the F4 sample was analyzed by LC-MS following reduction of the inter-chain disulfide bonds with dithiothreitol (DTT). The UV 214 nm reversed phase chromatography of the LC-MS analysis showed 2 major peaks, i.e., Peak 1 and peak 3 and a few minor peaks (). The major species under Peak 1 was determined to be the normal light chain of Mab-1 (observed mass 24034 Da, expected mass: 24034 Da). The deconvoluted mass spectrum under peak 3 displayed a series of peaks in the mass region of 27000-31000 Da (). The mass differences between these peaks are ∼162 Da, 203 Da or 291 Da, suggesting N-linked glycan structures differing in the number of hexose, HexNAc, or NeuAc monosaccharide residues, respectively. Based on the pattern of mass difference, these peaks were assigned as a Mab-1 HC fragment with various oligosaccharide structures, as labeled in .
Figure 3. UV 214 nm chromatogram of the reversed phase LC-MS analysis of the partially reduced F4 sample. (Peaks 1 and 3: see details in text. Peak 2: light chain with 2 unpaired cysteine residues; peak 4: intact heavy chain; peak 5: same species as in peak 3 and peak 4 but with 2 additional unpaired cysteine residues).
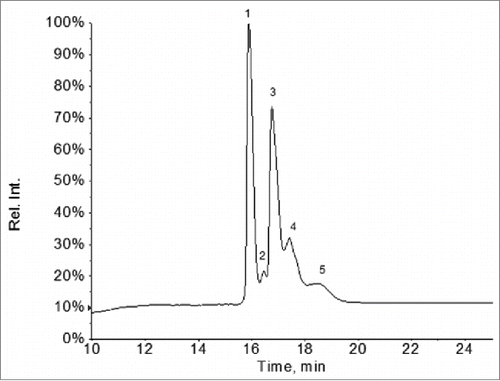
Figure 4. Deconvoluted mass spectrum of the ion species under peak 3 in in the LC-MS analysis of the partially reduced F4 sample.
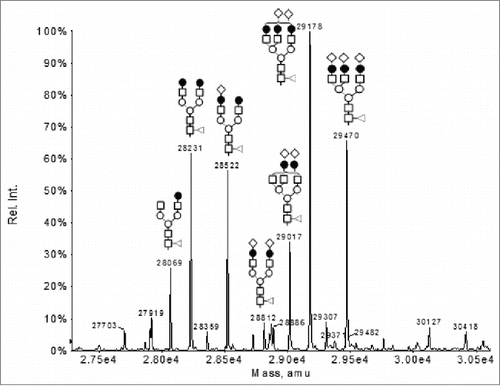
The mass of the non-glycosylated form of the fragment (i.e., the protein backbone) can be calculated by subtracting the observed mass of the glycosylated species with the theoretical mass of the corresponding N-linked oligosaccharide structure. For example, using the observed mass of the peak with G2F glycosylation (i.e., 28231 Da), the mass of the non-glycosylated protein backbone can be calculated to be 26461 Da by subtracting the observed mass (i.e., 28231 Da) with the theoretical mass of the G2F oligosaccharide (i.e., 1770 Da). This deduced mass of the non-glycosylated protein, i.e., 26461 Da did not match any segment of the Mab-1 amino acid sequence even after considering common modifications (e.g., oxidation, deamidation, pyroglutamate formation), thus suggesting the presence of an unusual structural modification in the unknown fragment. The observed masses of the minor peaks 2 and 5 in were 2 Da higher than the observed masses under peak 1 and peak 3 (data not shown), respectively, and, therefore, could be assigned as the respective species with 2 unpaired cysteine residues. The minor peak 4 in was assigned as the expected full-length HC of Mab-1.
To determine the overall mass of the unknown fragment, the F4 sample was also analyzed by LC-MS in its intact form, i.e., without the DTT reduction. The UV 214 nm reversed-phase chromatography of the LC-MS analysis showed a major peak, i.e., peak II and 2 minor peaks, i.e., peak I and peak III (). The predominant species under the minor peak I in was identified as the cysteinylated light chain of Mab-1 (observed mass: 24153 Da; expected mass 24153 Da). Low levels of cysteinylated light chain are commonly observed in recombinant mAb samples.Citation22 The minor peak III in was assigned as intact Mab-1 since it had the same retention time and mass spectrum as the intact Mab-1 in the normal Mab-1 control sample. This was expected since the F4 sample still contained ∼13% of the intact Mab-1. The deconvoluted mass spectrum under the main chromatography peak (peak II) showed a series of mass peaks in the range of 51000-55000 Da (). The mass differences between these peaks are ∼162 Da, 203 Da or 291 Da, suggesting N-linked glycan structures differing in the number of Hexose, HexNAc, or NeuAc monosaccharide residues, respectively. Based on the pattern of the mass difference, these peaks were assigned as the intact form of the aberrant IgG fragment with various oligosaccharide structures, as labeled in . By comparing the mass spectra shown in and , we found that the observed mass of each of the intact glycosylated species in was equal to the mass of the fragment peak with the same glycan form in the reduced sample in plus the mass of the Mab-1 light chain minus 2 Da (the mass change upon formation of a disulfide from 2 cysteine residues). For example, the mass of the G2F glycosylated fragment peak in (i.e., 28231 Da) plus the mass of the light chain (24034 Da) minus 2 Da equals to 52263 Da, which agreed with the observed mass of the G2F glycosylated peak in (i.e., 52261 Da) within the expected error of the mass spectrometer used for the analysis. This indicated that the unknown species in the aberrant IgG fragment was consisted of a Mab-1 light chain linked via disulfide to an unknown protein fragment of 26461 Da that was glycosylated with various oligosaccharide structures.
Figure 5. UV 214 nm chromatogram of the reversed-phase LC-MS analysis of the intact F4 sample.
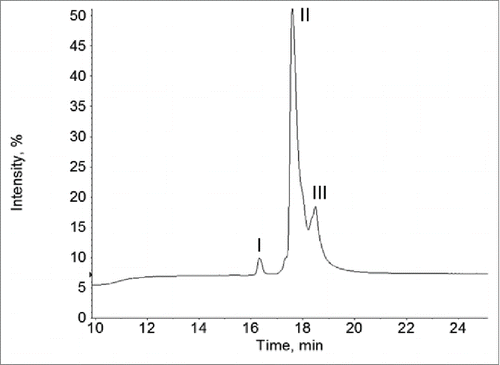
Figure 6. Deconvoluted mass spectrum under the peak II in in the LC-MS analysis of the intact F4 sample.
Characterization of the aberrant IgG fragment by LC-MS peptide mapping
To identify the unknown HC fragment, the F4 sample was analyzed by LC-MS peptide mapping following reduction, alkylation and proteolytic digestion with Lys-C. A purified Mab-1 sample containing the normal Mab-1 structure was analyzed alongside with the F4 sample for comparison. depicted the UV214 nm chromatography profiles of the Lys-C peptide maps of F4 and the normal Mab-1 control sample. While the 2 peptide maps have many common peaks, there are significant differences between the 2 samples. Several peaks observed in the peptide map of the expected Mab-1 sample disappeared or decreased in the F4 sample, indicated that some parts of the normal peptide sequence were missing or decreased in the F4 sample. On the other hand, a few new or increased peaks were observed in F4 compared to the normal Mab-1 sample. Most prominently, a distinct peak at 36.5 min was present in the Lys-C peptide map of the F4 sample (labeled peak x in ), but was missing in the normal sample (although there was a small peak at similar retention time detected in the normal Mab-1 sample, it contained a different mass, and therefore, was an unrelated species). The peak x contained a predominant MH22+ ion species at m/z 607.2812, corresponding to a monoisotopic mass of 1212.5478 Da, which did not match with any part of Mab-1 amino acid sequence. To identify the unknown peptides, the MH22+ ions at m/z 607.2812 were selected for collision-induced dissociation (CID) MS/MS analysis. The MS/MS spectrum of the m/z 607.2812 ion is depicted in . From the singly charged ion peaks at m/z 658.31, 773.33, 886.42 and 983.48 in the mass spectrum, a tentative sequence tag of D(L/I)P (if b ion series) or P(L/I)D (if y ion series) was suggested. However, neither D(L/I)P nor P(L/I)D was present in any part of the Mab-1 light chain or HC sequence, indicating that the unknown peptide could not be simply attributed to a posttranslational modification or a single amino acid mutation of the Mab-1 amino acid sequence. The MS/MS spectrum was also used to search against the National Center for Biotechnology Information (NCBI) non-redundant protein database using the Pro ID software (version 1.1 AB Sciex), but no meaningful hit was found, suggesting that it was a novel sequence that was not present in the protein database. Subsequently, we hypothesized that the unusual peptide might result from a reading frameshift event that was capable of producing a completely novel amino acid sequence. To evaluate this hypothesis, the DNA sequence of Mab-1 HC was translated in silico to a “frameshifted” amino acid sequence by shifting the normal reading frame either one nucleotide position toward the 5’ end (i.e., -1 frameshift), or one nucleotide position toward the 3’ end (i.e., +1 frameshift). The region of the DNA sequence coding for the Mab-1 HC amino acid sequence (210-254) and the corresponding amino acid sequences translated according to the in-frame (expected reading frame), the -1 frame or the +1 frame are displayed in . The HC translation was terminated after amino acid position 252 at a stop codon that resulted from the -1 frameshift. As shown in , Lys-C digestion of the -1 frameshift protein species would generate a C-terminal peptide TQGHPHDLPDP. The mass of this peptide, i.e., 1212.5523 Da, matched with the observed mass of the unknown peptide in the peak x in the Lys-C peptide map, i.e., 1212.5478 Da. Furthermore, as labeled in , de novo sequence obtained from the MS/MS spectrum of the MH22+ ions at m/z 607.2812 is consistent with the proposed -1 frameshift peptide TQGHPHDLPDP.
Figure 7. UV 214 nm chromatograms of peptide maps of the F4 sample and the normal Mab-1 sample digested with Lys-C following denaturation, reduction and alkylation. x indicates the unknown peak that was present in the F4 sample but not in the control sample.
Figure 8. CID MS/MS spectrum of the MH22+ ions under the peak x in the Lys-C peptide map of the F4 sample as shown in .
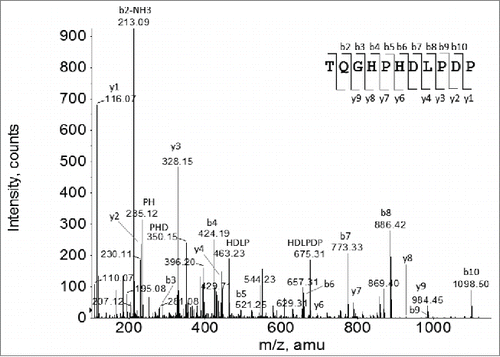
Figure 9. DNA sequence codons for the Mab-1 heavy chain amino acid sequence region (210-254) and the corresponding amino acid sequences translated according to the expected reading frame (i.e., in-frame), the -1 frame or the +1 frame. (* stop codon; the predicted C-terminal peptide of the -1 frameshift protein sequence after Lys-C digestion was underlined).

Next, the starting site of the frameshift and the entire altered sequence resulting from the -1 frameshift were determined. As shown in , if a -1 frameshift occurred at or before HC amino acid position 228, a stop codon would be generated at position 228, leading to a HC fragment (1-227). Since the data from peak x in the Lys-C peptide map () showed that the HC was actually terminated at position 252, we concluded that the -1 frameshift must occur somewhere after HC 228. On the other hand, an additional segment of amino acid sequence in the region of HC(236-243) corresponding to the expected -1 frameshift sequence, i.e., LPLPPKTQ was confirmed by LC-MS/MS data of the peptide map of the F4 sample using papain digestion (data not shown). Thus, it can be concluded that the -1 frameshift must occur within the amino acid sequence region of (229-235). According to the amino acid sequences illustrated in , digestion of the F4 sample with Lys-C would produce a peptide HC (218-241) that would span the region transitioning from the normal sequence to the -1 frameshift altered sequence.
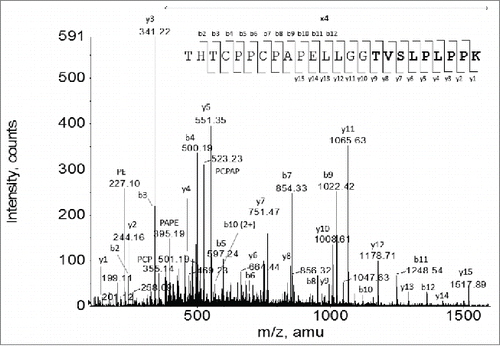
To determine the exact starting site of the -1 frameshift, all the sequences of the Lys-C peptide (218-241) corresponding to each different position in the region of (229-235) as the possible starting point of the -1 frameshift were generated, and the expected masses of the MH22+ or MH33+ ions for each of the peptide sequence were calculated (). The predicted ions were then searched using extracted ion chromatography (XIC) in the LC-MS Lys-C peptide map of the F4 sample. Only an XIC peak matching the mass of the Lys-C digestion peptide sequence THTCPPCPAPELLGGTVSLPLPPK was found (MH33+ ion observed m/z: 847.1044; expected m/z: 847.1064), thus indicating that the -1 frameshift started at amino acid residue Thr233. To confirm the amino acid sequence of the frameshifted peptide, the MH33+ ions at m/z 847.1 were selected as the precursor ions to acquire MS/MS data. The MS/MS spectrum confirmed the proposed peptide sequence THTCPPCPAPELLGGTVSLPLPPK (). Thus, it was determined that the altered peptide sequence by the -1 frameshift started at Thr233. However, as shown in and , it must be noted that the -1 frameshift could actually start in the DNA codons for Gly231, Gly232 since a -1 frameshift on those codons would not change the glycine to a different amino acid residue, and therefore, would also produce the same peptide sequence as observed.
Table 1 Expected sequences and corresponding masses of the ions of Lys-C Peptide (218-241) when assuming each position in the region of HC(229-235) as the possible starting point of the -1 frameshift (altered sequence from the -1 frameshift is underlined)
Figure 10. CID MS/MS spectrum of the MH33+ ion species corresponding to the Lys-C peptide HC (218-241) THTCPPCPAPELLGGTVSLPLPPK that spans across the starting site of the -1 frameshift. The altered amino acid sequence by the -1 frameshift is shown in a bold font.
The proposed -1 frameshift starting site was further confirmed by LC-MS peptide mapping of the F4 sample following Asp-N digestion. According to the proposed frameshift transition site, digestion of the F4 sample with Asp-N would produce a HC peptide (216-247) with the following sequence: DKTHTCPPCPAPELLGG TVSLPLPPKTQGHPH. Search of the Asp-N peptide map by XIC confirmed the presence of the predicted peptide (MH33+ ion observed m/z: 1147.2232; expected m/z: 1147.2465), thereby further confirming the proposed -1 frameshift sequence and the transition site.
Based on the above analyses, we determined that the unknown HC fragment consisted of the expected sequence (1-232) followed by a 20-mer amino acid sequence (233-252) resulting from the -1 frameshift, i.e., TVSLPLPPKTQGHPH. As a stop codon was introduced after P252, the aberrant HC fragment was terminated after Pro252.
According to the proposed -1 frameshift sequence, the average mass of the proposed aberrant HC (1-252) fragment was calculated to be 26460 Da, which is in good agreement with the protein backbone mass of the HC fragment deduced from the partially reduced LC-MS analysis, i.e., 26461 Da. This further confirmed the overall proposed sequence of the aberrant HC(1-252) fragment resulted from the -1 frameshift.
Characterization of the hinge region disulfide in the aberrant IgG fragment
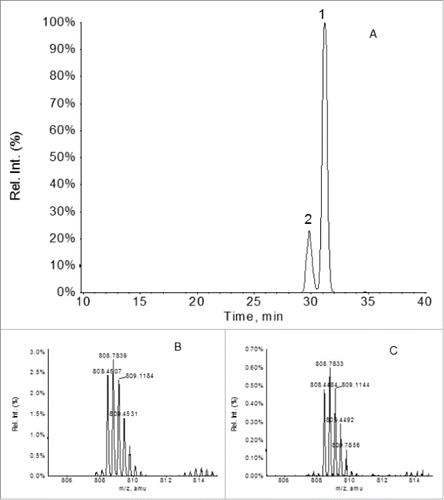
In a typical IgG1 molecule, the 2 cysteine residues in the HC hinge region, i.e., Cys221 and Cys224, form 2 pairs of inter-chain disulfide bonds with corresponding Cys221 and Cys224 residues from the other HC. However, since the aberrant IgG frameshift fragment contained only one light chain and one HC frameshift fragment (1-252) based on the LC-MS analyses, the hinge cysteine residues Cys221 and Cys224 in the aberrant IgG fragment could not possibly form the inter-HC disulfides as in the normal IgG1 structure because a second HC did not exist in the fragment. Instead, the hinge Cys221 and 224 must either form an intra-chain disulfide with each other or both remain as unpaired cysteine residues. To discern these two possible forms, the disulfide linkages in the novel IgG fragment were analyzed by LC-MS peptide mapping of the F4 sample following digestion by Lys-C under non-reduced conditions. As depicted in , a search of the non-reduced Lys-C peptide map for the 2 forms of the HC(218-241) peptide using their expected m/z of the MH33+ ions revealed only the mass of the peptide with an intra-chain disulfide (expected m/z: 808.4202; observed m/z: 808.4507 or 808.4484), but not the peptide with the 2 free cysteine residues (expected m/z: 809.0921). Interestingly, two XIC peaks, i.e., peak 1 and 2 in , were observed both containing the expected mass of the peptide (218-241) with the intra-chain hinge disulfide. This indicated that there were probably two different conformations of the intra-chain disulfide between the Cys221 and Cys224, most likely due to proline cis-trans isomerization. Based on these results, we concluded that the two hinge cysteine residues in the novel IgG fragment were connected to each other via an intra-chain disulfide.
Figure 11. XIC peaks of the MH33+ ions of the HC(218-241) peptide in which the Cys221 and Cys224 residues are connected by an intra-chain disulfide (expected m/z: 808.4202) (panel A) . The zoomed-in mass spectra of the MH33+ ions under the XIC peaks 1 and 2 are depicted in panes B and C, respectively.
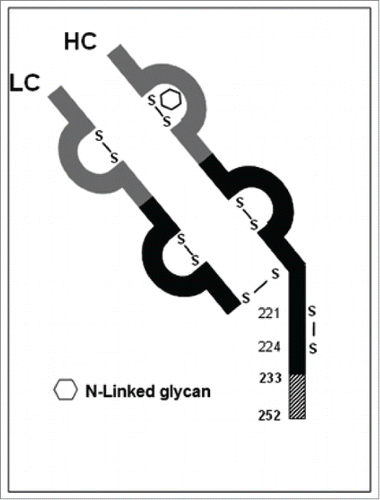
Overall structure of the aberrant IgG fragment
Based on the results obtained in the previous sections, an overall structure of the aberrant IgG fragments in cell line B is proposed (). The structure consists of a normal light chain linked to an aberrant HC fragment (1-252) in which the last 20 C-terminal amino acid residues resulted from a -1 frameshift event. The HC hinge region cysteine residues Cys221 and Cys224 were connected to each other via an intra-chain disulfide.
Figure 12. Sketch of the aberrant IgG fragment resulted from the -1 frameshift in Mab-1. IgG variable regions are shown in gray; IgG constant regions are shown in black; the novel amino acid sequence region HC (233-252) resulted from the -1 frameshift is shown as hatched box.
Discussion
During cell line generation for a recombinant IgG1 therapeutic mAb using GS-CHO cells, an aberrant IgG fragment was discovered. It was present at 1% level relative to the intact IgG as detected by SEC in only one of the “top 4” cell lines evaluated. The IgG fragment consisted of a normal light chain linked via a disulfide to an aberrant IgG HC fragment. The aberrant HC fragment consisted of the expected sequence (1-232) followed by a 20-mer novel peptide sequence resulting from a -1 frameshift.
Frameshifts have been observed in viral as well as cellular genes of all organisms, including E. coli and mammalian cells.Citation23,24 The reading frame can shift either one nucleotide position toward the 5’ end of the mRNA (-1 frameshift) or one nucleotide position toward the 3’ end of the mRNA (+1 frameshift). The major structural causes of frameshift include errors in DNA sequence, errors in mRNA sequence, and errors in the ribosome movement during translation.
On the DNA level, insertion of a nucleotide would lead to a -1 frameshift and deletion of a nucleotide would lead to a +1 frameshift. The insertion or deletion tends to occur more frequently on mono-nucleotide repeats.Citation25,26 The process can be explained by a DNA strand slippage mode.Citation26,27 According to the model, a template strand slippage would cause insertion of a nucleotide and a primer strand slippage would cause deletion of a nucleotide. It has been shown that G or C repeats are more prone to cause insertion or deletion than A or T repeats,Citation28 presumably due to the higher stability of the slippage intermediate.Citation29 One of the best-studied examples is insertion of G in a G7 repeat in the thymidine kinase (TK) gene of herpes simplex virus (HSV) as part of its elegant mechanism to evade the action of the anti-viral drug acyclovir (ACV) for treating HSV infection.Citation30 Such insertion causes a -1 frameshift, which would abolish expression of the full-length TK required to phosphorylate the ACV in order to activate its anti-viral activity.Citation31
On the RNA level, although the transcription error rate is considered much lower than those during DNA replication,Citation32 insertion or deletion of a nucleotide can also occur during transcription leading to a frameshift.Citation33,34,35 The insertion/deletion is believed to involve a slippage between the DNA and RNA strands similar to the Streisinger DNA slippage model, and also tends to occur more frequently on mononucleotide repeats. However, according to the reported examples to date, insertion/deletions during transcription appear to be more prone to occur on A or T repeats rather than G or C repeats,Citation33,34 a preference that is opposite to that in DNA replication. In addition to nucleotide insertion/deletion, aberrant splicing of the primary transcript can also lead to a frameshift if the normal reading frame was disrupted at the joint site.Citation18,36
On the translation level, erroneous movements of ribosomes can also lead to frameshifts.Citation23,24,37-41 Such frameshifts can be divided into programmed frameshift (PFS) and incidental frameshift.Citation31,42 Programmed frameshifts typically occur at higher efficiency (up to 100%) and often produce alternative functional proteins that form an integral part of the organism's physiology.Citation23 A programed frameshift signal is characterized by a slippery sequence on the mRNA where the frameshift actually takes place, and a stimulatory RNA structure such as stem loop, pseudoknot, or rare codons,Citation31,38,40 which are thought to cause the ribosome to pause allowing partition of the out-of-frame translation to occur.Citation43 For -1 PFS, the predominant slippery site is characterized by a heptanucleotide motif X XXX YYY Z (where X denotes any nucleotide, Y denotes A or U, and Z is A, U, or C).Citation23 Similarly, +1 PFS also involves a slippery sequence and a stimulatory RNA element, although the signal motifs for +1 PFS are much more diverse than those for -1 PFS.Citation23,40,41
Unlike PFS, the incidental frameshift does not require the presence of both of the slippery sequence and the stimulatory structure. Instead, it is driven by specific mRNA structure features such as rare codonsCitation44-48 or mononucleotide repeat of 6 to 9 guanine nucleotides (termed G-string).Citation31,42,49-52 The efficiency of incidental frameshift is usually low (i.e., <5%) although the efficiency of frame shifts at hungry codons can reach as high as 50%.Citation48 One of the most important examples of incidental frameshift is the frameshift on G-strings of 6 to 9 guanine nucleotides.Citation31,42,49-52 Frameshifts on G-strings have been extensively studied owing to their importance in the drug resistance mechanism of ACV.Citation31,49-52 As mentioned earlier, HSV mutants evade the anti-viral activity of ACV primarily by inserting a G in the G7 string in their TK gene. This leads to a -1 FS, and thereby abolishes the expression of full-length TK, which is required to activate the ACV.Citation31 On the other hand, the resultant G8 string in the HSV ACVr mutants can mediate low level of +1 translational FS, which would cancel out the -1 FS from the G insertion, and thereby restore expression of the full-length TK.Citation50,52 Although only low levels of full-length TK are expressed via the +1 FS in the HSV mutants, it is sufficient to permit HSV to reactivate from the latent state. The efficiency of +1 FS has been determined to be about 1% on G8,Citation52 but much lower on G7 and G6 strings.Citation50 While it has been shown that G7 can also mediate a lower level of -1 FS in addition to the +1 FS, G6 did not show any detectable level of –1 FS.Citation52 It was proposed that the frameshift on G-string was mediated by distorted RNA structures driven by interaction between the guanine residues from the G-string or from the rRNA (rRNA) via non-Watson-Crick base pairing.Citation50
As shown in , alteration of the expected amino acid sequence by the -1 frameshift in Mab-1 started at HC amino acid residue Thr233. On the surface, this appeared to suggest that the -1 frameshift started on the codon for the Thr233 residue, i.e., shifting the codon CCG (Pro233) to ACC (Thr233) (). However, inspection of the DNA sequence of MAb-1 HC in the region revealed that there was a GGGGGG repeat located immediately before the codon for Thr233 (). Since GGGGGG is a hotspot for nucleotide insertion/deletion as well as ribosomal frameshift, as discussed previously, the frameshift likely takes place on the GGGGGG repeat rather than the codon for Thr233. As illustrated in , theoretically there were 3 possible mechanisms of the -1 frameshift: 1) insertion of a G within the GGGGGG repeat at DNA level; 2) insertion of a G within the GGGGGG at RNA level; or 3) a shift of the ribosome one nucleotide position toward the 5’ direction at the codon GGG (Gly231) or GGA (Gly232) during translation. Although none of the 3 possible mechanisms could be completely ruled out, it appeared that insertion of a nucleotide (likely G) within the GGGGGG repeat at DNA level is more likely than the other 2 mechanisms since insertion at RNA level has only been reported for A/T but not G/C repeats,Citation33,34 and frameshifts on G-string are predominately +1 rather than -1 frameshift.Citation50,52 A homology search of the Mab-1 frameshifted peptide sequence SCDKTHTCPPCPAPELLGGTVSLPLPPKTQGHPHDLPDP in the NCBI Genbank database (Cur_NR_Prot)Citation53 using BLASTCitation54 found that it was partially shared by sequences of 3 IgG HC fragments in mAb samples prepared with peripheral blood lymphocytes of patients.Citation55
Figure 13. Three possible mechanisms for the -1 frameshift observed in Mab-1. Reading frames are denoted by short lines under each of the codons. -1 shifted frames and corresponding amino acid residues are shown in a bold font. Inserted nucleotides are shown in a bold font and denoted with arrows. The normal in-frame translations are shown in panel A. The three possible -1 frameshift mechanisms are depicted in panels B, C and D, respectively. Only the heavy chain amino acid sequence region 226-240 encompassing the frameshift starting site and the corresponding DNA or mRNA sequences are depicted.

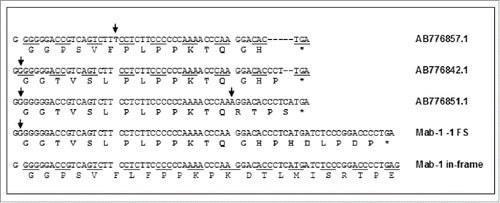
The IgG HC fragments, i.e., BAN63135.1, BAN63144.1 and BAN63150.1, are predicted from the submitted cDNA sequences in GenBank, i.e., AB776842.1, AB776851.1 and AB776857.1, respectively. shows comparison of the cDNA and amino acid sequences of the 3 IgG fragments from Genbank with the DNA sequence (with an insertion of G in the G6-string) and the amino acid sequences in the -1 frameshift region of Mab-1. In BAN63135.1, BAN63144.1, the frameshifts were both initiated by an insertion of G in the GGGGGG region, while in BAN63150.1, the frameshift was caused by insertion of a T in a region that contains only 2 consecutive T nucleotides. As shown in , while the stop codon in the frameshift in Mab-1 was solely resulted from a G insertion, generation of the stop codons in the 3 IgG fragments from GenBank involved additional insertion or deletion of nucleotides. Since no other information other than the cDNA sequence and the predicted peptide sequences were available in Genbank, it was unclear what the underlying mechanisms are and what biological significance may be for those IgG HC fragments published in the GenBank database. Nevertheless, the sequences AB776842.1 and AB776851.1 show that the insertion of G in the GGGGGG DNA sequence indeed could occur in human IgG molecules and thereby support our hypothesis that the -1 frameshift observed in Mab-1 was probably also caused by an insertion of G at DNA level. To definitively confirm whether the frameshift in Mab-1 took place at DNA, RNA or ribosome level, DNA and mRNA (or cDNA) sequencing data would be necessary. However, due to the low level of the observed frameshift variant, we did not attempt to sequence the DNA or cDNA of Mab-1 in cell line B, so the exact mechanism of the -1 frameshift remains to be confirmed. Nevertheless, since GGGGGG is the most likely structural root cause of the frameshift regardless of which mechanism, breaking the GGGGGG repeat by replacing the GGG codon with other redundant codons for Gly, e.g., GGX (X=A ,T or C), should eliminate the -1 frameshift.
Figure 14. Comparison of the DNA sequence and the amino acid sequence in the -1 frameshift region of Mab-1with the cDNA and amino acid sequences of 3 IgG fragments found in Genebank by BLAST homology search. The frameshifted amino acid residues are shown in a bold font. Inserted nucleotides are shown in a bold font and denoted with arrows. Deleted nucleotides are denoted with short dashes (-).
It has been found that the G6 string has been preferentially avoided in gene sequences of various species, most remarkably in mammalian cells, suggesting that the G6 string might have been selected against during evolution due to its susceptibility to errors at DNA, RNA or translational level.Citation25 On the other hand, G6 string is conserved in the DNA sequence coding for the human IgG constant region near the hinge region. This leads to an interesting question: is conservation of GGGGGG motif in the IgG constant region merely a coincidence or is there underlying unknown biological significance?
A few examples of frameshift variants in recombinant proteins expressed in E. coli have been reported, all of which were caused by single or tandem repeats of rare arginine codons.Citation44-48 Recently, an example of out-of-frame expression as a result of DNA sequence rearrangement was reported in IgG produced in CHO.Citation18 To our knowledge, this study represents the first example of a pure frameshift variant observed in recombinant proteins expressed in a mammalian cell line, CHO in this case. Since the GGGGGG nucleotide motif is in the constant region common to human IgGs (except for IgG2 isotype), it may be observed in other mAbs.
Due to the completely altered amino acid sequence caused by the -1 frameshift, the frameshift fragments are likely to have different biological activities and pharmacokinetic profile compared with that of the unmodified products. Perhaps more importantly, the novel amino acid sequence that resulted from the frameshift may cause concerns of potential immunogenicity to patients due to its “foreignness” to the human immune system. In order to minimize the potential risks associated with the frameshift variant and to ensure consistent high product quality, it was decided to eliminate the problematic cell line B as a possibility for the production cell line despite its otherwise desirable attributes. Instead, another one of the “top” cell lines, in which the frameshift was not detected by SEC, was chosen as the production cell line to manufacture Mab-1. Characterization of frameshift variants poses certain unique challenges as the normal database search tool is not readily available for identifying the frameshifted sequence and for determining the site of the frameshift. The hypothesis-driven analytical approaches utilized in this report may be valuable for rapid identification and characterization of frameshift variants in the fast-paced environment of biotherapeutics development. This study underscores the complexity of the biological expression system for producing recombinant proteins and the importance of thorough structural characterization using modern characterization techniques such as MS to reduce potential safety risk and to ensure consistent high quality of biopharmaceutical products.
Materials and methods
Chemical reagents
8 M guanidine hydrochloride (#24115), DTT (#20291), formic acid (#28905) and trifluoroacetic acid (TFA) (#28904) were purchased from Pierce. Sequencing grade modified trypsin was purchased from Promega (#V528A). MS-grade lysyl endopeptidase (Lys-C) was purchased from Wako Chemicals USA (#125-05061). Sequencing grade endoproteinase Asp-N (#11054589001) and papain (#10108014001) were purchased from Roche Applied Science. PNGAse F was purchased from QA Bio (#E-PNG01). HPLC grade water and acetonitrile for preparation of the mobile phases for the LC-MS analysis were purchased from Burdick and Jackson. Iodoacetamide (#I1149-25g) and all other chemicals were purchased from Sigma–Aldrich.
Protein material
The recombinant mAb used in this study (Mab-1) is an IgG1 humanized mAb containing 2 N-linked glycosylation sites on each HC, one located in the Fc region, the other in the Fab region. Mab-1 was produced in a CHO K1SV cell line and purified at Eli Lilly and Company. A stressed Mab-1 sample was prepared by incubating the purified Mab-1 in 10 mM Tris hydrochloride buffer, pH 9 at 40oC for 8 weeks.
Cell line generation
The light chain and HC genes of Mab-1 were inserted into an expression plasmid that was designed with all the elements of the GS-Gene Expression System™ (Lonza Biologics). The cell line generation process is similar to what has been described previouslyCitation56 except that GS-CHO cells instead of the GS gene knock-out CHO cells were used in this study. The productivity and product quality of the “top 4” clonally-derived cell lines were further evaluated in 5-L bioreactors in order to determine the final production cell line. The bioreactor cell cultures for the evaluated cell lines were harvested after 14 days, and the antibody was purified using a Protein-A affinity chromatography column.
SEC analysis
Samples were analyzed using a Waters 2695 HPLC system with a 2487 UV detector on a TSK Gel G3000SWXLSEC column (7.8 x 300 mm, 5 μm, 250 Å; Tosoh Bioscience, #08541). The column was eluted isocratically with a buffer consisting of 50 mM sodium phosphate, 150 mM NaCl, pH 7.5 at a flow rate of 0.5 mL/min at ambient temperature. The chromatography was detected by UV absorbance at 214 nm wavelength. About 6 μg of protein was injected onto the column in each analysis.
Preparation of partially reduced samples for top-down LC-MS analyses
An aliquot of the sample containing 27 μg of protein was diluted into 100 μL of 0.1M Tris hydrochloride buffer (pH 8.0) and then reduced with 2 μL of 324 mM DTT at ambient temperature for 25 minutes immediately prior to the LC-MS analysis.
Digestion of samples for reduced peptide mapping
An aliquot of sample containing 20 μg of protein was evaporated to dryness in a Speedvac (Savant, Model AES1010) without heating. The sample was reconstituted in 9 μL of 6 M guanidine hydrochloride in 250 mM Tris hydrochloride buffer (pH 8.0) and mixed with 0.6 μL of 324 mM DTT. The sample was incubated at 55°C for 15 min to denature the protein and to reduce the disulfides. The sample was mixed with 0.8 μL of 500 mM iodoacetamide in water and incubated at ambient temperature in dark for 20 min. The sample was diluted to a final volume of 100 μL with 50 mM Tris hydrochloride buffer (pH 8.0) and digested with either 0.4 μg of Lys-C or 0.4 μg of Asp-N at 37°C for 4 hours. The digestion was stopped by adding 2.5 μL of 20% TFA in water to the sample.
Digestion of samples by papain
An aliquot of sample containing 60 μg of the protein was digested with 1 μL of papain (1 μg/μL) in a buffer containing 20 mM sodium citrate, 50 mM NaCl, 5 mM cysteine, pH 5.0 at 37°C for 2 hours. The sample was then mixed with 20 μL of 1 M Tris hydrochloride buffer (pH8) and 1 μL of 324 mM DTT and incubated at ambient temperature for 15 minutes to reduce the inter-chain disulfides. Subsequently 2 μL of 500 mM iodoacetamide was added to alkylate the reduced cysteine residues.
Digestion of samples for non-reduced peptide mapping
An aliquot of sample containing 20 μg of protein was evaporated to dryness in a Speedvac without heating. The sample was reconstituted in 9 μL of 6 M guanidine hydrochloride in 250 mM Tris hydrochloride buffer (pH 8.0) and incubated at 55oC for 15 min to denature the protein. The sample was then diluted to a final volume of 100 μL with 50 mM Tris hydrochloride buffer (pH 8.0) and digested with either 0.4 μg of Lys-C or 0.4 μg of Asp-N at 37oC for 4 hours. The digestion was stopped by adding 2.5 μL of 20% TFA in water to the sample.
Top-down LC-MS analysis
The intact samples or the partially reduced sample was analyzed on an Agilent 1100 HPLC system coupled with a QStar XL Quadrapole TOF hybrid mass spectrometer (Applied Biosystems). A polymer-based reversed phase column (5 μm particle, 1000 Å pore, 2.1x50 mm; Polymer laboratory, #PLRP-S) was used for the analyses. The column was maintained at 65oC and eluted using mobile phase A (0.05% TFA and 0.1% formic acid in water) and mobile phase B (0.05% TFA and 0.1% formic acid in acetonitrile) with a linear gradient of 10-40% mobile phase B over a period of 20 min at a flow rate of 0.25 mL/min. For the LC-MS analysis of the intact protein in the non-reduced sample, 12 μg of the protein was injected onto the column for each analysis. For the LC-MS analysis of the partially reduced sample, 21 μg of the sample was injected onto the column for each analysis. The mass spectra were deconvoluted using the Protein Reconstruct tool of the Analyst 1.1 software (Applied Biosystems). The major parameters of the mass spectrometer for the analysis were as follows: capillary voltage 4.5 kV; capillary temperature: 450oC; Declustering potential: 80 V; Focusing potential: 300 V; m/z range for MS data acquisition: 600-3500 Da.
LC-MS peptide mapping
The reduced/alkylated or non-reduced samples after digestion with Lys-C or Asp-N were analyzed on an Agilent 1100 capillary HPLC system coupled with a QStar XL Quadrapole TOF hybrid mass spectrometer (Applied Biosystems). A C18 reversed phase column (5 μm particle, 300 Å pore, 0.5x150 mm; Grace Vydac, #218MS5.515) was used for the analyses. The column was maintained at 40oC and eluted using mobile phase A (0.05% TFA in water) and mobile phase B (0.04% TFA acetonitrile) with a linear gradient of 2-40% mobile phase B over a period of 120 min at a flow rate of 0.02 mL/min. For additional LC-MS/MS analyses, 0.1% formic acid in water was used as mobile phase A and 0.1% formic acid in acetonitrile was used as mobile phase B. The MS/MS data was acquired using the information-dependent acquisition (IDA) function of the Analyst software. Approximately 1.6 μg of the digested protein sample was injected onto the column in each analysis. The major parameters of the mass spectrometer for the analysis were as follows: capillary voltage 4.5 kV; capillary temperature: 150oC; Declustering potential: 35 V; Focusing potential: 250 V; m/z range for MS data acquisition: 250-2000 Da; m/z range for MS/MS data acquisition: 100-2000 Da; Precursor ion mass tolerance range: ±100 mmu; Collision energy: rolling collision energy. The MS and MS/MS data was processed with the Analyst software (Applied Biosystems).
Disclosure of potential conflicts of interest
Zhirui Lian and Tongtong Wang are employees of Eli Lilly and Company therefore receive salaries and may own Eli Lilly stock or stock options. Qindong Wu is a former employee of Eli Lilly and Company and may hold Eli Lilly stock.
Acknowledgments
We thank Jonathan Lawrence for isolating the enriched frameshift fractions using the CEX chromatography for this work. We thank Christopher Frye, Luhong He, Lihua Huang and Bryan Harmon for critical review of the manuscript and helpful discussions. We also thank Michael R De Felippis and Steven Maple for their guidance and support.
References
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol 2004: 22:1393–8; PMID:15529164; http://dx.doi.org/10.1038/nbt1026
- Matasci M, Hacker DL, Baldi L, Wurm FM. Recombinant therapeutic protein production in cultivated mammalian cells: current status and future prospects. Drug Discov Today: Technol 2008; 5(2):e37-42; PMID:24981089; http://dx.doi.org/10.1016/j.ddtec.2008.12.003
- Harris RJ, Murnane AA, Utter SL, Wagner KL, Cox ET, Polastri GD, Helder JC, Sliwkowski MB. Assessing genetic heterogeneity in production cell lines: detection by peptide mapping of a low level Tyr to Gln sequence variant in a recombinant antibody. Nature Biotechnol 1993; 11(11):1293–7; PMID:7764191; http://dx.doi.org/10.1038/nbt1193-1293
- Dorai H, Sauerwald T, Campbell A, Kyung Y, Goldstein J, Magill A, Lewis M, Tang Q, Jan D, Ganguly S, Moore G. A case study in rapid detection of mutation in mammalian production cell lines. BioProcess Int 2007; 5(8):66–72
- Guo D, Gao A, Michels DA, Feeney L, Eng M, Chan B, Laird MW, Zhang B, Yu XC, Joly J, Snedecor B, Shen A. Mechanisms of unintended amino acid sequence changes in recombinant monoclonal antibodies expressed in Chinese Hamster Ovary (CHO) cells. Biotechnol Bioeng. 2010; 107:163–71; PMID:20506532; http://dx.doi.org/10.1002/bit.22780
- Que AH, Zhang B, Yang Y, Zhang J, Derfus G, Amanullah A. Sequence variant analysis using peptide mapping by LC-MS/MS. BioProcess Int 2010; 8:52–60
- Ren D, Zhang J, Pritchett R, Liu H, Kyauk J, Luo J, Amanullah A. Detection and identification of a serine to arginine sequence variant in a therapeutic monoclonal antibody. J Chromatogr B 2011; 879(27):2877–84; http://dx.doi.org/10.1016/j.jchromb.2011.08.015
- Zhang T, Huang Y, Chamberlain S, Romeo T, Zhu-Shimoni J, Hewitt D, Zhu M, Hatta V, Mauger B, Kao YH. Identification of a single base-pair mutation of TAA (Stop codon)→ GAA (Glu) that causes light chain extension in a CHO cell derived IgG1. mAbs 2012; 4(6):694–700; PMID:23018810; http://dx.doi.org/10.4161/mabs.22232
- Fu J, Bongers J, Tao L, Huang D, Ludwig R, Huang Y, Qian Y, Basch J, Goldstein J, Krishnan R, You L, Li ZJ, Russell RJ. Characterization and identification of alanine to serine sequence variants in an IgG4 monoclonal antibody produced in mammalian cell lines. J Chromatogr B 2012; 908:1–8; http://dx.doi.org/10.1016/j.jchromb.2012.09.023
- Yang Y, Strahan A, Li C, Shen A, Liu H, Ouyang J, Kata V, Francissen K, Zhang B. Detecting low level sequence variants in recombinant monoclonal antibodies. Mabs 2010; 2:285–98; PMID:20400866; http://dx.doi.org/10.4161/mabs.2.3.11718
- Zeck A, Regula JT, Larraillet V, Mautz B, Popp O, Göpfert U, Wiegeshoff F, Vollertsen UEE, Gorr IH, KollH, Papadimitriou A. Low level sequence variant analysis of recombinant proteins: an optimized approach. PloS one 2012; 7(7):e40328; PMID:22792284; http://dx.doi.org/10.1371/journal.pone.0040328
- Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, Huang YM, Foley SF, Garber E, Pedseson N, Meier W. Discovery and investigation of misincorporation of serine at asparagine positions in recombinant proteins expressed in Chinese hamster ovary cells. J Biol Chem 2009; 284(47):32686–94; PMID:19783658; http://dx.doi.org/10.1074/jbc.M109.059360
- Yu XC, Borisov OV, Alvarez M., Michels DA, Wang YJ, Ling V. Identification of codon-specific serine to asparagine mistranslation in recombinant monoclonal antibodies by high-resolution mass spectrometry. Anal Chem 2009; 81:9282–90; PMID:19852494; http://dx.doi.org/10.1021/ac901541h
- Feeney L, Carvalhal V, Yu XC, Chan B, Michels DA, Wang YJ, Shen A, Ressl J, Dusel B, Laird MW. Eliminating tyrosine sequence variants in CHO cell lines producing recombinant monoclonal antibodies. Biotechnol Bioeng 2013; 110(4):1087–97; PMID:23108857; http://dx.doi.org/10.1002/bit.24759
- Raina M, Moghal A, Kano A, Jerums M, Schnier PD, Luo S, Deshpande R, Bondarenko PV, Lin H, Ibba M. Reduced amino acid specificity of mammalian tyrosyl-tRNA synthetase is associated with elevated mistranslation of Tyr codons. J Biol Chem 2014; 289(25):17780–90; PMID:24828507; http://dx.doi.org/10.1074/jbc.M114.564609
- Zhang Z, Shah B, Bondarenko P. GU and certain wobble position mismatches as possible main causes of amino acid misincorporations. Biochemistry 2013; 52(45):8165–76; PMID:24128183; http://dx.doi.org/10.1021/bi401002c
- Wan M, Shiau FY, Gordon W, Wang GY. Variant antibody identification by peptide mapping. Biotechnol Bioeng 1999; 62:485–8; PMID:9921157; http://dx.doi.org/10.1002/(SICI)1097-0290(19990220)62:4%3c485::AID-BIT12%3e3.0.CO;2-E
- Scott RA, Rogers R, Balland A, Brady LJ. Rapid identification of an antibody DNA construct rearrangement sequence variant by mass spectrometry. mAbs 2013; 6, no. 6:1453–63; http://dx.doi.org/10.4161/mabs.36222
- ICH guidaline Q6B: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products.
- ICH guideline Q5E: Comparability of Biotechnological/Biological Products subject to Changes in their Manufacturing Process.
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs 2011; 3(3):253–63; PMID:21487244; http://dx.doi.org/10.4161/mabs.3.3.15608
- Lu C, Liu D, Liu H, Motchnik P. Characterization of monoclonal antibody size variants containing extra light chains. Mabs 2013; 5(1):102–13; PMID:23255003; http://dx.doi.org/10.4161/mabs.22965
- Ketteler R. On programmed ribosomal frameshifting: the alternative proteomes. Front Genet 2012; 3(242):1–10; PMID:22303408
- Namy O, Rousset JP, Napthine S, Brierley I. Reprogrammed genetic decoding in cellular gene expression. Mol Cell 2004; 13(2):157–68; PMID:14759362; http://dx.doi.org/10.1016/S1097-2765(04)00031-0
- Gu T, Tan S, Gou X, Araki H, Tian D. Avoidance of long mononucleotide repeats in codon pair usage. Genetics 2010; 186(3):1077–84; PMID:20805553; http://dx.doi.org/10.1534/genetics.110.121137
- Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: structural biology of indel mutations. Trends in Biochemical Sciences 2006; 31(4):206–14; PMID:16545956; http://dx.doi.org/10.1016/j.tibs.2006.02.004
- Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M. Frameshift mutations and the genetic code. Cold Spring Harbor Symposia on Quantitative Biology 1996; 31:77–84; http://dx.doi.org/10.1101/SQB.1966.031.01.014
- Gragg H, Harfe BD, Jinks-Robertson S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol Cell Biol 2002; 22(24):8756–62; PMID:12446792; http://dx.doi.org/10.1128/MCB.22.24.8756-8762.2002
- Sagher D, Hsu A, Strauss B. Stabilization of the intermediate in frameshift mutation. Mutat Res 1999; 423(1):73–7; http://dx.doi.org/10.1016/S0027-5107(98)00227-9
- Hwang CB, Chen HJH. An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase. Gene 1995; 152(2):191–3; PMID:7835698; http://dx.doi.org/10.1016/0378-1119(94)00712-2
- Griffiths A. Slipping and sliding: frameshift mutations in herpes simplex virus thymidine kinase and drug-resistance. Drug Resist Updat 2011; 14:251–9; PMID:21940196; http://dx.doi.org/10.1016/j.drup.2011.08.003
- Rosenberger RF, Foskett G. An estimate of the frequency of in vivo transcriptional errors at a nonsense codon in Escherichia coli. Mol Gen Genet 1981; 183:561–3; PMID:7038382; http://dx.doi.org/10.1007/BF00268784
- Larsen B, Wills NM, Nelson C, Atkins JF, Gesteland RF. Nonlinearity in genetic decoding. Homologous DNA replicase genes use alternatives of transcriptional slippage or translational frameshifting. Proc Natl Acad Sci USA 2000; 97:1683–8; PMID:10677518
- Wagner LA, Weiss RB, Driscoll R, Dunn DS, Gesteland RF. Transcriptional slippage occurs during elongation at runs of adenine or thymine in Escherichia coli. Nucleic Acids Res 1990; 18(12):3529–35; PMID:2194164
- Baranov PV, Hammer AW, Zhou J, Gesteland RF, Atkins JF. Transcriptional slippage in bacteria: distribution in sequenced genomes and utilization in IS element gene expression. Genome Biol 2005; 6(3):R25; PMID:15774026
- Levitin F, Baruch A, Weiss M, Stiegman K, Hartmann ML, Yoeli-Lerner M, Ziv R, Zrihan-Licht S, Shina S, Gat A, et al. A novel protein derived from the MUC1 gene by alternative splicing and frameshifting. J Biol Chem 2005; 280(11):10655–63; PMID:15623537
- Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988; 331:280–3; PMID:3336440
- Farabaugh PJ. Programmed translational frameshifting. Microbiol Rev 1996; 60(1):103–34; PMID:8852897
- Gesteland RF, Weiss RB, Atkins JF. Recoding: reprogrammed genetic decoding. Science 1992; 257(5077):1640–1; PMID:1529352
- Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene 2002; 286(2):187–201; PMID:11943474
- Dinman JD. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip Rev RNA 2012; 3(5):661–73; PMID:22715123
- Zook MB, Howard MT, Sinnathamby G, Atkins JF, Eisenlohr LC. Epitopes derived by incidental translational frameshifting give rise to a protective CTL response. J Immunol 2006; 176(11):6928–34; PMID:16709853
- Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 2006; 441(7090):244–7; PMID:16688178
- Yoon SI, Walter MR. Identification and characterization of a+ 1 frameshift observed during the expression of Epstein-Barr virus IL-10 in Escherichia coli. Protein Expr Purif 2007; 53(1):132–7; PMID:17224278
- McNulty DE, Claffee BA, Huddleston MJ, Kane JF. Mistranslational errors associated with the rare arginine codon CGG in Escherichia coli. Protein Expr Purif 2003; 27(2):365–74; PMID:12597898
- Lainé S, Thouard A, Komar AA, Rossignol JM. Ribosome can resume the translation in both +1 or− 1 frames after encountering an AGA cluster in Escherichia coli. Gene 2008; 412(1):95–101
- Kerrigan JJ, McNulty DE, Burns M, Allen KE, Tang X, Lu Q,Trulli JM, Johanson KO, Kane JF. Frameshift events associated with the lysyl-tRNA and the rare arginine codon, AGA, in Escherichia coli: A case study involving the human Relaxin 2 protein. Protein Expr Purif 2008; 60(2):110–6; PMID:18474430
- Spanjaard RA, Van Duin J. Translation of the sequence AGG-AGG yields 50% ribosomal frameshift. Proc Natl Acad Sci USA 1988; 85(21):7967–71
- Hwang CB, Horsburgh B, Pelosi E, Roberts S, Digard P, Coen DM. A net +1 frameshift permits synthesis of thymidine kinase from a drug-resistant herpes simplex virus mutant. Proc Natl Acad Sci USA 1994; 91:5461–5; PMID:8202508
- Horsburgh BC, Kollmus H, Hauser H, Coen DM. Translational recoding induced by G-rich mRNA sequences that form unusual structures. Cell 1996; 86:949–59; PMID:8808630
- Griffiths A, Link MA, Furness CL, Coen DM. (2006). Low-level expression and reversion both contribute to reactivation of herpes simplex virus drug-resistant mutants with mutations on homopolymeric sequences in thymidine kinase. J Virol 2006; 80:6568–74; PMID:16775343
- Pan D, Coen DM. Net -1 frameshifting on a noncanonical sequence in a herpes simplex virus drug-resistant mutant is stimulated by nonstop mRNA. Proc Natl Acad Sci USA 2012; 109:14852–7
- Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2013; 41(D1):D36–D42; PMID:23193287
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids Res 1997; 25(17):3389–402; PMID:9254694
- Fujiyama K, Sasaki T, Ramasoota P, Ikuta K. Genbank (direct submission) 2013; Accession no: AB776842.1, AB776851.1 and AB776857.1
- Fan L, Kadura I, Krebs LE, Hatfield CC, Shaw MM, Frye CC. Improving the efficiency of CHO cell line generation using glutamine synthetase gene knockout cells. Biotechnol Bioeng 2012; 109(4):1007–15; PMID:22068567