
ABSTRACT
Therapeutic monoclonal antibodies (mAbs) are glycoproteins produced by living cell systems. The glycan moieties attached to the proteins can directly affect protein stability, bioactivity, and immunogenicity. Therefore, glycan variants of a glycoprotein product must be adequately analyzed and controlled to ensure product quality. However, the inherent complexity of protein glycosylation poses a daunting analytical challenge. This review provides an update of recent advances in glycan analysis, including the potential utility of lectin-based microarray for high throughput glycan profiling. Emphasis is placed on comparison of the major types of analytics for use in determining unique glycan features such as glycosylation site, glycan structure, and content.
Abbreviations
| AE | = | anion-exchange |
| CE | = | capillary electrophoresis |
| cIEF | = | capillary isoelectric focusing |
| ESI | = | electrospray ionization |
| HILIC | = | hydrophilic interaction liquid chromatography |
| HPAEC-PAD | = | high-performance anion-exchange chromatography with pulsed amperometric detection |
| HPLC | = | high-performance liquid chromatography |
| ICH | = | International Conference on Harmonization |
| IEF | = | isoelectric focusing |
| IEX | = | ion-exchange |
| IM | = | ion-mobility |
| MALDI | = | matrix-assisted laser desorption/ionization |
| MS | = | mass spectrometry |
| MS/MS | = | tandem mass spectrometry |
| PAGE | = | polyacrylamide gel electrophoresis |
| PGC | = | porous graphitic carbon |
| RP | = | reversed-phase |
| SEC | = | size-exclusion chromatography |
Introduction
Many biopharmaceuticals are glycosylated proteins (also named glycoproteins) produced by living cell systems. These include monoclonal antibodies (mAbs) and other recombinant protein products (e.g., fusion proteins, growth factors, cytokines, therapeutic enzymes, and hormones), which are approved (see examples in A–B and supplement I) or under development as treatments for cancer, autoimmune diseases, and other life-threatening conditions. Appropriate glycosylation of a therapeutic protein is critical for product solubility, stability, pharmacokinetics and pharmacodynamics (PK/PD), bioactivity, and safety (e.g., immunogenicity).Citation1 It is well documented that protein glycosylation not only increases protein stability in vitro, but also protects proteins from proteolytic degradation in vivo.Citation2–5 In this regard, non-glycosylated erythropoietin is known to be more susceptible to denaturation or degradation induced by chemicals, pH changes or heating conditions compared to the fully glycosylated form.Citation6
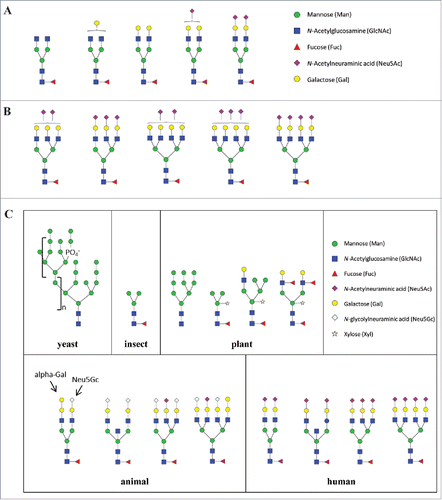
Figure 1. Examples of N-glycans found in recombinant glycoproteins. Shown are typical glycan structures for therapeutic humanized IgG1 mAb (A), recombinant human erythropoietin (B), and N-glycans produced in the commonly used expression systems (C) (derived from Ghaderi D et al. 2012).Citation115
Protein glycosylation can also influence PK/PD properties of therapeutic glycoproteins.Citation2,7 There is evidence that partially glycosylated proteins, which usually contain terminal galactose, have much shorter circulatory lifetimes compared to fully glycosylated proteins with terminal sialic acid. This is mainly due to the binding of galactose with hepatic asialoglycoprotein receptors expressed on hepatocytes, which promotes hepatic clearance of the partially glycosylated protein.Citation8 Other lectin-like receptors with binding specificity to terminal mannose, N-acetyl-glucosamine (GlcNAc) or fucose also contribute to clearance of glycoproteins containing these glycans.Citation2 Additionally, IgG Fc glycosylation is critical to many antibody effector functions through modulating Fc-FcγR interactions.Citation9–12 Human FcγR family includes activating (FcγRIa, FcγRIIa, and FcγRIIIa) and inhibitory (FcγRIIb) receptors. Fc glycosylation plays important roles in modulating antibody binding affinities with FcγRs or C1q on effector cells, and thus affects immune effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) ().Citation13 Optimization of Fc glycosylation has been explored to enhance the ADCC activity of therapeutic mAbs.Citation14
Table 1. Potential impacts of Fc glycosylation on therapeutic mAbs.Citation13
Protein glycosylation is a complex post-translational modification (PTM) involving attachment of glycans at specific sites on a protein, most commonly at Asn (N-linked) or Ser/Thr (O-linked) residues. The N-linked glycosylation occurs at the consensus sequence of Asn-X-Ser/Thr (where X is any amino acid except proline). By contrast, O-linked glycans are usually attached to serine (Ser) or threonine (Thr) residues via an α-O-glycosidic bond formed between N-acetylgalactosamine (GalNAc) and the hydroxyl group (-OH) of Ser/Thr (murine α-GalNAc O-glycans).Citation15 Protein N-glycosylation starts in the endoplasmic reticulum (ER), where a glycan chain (Glc3Man9GlcNAc2) is added to Asn of the Asn-X-Ser/Thr sequon. The glycan chain is then trimmed by various glycosidases and the resultant glycoprotein is transported to the Golgi apparatus for further modifications. Within the Golgi, protein glycans are subjected to a multi-step trimming and modification process, which is catalyzed by various glycan-processing enzymes, resulting in diverse N-glycan structures such as high-mannose, complex, and hybrid glycans.Citation16 Protein O-glycan biosynthesis is initiated in the Golgi by directly transferring GalNAc from UDP-GalNAc to Ser/Thr residues.Citation17 The O-GalNAc precursor undergoes further derivations, thereby producing up to 8 O-GalNAc glycan core structures with 4 (core 1, 2, 3 and 4) of them commonly seen in mammals.Citation18
To add complexity, protein glycosylation is influenced by the type of host cells and fluctuations in fermentation conditions (e.g., media, pH, temperature, agitation).Citation19 Therapeutic glycoproteins can be produced by mammalian cells, yeast strains, plant cells, or genetically modified animals in which each host system has its own unique glycosylation machinery that produces proteins with distinct glycan patterns (C and Supplement I). For instance, mammalian cells (e.g., Chinese hamster ovary cells) are widely used for production of therapeutic glycoproteins containing human-like glycans. However, mammalian cell-expressed proteins may also contain minor non-human glycans such as N-glycolylneuraminic acid (Neu5Gc) and terminal α1-3-Galactose (α-Gal) modification (C). By contrast, yeast strains usually express proteins with high content of mannose (up to 100 units). Other platforms for expressing therapeutic glycoproteins include engineered plant cells and genetically modified animals.Citation20,21 However, these systems may produce proteins with non-human glycan variants such as xylose, Neu5Gc or terminal α-Gal, which are known to be immunogenic.Citation21 Due to the complexity of the glycosylation process, glycoproteins produced by living cell systems usually contain macro- and micro-heterogeneity in terms of glycosylation patterns. Macro-heterogeneity refers to the variability of glycosylation sites and glycan numbers while micro-heterogeneity concerns glycan structural variations at a specific site. Both macro- and micro- heterogeneities contribute to diversity of glycoforms, which share identical amino acid backbones, but may be different in glycosylation sites, glycan contents or structures.Citation22
Analytics for therapeutic protein glycosylation
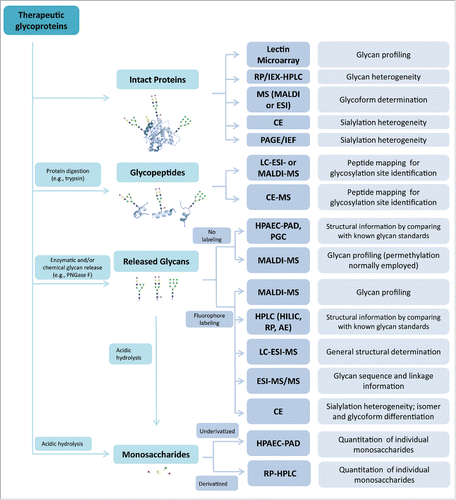
The multiple levels of glycan heterogeneity pose a daunting analytical challenge. In the development of therapeutic glycoproteins, especially biosimilar products,Citation23 glycan analysis usually involves the use of complementary methods for assessing specific glycosylation attributes, including glycosylation site, glycan structure and abundance.Citation24–26 The analytical methods, which include high-performance liquid chromatography (HPLC), capillary electrophoresis (CE), mass spectrometry (MS), isoelectric focusing (IEF), and lectin-based microarray, are grouped and discussed according to their utility in analyzing intact glycoproteins, glycopeptides, released glycans, and monosaccharides ().
Figure 2. Overview of analytics for assessing glycans. The commonly used methods for glycan analysis are divided into 4 main groups per their applications in analyzing intact proteins, glycopeptides, released glycans or monosaccharides (see detail in the text). These methods are usually used in combination to determine glycan profiles, glycan structures and heterogeneity, glycosylation site(s) and the content of specific glycans.
Glycan profiling of intact glycoproteins
Intact glycoproteins can be directly analyzed for general glycan patterns and glycan heterogeneity. In this regard, electrospray ionization - time of flight (ESI-TOF) MS coupled with reversed-phase (RP) HPLC or size-exclusion chromatography (SEC) have been commonly used.Citation27 RPLC-MS provides better chromatographic separation of protein variantsCitation27 and has shown ability in the detection of intact mAb with 10 ppm accuracy (approx. 150,000 ± 1.5 Da), but the procedures require high column temperatures (60–80°C).Citation22,28,29 SEC-MS using either a non-denaturing or denaturing mobile phase is operated at room temperature and produces good quality of mass spectra, but SEC resolution is relatively lower than RP.Citation30-32 The LC-ESI-MS approach has been applied to glycoform profiling of more complex therapeutic proteins such as erythropoietin, which contains one O- and 3 N-glycosylation sites.Citation33 While traditional ESI-TOF-MS analysis of even more heterogeneous glycoproteins such as darbepoetin alfa (known to contain 5 N-glycosylation sites) under denaturing conditions remains challenging, the recently developed Orbitrap ExactiveTM Plus high-resolution mass spectrometer enables accurate determination of highly complex glycan profiles at the native intact protein level. This technology takes advantage of the substantially fewer charges on a given protein when ionized in an aqueous ammonium acetate buffer, where the protein retains more folded “native” structure.Citation34,35 In addition to ESI-MS, matrix-assisted laser desorption ionization (MALDI) MS can be used for fast glycan analysis of small intact proteins, but with a lower mass accuracy.Citation36
To increase the sensitivity of MS analysis of mAbs, one approach has been to reduce a mAb into individual heavy and light chains.Citation28,37 For this purpose, commonly used reducing reagents include dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), β-mercaptoethanol (β-ME) and mercaptoethylamine (MEA).Citation38 Alternatively, a mAb can be cleaved into smaller fragments using selective proteases such as papain, pepsin, IdeZ, and IdeS.Citation39,40 Papain cleaves IgG above the hinge region into 3 fragments, 2 Fab and one Fc, while pepsin cleaves IgG below the hinge region to produce F(ab’)2 fragment and a degraded Fc fragment. IdeZ and IdeS both cleave IgG to yield a F(ab’)2 fragment and an intact Fc fragment.
The heterogeneity of glycoforms can also be analyzed by CE-MS.Citation41–43 Additionally, protein sialylation heterogeneity can be analyzed at the intact glycoprotein level using charge-based electrophoresis such as IEF,Citation41,44 CE,Citation45 capillary isoelectric focusing (cIEF),Citation41,46 or ion-exchange (IEX) chromatography.Citation43,47 These approaches are commonly used for quality control testing;Citation48 however, although IEF and IEX are traditional methods, they are incompatible with MS and time-consuming. CE and cIEF are emerging technologies with attractive features such as high speed, high resolution, and compatibility with MS, but their drawback is that the capillaries tend to adsorb intact proteins.Citation49
Analysis of glycopeptides
Characterization of protein glycosylation site(s) and occupancy is usually achieved at the glycopeptide level using ESI-MS or MALDI-MS.Citation50,51 This approach is also important for characterization of O-glycans because it has been difficult in achieving quantitative O-glycan release (US. Pharmacopoeial Convention (USP) general chapter <1084 >).Citation52,53 Other PTMs can also be detected through peptide analysis. The analytical procedures start with digestion of glycoprotein into suitably sized glycopeptides by a highly specific protease such as trypsin, Lys-C, or Glu-C based on the sequence of the protein as well as the glycan modifications close to the peptide backbone. A combination of multiple proteases may be necessary in specific cases like erythropoietin.Citation54
Following digestion and prior to MS analysis, enrichment of glycopeptides by HPLC is often required in order to overcome ion suppression of glycopeptide signals by regular peptide signals.Citation55 This can be achieved by coupling HPLC with ESI-MS in an online manner, or with MALDI-MS in an offline manner, where the LC-ESI-MS with online separation is a more efficient approach, and thus is most widely used. As for traditional RPLC-MS analysis using C18 columns, detection of low-abundant glycopeptides may be challenging due to the ion suppression of co-eluting glycopeptides with high abundance. Hydrophilic interaction liquid chromatography (HILIC) provides better separation of hydrophilic glycopeptides as well as regular peptides.Citation56 However, glycopeptide isomers with the same mass cannot be distinguished by this approach. A novel nano-LC-MS method was recently introduced to differentiate glycopeptide isomers by using microfluidic chip-based capillaries packed with porous graphitic carbon (PGC) stationary phase, which can resolve isomeric glycopeptides containing very short peptide moieties (as short as 3 residues) produced by unique and nonspecific digestion enzyme pronase E.Citation57 On the other hand, a high-throughput work flow for monitoring Fc-glycosylation during fermentation was tested where the IgGs were purified by protein A immobilized on 96-well plates, and the tryptic glycopeptides were extracted from the plates by HILIC beads followed by ESI-MS analysis without HPLC separation of the extracted glycopeptides.Citation58
Compared to ESI, MALDI is more suitable for rapid glycopeptide profiling because of the relatively simple mass spectra resulting from the singly charged ions, but its drawback is related to degradation of the underivatized sialylated glycopeptides due to in-source and post-source decay,Citation59 which could be avoided by sialic acid derivatization prior to MALDI-MS analysis.Citation60
Tandem MS analysis of glycopeptides allows determination of glycosylation site(s) and glycan composition present on a glycopeptide. The recently developed in-source electron-transfer dissociation (ETD), which predominately cleaves the peptide bonds, can be used to identify the glycosylation site(s) of glycoproteins.Citation61 Classical collision-induced dissociation (CID) method, on the other hand, mainly cleaves the glycan moiety attached to a glycopeptide, which generates information about carbohydrate composition and sequence.Citation50,54 The combination of these methods in one LC-MS experiment could provide an efficient sequencing of glycopeptides.Citation61,62
CE-MS is also a useful tool in determining site-specific glycan micro-heterogeneity of glycoproteins at the glycopeptide level.Citation63 In traditional RPLC-MS, highly hydrophilic peptides may have insufficient interaction with the RP chromatography matrix, and thus get lost during the loading and desalting step. In this case, CE-MS can be a good alternative to detect these small hydrophilic peptides and achieve complete sequence coverage.Citation63,64
Analysis of released glycans
N-linked glycans on a glycoprotein can be released by an amidase such as peptide-N-glycosidase F (PNGase F). O-linked glycans are commonly released through reductive alkaline β-elimination.Citation65 However, it is difficult to achieve complete glycan release and the unreleased glycans are left undetermined. Released glycans can be rapidly profiled by MALDI-TOF MS where glycan permethylation is usually employed to improve ionization efficiency and to stabilize sialylated glycans during simultaneous analysis of neutral and sialylated glycans in the positive-ion mode.Citation27,66,67 Moreover, with linkage-specific sialic acid esterification, not only the sialic acids can be stabilized, but different sialic acid linkages (α2–3 and α2–6) can also be distinguished by MALDI-MS.Citation68
To facilitate HPLC- and MS-based glycan analysis, released glycans are usually fluorescently labeled through reductive amination. The two most commonly used fluorescent tags are 2-aminobenzamide (2-AB) and 2-aminobenzoic acid (2-AA).Citation69 2-AB lacks negative charges and is used as a gold standard for HPLC approaches with extensive database established, but shows low ESI ionization efficiency. 2-AA carries one negative charge, and thus is suitable for CE and MALDI analysis.Citation69 A recent study indicated that procainamide [4-amino-N-(2-diethylaminoethyl) benzamide]-labeled N-glycans show enhanced ESI ionization efficiency and fluorescence intensity comparing to 2-AB-labeled glycans, and thus improve identification of low abundance N-glycans.Citation70 Prior to HPLC and MS analysis, purification steps are usually required to remove excess tags and salts, which include HILIC, RP, PGC, or gel filtration approaches based on the labeling method.Citation69
The labeled glycans can be separated using chromatographic approaches such as HILIC, RP-HPLC, or anion exchange HPLC (AE-HPLC). HILIC has shown better resolution in separating glycans over other HPLC methods.Citation71–73 The elution profile of 2-AB-labeled glycans is compared against a 2-AB-labeled dextran ladder (glucose homopolymer), yielding an estimation of the number of glucose units of each species.Citation67,74 This information is then used to calculate the abundance of individual monosaccharides, which allows prediction of glycan structure.Citation75 On the other hand, N-glycans without fluorescence-labeling can be separated by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) or PGC chromatography,Citation74,76 but with limited robustness and reproducibility.Citation73
Released glycans, upon fluorescence labeling, can also be determined using CE at high sensitivity and resolution. The fluorescent tag 8-aminopyrene-1,3,6-trisulfonate (APTS) is widely used for CE analysis of oligosaccharides;Citation77,78 sometimes other labeling reagents such as 2-AA are also used.Citation79 This approach offers a unique ability to differentiate isomeric glycan structures.Citation77–81 However, only a limited number of glycan standards is available for use in making the assignment of a glycan structure to a corresponding CE peak signal. In this case, sequential exoglycosidase digestion of oligosaccharide followed by CE analysis has the potential to provide detailed carbohydrate sequence information.Citation81,82 When hyphenated with MS, CE-MS proves to be useful in peptide mapping and glycoform characterization;Citation42,64,83–85 however, its application in oligosaccharide analysis is somewhat limited.Citation86,87 A recent study compared 7 orthogonal methods (e.g., HILIC, CE, and HPAEC-PAD) in analyzing released Fc glycans, which showed similar results for glycan patterns and content.Citation88
Glycan structure assessment is challenging due to the isomeric and branched nature of oligosaccharides. The traditional approach involves the use of exoglycosidases to selectively and sequentially release terminal monosaccharides, which produces trimmed glycans to be analyzed by HILIC, CE or MALDI-MS. The structure of the originating glycans can be postulated according to respective shifts in glucose units, migration time and mass following each enzymatic digestion.Citation89 These approaches are valuable for new glycan structure analysis, but they involve a lengthy procedure and require highly pure enzymes to achieve cleavage at the desired sites. Recently, LC-ESI-CID-MS/MS was applied to glycan sequencing, which showed improved speed and sensitivity.Citation90 To determine the linkages in a glycan by MS/MS, cleavage across the glycosidic ring (cross-ring fragmentation) is required, which can be done by ESI-QTOF MS in a negative-ion mode under low energy CID conditions.Citation91–93 However, MS generally cannot resolve the structural isomers, or determine the 3-dimensional structure of the glycans. To fill this gap, nuclear magnetic resonance (NMR) and X-ray crystallography are employed.Citation94,95 In fact, NMR remains the only technique that can determine anomericity and linkage information in a new glycan structure,Citation67 but it requires relatively large amounts of purified glycans. A newly developed ion-mobility (IM) MS technique, which separates ions in gas phase based on size, shape, and charge, was shown to effectively detect glycan isomers.Citation96
Analysis of monosaccharides
Quantification of released monosaccharide provides information on the content of a specific-terminal monosaccharide such as sialic acid, mannose-6-phosphate (M6P), GlcNAc, or O-linked monosaccharides.Citation97–99 Sialic acid residues can be selectively released by mild acid hydrolysis or by an enzyme neuraminidase (USP<1084>). Released sialic acids are normally quantitated by HPAEC-PAD without derivatization.Citation100 The absolute amount of sialic acids can be determined by HPAEC-PAD using commercially available sialic acid standards,Citation25 e.g., 3-deoxy-D-glycero-D-galacto-nonulosonic acid (USP<1084>). Alternatively, released sialic acids can be labeled with a fluorescent tag 1,2-diamino-4,5-methylenedioxybenzene followed by RP-HPLC analysis.Citation101
To analyze the composition of monosaccharides other than sialic acids, these monosaccharides are commonly released by strong acid hydrolysis and separated by HPLC.Citation102 Specifically, trifluoracetic acid (TFA) is widely used for hydrolysis of glycans after selective release of sialic acids, and released amino monosaccharides are normally re-N-acetylated.Citation67,103 Common problems associated with acid hydrolysis are incomplete glycan cleavage and instability of the released monosaccharides under the acidic conditions.Citation102 Identification and quantification of non-sialic acid monosaccharides are normally conducted by HPAEC-PAD without derivatization, and 2-deoxy-glucose can be used as standard for these neutral monosaccharides (USP<1084>).Citation104
Lectin array-based glycan analysis
Lectins, which are a group of glycan binding proteins (GBPs) that are naturally occurring in plants and many other species, are known to selectively interact with glycan molecules either released or attached to a peptide backbone. This unique property has been explored in the development of analytics for glycan determination. Several relevant platforms have been reported in which lectin-based microarray has demonstrated a utility in capturing glycan profiles of therapeutic glycoproteins.Citation105–109
In the design of a lectin microarray, specified lectins are immobilized onto a solid phase (e.g., glass chips) that is activated either chemically (e.g., epoxy, NHS ester, gold or amino group) or biochemically (e.g., streptavidin).Citation105,107 After lectin coating, residual active groups on the solid surface are blocked by appropriate agents (e.g., Tris buffer or bovine serum albumin). When a glycoprotein is applied onto the lectin chip, the binding events detected at specific lectin spots provide a picture of glycan variants that are likely present in the sample. In conventional microarray analysis, washing procedures are required to remove unbound glycoproteins from the lectin chips. Such operation procedures will disrupt the steady-state interactions between lectins and glycans because lectin-glycan binding is generally weak (Kd∼10−4–10–7 M) compared to high binding affinity of antibody-antigen and biotin-streptavidin complexes. The recently developed evanescent-field activated fluorescence system allows real-time detection of lectin-glycan interactions at equilibrium without the need of washing procedures.Citation106,110,111 Such a platform has been used in assessing glycan variants of purified glycoproteins, crude extracted membrane proteins,Citation112 and live cell plasma membrane proteins.Citation113 A recent study compared 3 chromatographic methods (HPAEC-PAD, 2-AA HILIC, 2-AB HILIC) and a lectin microarray method for glycan profiling of a therapeutic mAb.Citation114 The lectin-based assays identified “glycan structural classes” in the 4 mAb lots, which were generally consistent with those detected by other classical methods.
Our laboratory has recently tested over a dozen Food and Drug Administration-approved therapeutic glycoproteins (including 9 mAbs) using commercial lectin chips that contain 45 distinct lectins (GlycoTechnica Ltd.). The derived glycan profiles (e.g., glycan variants and relative content) are generally consistent with the known glycosylation patterns of each product (data not shown). For example, a recombinant human glycoprotein shows strong binding signal at phytohemagglutinin-L (PHAL)-coated spots, confirming the presence of tri-/tetra-antennary glycan structures in the sample (A). No binding signal was detected for a recombinant IgG1 monoclonal antibody that is known to be deficient of such glycan structure. In another case, we found that the lectin-based assay was able to distinguish terminal sialylation variants (e.g., α2–3- vs. α2–6-sialylation) (B). Our data show promise for lectin microarrays in profiling glycan variants that are commonly present in therapeutic proteins. Compared to MS-based methods, which usually involve multiple sample processing steps, a lectin microarray directly works on intact glycoproteins with only minimal alteration to a testing sample (e.g., fluorescent labeling). The lectin microarray, when coupled with a sophisticated detection system, appears to provide a high throughput platform for rapid screening of glycan profiles of therapeutic proteins. However, the lectin-based microarray still faces major challenges before being adopted as a tool for characterization of therapeutic glycoproteins. In this regard, most commercial lectin microarrays utilize lectins from natural sources (e.g., plants) that are expected to display weak binding affinities to a spectrum of glycans. This phenomenon complicates interpretation of the detected binding events with a specific lectin molecule. Nonetheless, the lectin microarray platform appears to allow high throughput screening of the presence or absence of specific glycan variants in glycoprotein samples.
Figure 3. Lectin microarray analysis of glycans. (A). A recombinant human glycoprotein showed strong binding signal at PHAL coated spots (left panel), confirming the presence of tri- and/or tetra-antennary glycan structures in the testing sample. By contrast, no binding signal was detected for another mAb lacking such glycans (right panel); (B) Two therapeutic mAbs, which were known to contain α2–3- or α2–6-sialylation, respectively produced signals at α2–3-sialic acid binding lectin MAL (left panel) and α2–6-sialic acid binding lectins (SNA, SSA, TJA-I)(right panel).

Perspectives
The complexity of protein glycosylation poses a daunting analytical challenge in the development of therapeutic glycoproteins (e.g., mAbs). Orthogonal methods are required to characterize specific features: 1) the carbohydrate content (neutral sugars, amino sugars, and sialic acids); 2) the structure of the carbohydrate chains, the oligosaccharide pattern (antennary profile), and 3) the glycosylation site(s) (ICH guidance Q6B). The selection of appropriate methods for a specific glycoprotein will depend on the purpose of testing and the ability of individual method ( and ). IEF, IEX, or CE alone or in combination is commonly used to monitor heterogeneity in sialic acids on intact glycoproteins. HPLC is widely used to quantify the amounts of released oligosaccharides. MS coupled with HPLC remains a powerful tool in the characterization of glycosylation site(s) occupancy and carbohydrate structures. However, those oligosaccharide-based methods involve release of glycans from a glycoprotein. When conducting these assays, cautions must be taken to ensure that glycans are effectively released and recovered using appropriate procedures. Ideally, glycan analysis should involve procedures that only minimally alter the test samples. In this regard, lectin microarray appears to directly measure glycan variations in an intact protein without the need of clipping glycans from the protein backbone. Such a platform could be used as a complementary tool for characterization of protein glycosylation.
Table 2. Comparison of analytical methods for glycoprotein characterization.
References
- Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci 2009; 30:356–62; PMID:19552968; http://dx.doi.org/10.1016/j.tips.2009.04.007
- Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs 2010; 24:9–21; PMID:20055529; http://dx.doi.org/10.2165/11530550-000000000-00000
- Sola RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci 2009; 98:1223–45; PMID:18661536; http://dx.doi.org/10.1002/jps.21504
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 2011; 3:568–76; PMID:22123061; http://dx.doi.org/10.4161/mabs.3.6.17922
- Alsenaidy MA, Okbazghi SZ, Kim JH, Joshi SB, Middaugh CR, Tolbert TJ, Volkin DB. Physical stability comparisons of IgG1-Fc variants: effects of N-glycosylation site occupancy and Asp/Gln residues at site Asn 297. J Pharm Sci 2014; 103:1613–27; PMID:24740840; http://dx.doi.org/10.1002/jps.23975
- Narhi LO, Arakawa T, Aoki KH, Elmore R, Rohde MF, Boone T, Strickland TW. The effect of carbohydrate on the structure and stability of erythropoietin. J Biol Chem 1991; 266:23022–6; PMID:1744097
- Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet 2012; 13:307–35; PMID:22970722; http://dx.doi.org/10.1146/annurev-genom-090711-163739
- Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J Biol Chem 1971; 246:1461–7; PMID:5545089
- Stadlmann J, Pabst M, Altmann F. Analytical and Functional Aspects of Antibody Sialylation. J Clin Immunol 2010; 30 Suppl 1:S15–9; PMID:20390325
- Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci U S A 2013; 110:9868–72; PMID:23697368; http://dx.doi.org/10.1073/pnas.1307864110
- Bohm S, Schwab I, Lux A, Nimmerjahn F. The role of sialic acid as a modulator of the anti-inflammatory activity of IgG. Semin Immunopathol 2012; 34:443–53; PMID:22437760; http://dx.doi.org/10.1007/s00281-012-0308-x
- Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun 2012; 12:13; PMID:22896758
- Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015; 25(12):1325–34; PMID:26263923
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs 2012; 4:419–25; PMID:22699226; http://dx.doi.org/10.4161/mabs.20996
- Schachter H. The joys of HexNAc. The synthesis and function of N- and O-glycan branches. Glycoconj J 2000; 17:465–83; PMID:11421343; http://dx.doi.org/10.1023/A:1011010206774
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 1985; 54:631–64; PMID:3896128; http://dx.doi.org/10.1146/annurev.bi.54.070185.003215
- Ten Hagen KG, Fritz TA, Tabak LA. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2003; 13:1R–16R; PMID:12634319; http://dx.doi.org/10.1093/glycob/cwg007
- Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol 2008; 70:431–57; PMID:17850209; http://dx.doi.org/10.1146/annurev.physiol.70.113006.100659
- Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009; 19:936–49; PMID:19494347; http://dx.doi.org/10.1093/glycob/cwp079
- Ma JK, Drake PM, Christou P. The production of recombinant pharmaceutical proteins in plants. Nat Rev Genet 2003; 4:794–805; PMID:14526375; http://dx.doi.org/10.1038/nrg1177
- Houdebine LM. Production of pharmaceutical proteins by transgenic animals. Comp Immunol Microbiol Infect Dis 2009; 32:107–21; PMID:18243312; http://dx.doi.org/10.1016/j.cimid.2007.11.005
- Beck A, Debaene F, Diemer H, Wagner-Rousset E, Colas O, Dorsselaer AV, Cianferani S. Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J Mass Spectrom 2015; 50:285–97; PMID:25800010; http://dx.doi.org/10.1002/jms.3554
- Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs 2013; 5:621–3; PMID:23924791; http://dx.doi.org/10.4161/mabs.25864
- Beck A, Wagner-Rousset E, Ayoub D, Van DA, Sanglier-Cianferani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715–36; PMID:23134362; http://dx.doi.org/10.1021/ac3032355
- Lingg N, Zhang P, Song Z, Bardor M. The sweet tooth of biopharmaceuticals: importance of recombinant protein glycosylation analysis. Biotechnol J 2012; 7:1462–72; PMID:22829536; http://dx.doi.org/10.1002/biot.201200078
- Dotz V, Haselberg R, Shubhakar A, Kozak RP, Falck D, Rombouts Y, Reusch D, Somsen GW, Fernandes DL, Wuhrer M. Mass spectrometry for glycosylation analysis of biopharmaceuticals. TrAC Trends Analyt Chem 2015; 73:1–9; http://dx.doi.org/10.1016/j.trac.2015.04.024
- Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147–76; PMID:18720354; http://dx.doi.org/10.1002/mas.20190
- Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, Chen W, Skilton SJ, Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2:379–94; PMID:20458189; http://dx.doi.org/10.4161/mabs.11986
- Ayoub D, Jabs W, Resemann A, Evers W, Evans C, Main L, Baessmann C, Wagner-Rousset E, Suckau D, Beck A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 2013; 5:699–710; PMID:23924801; http://dx.doi.org/10.4161/mabs.25423
- Xu CF, Zang L, Weiskopf A. Size-exclusion chromatography-mass spectrometry with m-nitrobenzyl alcohol as post-column additive for direct characterization of size variants of monoclonal antibodies. J Chromatogr B Analyt Technol Biomed Life Sci 2014; 960:230–8; PMID:24835510; http://dx.doi.org/10.1016/j.jchromb.2014.04.023
- Liu H, Gaza-Bulseco G, Chumsae C. Analysis of reduced monoclonal antibodies using size exclusion chromatography coupled with mass spectrometry. J Am Soc Mass Spectrom 2009; 20:2258–64; PMID:19786356; http://dx.doi.org/10.1016/j.jasms.2009.08.015
- Brady LJ, Valliere-Douglass J, Martinez T, Balland A. Molecular mass analysis of antibodies by on-line SEC-MS. J Am Soc Mass Spectrom 2008; 19:502–9; PMID:18258452; http://dx.doi.org/10.1016/j.jasms.2007.12.006
- Harazono A, Hashii N, Kuribayashi R, Nakazawa S, Kawasaki N. Mass spectrometric glycoform profiling of the innovator and biosimilar erythropoietin and darbepoetin by LC/ESI-MS. J Pharm Biomed Anal 2013; 83:65–74; PMID:23708432; http://dx.doi.org/10.1016/j.jpba.2013.04.031
- Rosati S, Rose RJ, Thompson NJ, vanGÇàDuijn E, Damoc E, Denisov E, Makarov A, Heck AJR. Exploring an Orbitrap Analyzer for the Characterization of Intact Antibodies by Native Mass Spectrometry. Angewandte Chemie International Edition 2012; 51:12992–6; http://dx.doi.org/10.1002/anie.201206745
- Rosati S, van den Bremer ET, Schuurman J, Parren PW, Kamerling JP, Heck AJ. In-depth qualitative and quantitative analysis of composite glycosylation profiles and other micro-heterogeneity on intact monoclonal antibodies by high-resolution native mass spectrometry using a modified Orbitrap. MAbs 2013; 5:917–24; PMID:23995615; http://dx.doi.org/10.4161/mabs.26282
- Thompson NJ, Rosati S, Rose RJ, Heck AJ. The impact of mass spectrometry on the study of intact antibodies: from post-translational modifications to structural analysis. Chem Commun (Camb) 2013; 49:538–48; PMID:23183499; http://dx.doi.org/10.1039/C2CC36755F
- Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim DI, Lee SY, Chang SJ. Physicochemical characterization of Remsima. MAbs 2014; 6:1163–77; PMID:25517302; http://dx.doi.org/10.4161/mabs.32221
- Crivianu-Gaita V, Romaschin A, Thompson M. High efficiency reduction capability for the formation of Fab׳ antibody fragments from F(ab)2 units. Biochem Biophysics Rep 2015; 2:23–8; http://dx.doi.org/10.1016/j.bbrep.2015.04.004
- Brezski RJ, Jordan RE. Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs 2010; 2:212–20; PMID:20400859; http://dx.doi.org/10.4161/mabs.2.3.11780
- Janin-Bussat MC, Tonini L, Huillet C, Colas O, Klinguer-Hamour C, Corvaia N, Beck A. Cetuximab Fab and Fc N-glycan fast characterization using IdeS digestion and liquid chromatography coupled to electrospray ionization mass spectrometry. Methods Mol Biol 2013; 988:93–113; PMID:23475716; http://dx.doi.org/10.1007/978-1-62703-327-5_7
- Zhao SS, Chen DD. Applications of capillary electrophoresis in characterizing recombinant protein therapeutics. Electrophoresis 2014; 35:96–108; PMID:24123141; http://dx.doi.org/10.1002/elps.201300372
- Haselberg R, de Jong GJ, Somsen GW. Low-flow sheathless capillary electrophoresis-mass spectrometry for sensitive glycoform profiling of intact pharmaceutical proteins. Anal Chem 2013; 85:2289–96; PMID:23323765; http://dx.doi.org/10.1021/ac303158f
- Thakur D, Rejtar T, Karger BL, Washburn NJ, Bosques CJ, Gunay NS, Shriver Z, Venkataraman G. Profiling the glycoforms of the intact α subunit of recombinant human chorionic gonadotropin by high-resolution capillary electrophoresis-mass spectrometry. Anal Chem 2009; 81:8900–7; PMID:19817480; http://dx.doi.org/10.1021/ac901506p
- Reichel C, Thevis M. Gel electrophoretic methods for the analysis of biosimilar pharmaceuticals using the example of recombinant erythropoietin. Bioanalysis 2013; 5:587–602; PMID:23425274; http://dx.doi.org/10.4155/bio.13.9
- de Kort BJ, de Jong GJ, Somsen GW. Profiling of erythropoietin products by capillary electrophoresis with native fluorescence detection. Electrophoresis 2012; 33:2996–3001; PMID:23002011; http://dx.doi.org/10.1002/elps.201200303
- Suba D, Urbanyi Z, Salgo A. Capillary isoelectric focusing method development and validation for investigation of recombinant therapeutic monoclonal antibody. J Pharm Biomed Anal 2015; 114:53–61; PMID:26025812; http://dx.doi.org/10.1016/j.jpba.2015.04.037
- Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011; 29:310–2; PMID:21478841; http://dx.doi.org/10.1038/nbt.1839
- Federici M, Lubiniecki A, Manikwar P, Volkin DB. Analytical lessons learned from selected therapeutic protein drug comparability studies. Biologicals 2013; 41:131–47; PMID:23146362; http://dx.doi.org/10.1016/j.biologicals.2012.10.001
- Staub A, Guillarme D, Schappler J, Veuthey JL, Rudaz S. Intact protein analysis in the biopharmaceutical field. J Pharm Biomed Anal 2011; 55:810–22; PMID:21334842; http://dx.doi.org/10.1016/j.jpba.2011.01.031
- Bongers J, Devincentis J, Fu J, Huang P, Kirkley DH, Leister K, Liu P, Ludwig R, Rumney K, Tao L, et al. Characterization of glycosylation sites for a recombinant IgG1 monoclonal antibody and a CTLA4-Ig fusion protein by liquid chromatography-mass spectrometry peptide mapping. J Chromatogr A 2011; 1218:8140–9; PMID:21978954; http://dx.doi.org/10.1016/j.chroma.2011.08.089
- Gong B, Cukan M, Fisher R, Li H, Stadheim TA, Gerngross T. Characterization of N-linked glycosylation on recombinant glycoproteins produced in Pichia pastoris using ESI-MS and MALDI-TOF. Methods Mol Biol 2009; 534:213–23; PMID:19277549
- Huang LJ, Lin JH, Tsai JH, Chu YY, Chen YW, Chen SL, Chen SH. Identification of protein O-glycosylation site and corresponding glycans using liquid chromatography-tandem mass spectrometry via mapping accurate mass and retention time shift. J Chromatogr A 2014; 1371:136–45; PMID:25456591; http://dx.doi.org/10.1016/j.chroma.2014.10.046
- Houel S, Hilliard M, Yu YQ, McLoughlin N, Martin SM, Rudd PM, Williams JP, Chen W. N- and O-glycosylation analysis of etanercept using liquid chromatography and quadrupole time-of-flight mass spectrometry equipped with electron-transfer dissociation functionality. Anal Chem 2014; 86:576–84; PMID:24308717; http://dx.doi.org/10.1021/ac402726h
- Kolarich D, Jensen PH, Altmann F, Packer NH. Determination of site-specific glycan heterogeneity on glycoproteins. Nat Protoc 2012; 7:1285–98; PMID:22678432; http://dx.doi.org/10.1038/nprot.2012.062
- Ongay S, Boichenko A, Govorukhina N, Bischoff R. Glycopeptide enrichment and separation for protein glycosylation analysis. J Sep Sci 2012; 35:2341–72; PMID:22997027; http://dx.doi.org/10.1002/jssc.201200434
- Desaire H. Glycopeptide analysis, recent developments and applications. Mol Cell Proteomics 2013; 12:893–901; PMID:23389047; http://dx.doi.org/10.1074/mcp.R112.026567
- Hua S, Nwosu CC, Strum JS, Seipert RR, An HJ, Zivkovic AM, German JB, Lebrilla CB. Site-specific protein glycosylation analysis with glycan isomer differentiation. Anal Bioanal Chem 2012; 403:1291–302; PMID:21647803; http://dx.doi.org/10.1007/s00216-011-5109-x
- Reusch D, Haberger M, Selman MH, Bulau P, Deelder AM, Wuhrer M, Engler N. High-throughput work flow for IgG Fc-glycosylation analysis of biotechnological samples. Anal Biochem 2013; 432:82–9; PMID:23026777; http://dx.doi.org/10.1016/j.ab.2012.09.032
- Harrison R, Hitchen PG, Panico M, Morris HR, Mekhaiel D, Pleass RJ, Dell A, Hewitt JE, Haslam SM. Glycoproteomic characterization of recombinant mouse α-dystroglycan. Glycobiology 2012; 22:662–75; PMID:22241827; http://dx.doi.org/10.1093/glycob/cws002
- Reusch D, Haberger M, Falck D, Peter B, Maier B, Gassner J, Hook M, Wagner K, Bonnington L, Bulau P, et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles-Part 2: Mass spectrometric methods. MAbs 2015; 7:732–42; PMID:25996192; http://dx.doi.org/10.1080/19420862.2015.1045173
- Scott NE, Parker BL, Connolly AM, Paulech J, Edwards AV, Crossett B, Falconer L, Kolarich D, Djordjevic SP, Hojrup P, et al. Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol Cell Proteomics 2011; 10:M000031-MMCP201; PMID:20360033; http://dx.doi.org/10.1074/mcp.M000031-MCP201
- Jiang H, Wu SL, Karger BL, Hancock WS. Characterization of the glycosylation occupancy and the active site in the follow-on protein therapeutic: TNK-tissue plasminogen activator. Anal Chem 2010; 82:6154–62; PMID:20552988; http://dx.doi.org/10.1021/ac100956x
- Gahoual R, Burr A, Busnel JM, Kuhn L, Hammann P, Beck A, Francois YN, Leize-Wagner E. Rapid and multi-level characterization of trastuzumab using sheathless capillary electrophoresis-tandem mass spectrometry. MAbs 2013; 5:479–90; PMID:23563524; http://dx.doi.org/10.4161/mabs.23995
- Whitmore CD, Gennaro LA. Capillary electrophoresis-mass spectrometry methods for tryptic peptide mapping of therapeutic antibodies. Electrophoresis 2012; 33:1550–6; PMID:22736356; http://dx.doi.org/10.1002/elps.201200066
- Wada Y, Dell A, Haslam SM, Tissot B, Canis K, Azadi P, Backstrom M, Costello CE, Hansson GC, Hiki Y, et al. Comparison of methods for profiling O-glycosylation: Human Proteome Organisation Human Disease Glycomics/Proteome Initiative multi-institutional study of IgA1. Mol Cell Proteomics 2010; 9:719–27; PMID:20038609; http://dx.doi.org/10.1074/mcp.M900450-MCP200
- Sanchez-De Melo I, Grassi P, Ochoa F, Bolivar J, Garcia-Cozar FJ, Duran-Ruiz MC. N-glycosylation profile analysis of Trastuzumab biosimilar candidates by Normal Phase Liquid Chromatography and MALDI-TOF MS approaches. J Proteomics 2015; 127:225–33; PMID:25907685; http://dx.doi.org/10.1016/j.jprot.2015.04.012
- Marino K, Bones J, Kattla JJ, Rudd PM. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol 2010; 6:713–23; PMID:20852609; http://dx.doi.org/10.1038/nchembio.437
- Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal Chem 2014; 86:5784–93; PMID:24831253; http://dx.doi.org/10.1021/ac500335t
- Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M. Glycan labeling strategies and their use in identification and quantification. Anal Bioanal Chem 2010; 397:3457–81; PMID:20225063; http://dx.doi.org/10.1007/s00216-010-3532-z
- Kozak RP, Tortosa CB, Fernandes DL, Spencer DI. Comparison of procainamide and 2-aminobenzamide labeling for profiling and identification of glycans by liquid chromatography with fluorescence detection coupled to electrospray ionization-mass spectrometry. Anal Biochem 2015; 486:38–40; PMID:26079702; http://dx.doi.org/10.1016/j.ab.2015.06.006
- Melmer M, Stangler T, Schiefermeier M, Brunner W, Toll H, Rupprechter A, Lindner W, Premstaller A. HILIC analysis of fluorescence-labeled N-glycans from recombinant biopharmaceuticals. Anal Bioanal Chem 2010; 398:905–14; PMID:20640408; http://dx.doi.org/10.1007/s00216-010-3988-x
- Shang TQ, Saati A, Toler KN, Mo J, Li H, Matlosz T, Lin X, Schenk J, Ng CK, Duffy T, et al. Development and application of a robust N-glycan profiling method for heightened characterization of monoclonal antibodies and related glycoproteins. J Pharm Sci 2014; 103:1967–78; PMID:24840237; http://dx.doi.org/10.1002/jps.24004
- Melmer M, Stangler T, Premstaller A, Lindner W. Comparison of hydrophilic-interaction, reversed-phase and porous graphitic carbon chromatography for glycan analysis. J Chromatogr A 2011; 1218:118–23; PMID:21122866; http://dx.doi.org/10.1016/j.chroma.2010.10.122
- Pabst M, Altmann F. Glycan analysis by modern instrumental methods. Proteomics 2011; 11:631–43; PMID:21241022; http://dx.doi.org/10.1002/pmic.201000517
- Royle L, Dwek RA, Rudd PM. Determining the structure of oligosaccharides N- and O-linked to glycoproteins. Curr Protoc Protein Sci 2006; 12:12.6.1-12.6.45; Chapter 12:Unit; PMID:18429295
- Grey C, Edebrink P, Krook M, Jacobsson SP. Development of a high performance anion exchange chromatography analysis for mapping of oligosaccharides. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:1827–32; PMID:19482525; http://dx.doi.org/10.1016/j.jchromb.2009.05.003
- Guttman A. High-resolution carbohydrate profiling by capillary gel electrophoresis. Nature 1996; 380:461–2; PMID:8602248; http://dx.doi.org/10.1038/380461a0
- Guttman A, Chen FT, Evangelista RA. Separation of 1-aminopyrene-3,6,8-trisulfonate-labeled asparagine-linked fetuin glycans by capillary gel electrophoresis. Electrophoresis 1996; 17:412–7; PMID:8900952; http://dx.doi.org/10.1002/elps.1150170221
- Kamoda S, Ishikawa R, Kakehi K. Capillary electrophoresis with laser-induced fluorescence detection for detailed studies on N-linked oligosaccharide profile of therapeutic recombinant monoclonal antibodies. J Chromatogr A 2006; 1133:332–9; PMID:16945378; http://dx.doi.org/10.1016/j.chroma.2006.08.028
- Mechref Y, Muzikar J, Novotny MV. Comprehensive assessment of N-glycans derived from a murine monoclonal antibody: a case for multimethodological approach. Electrophoresis 2005; 26:2034–46; PMID:15841499; http://dx.doi.org/10.1002/elps.200410345
- Ma S, Nashabeh W. Carbohydrate analysis of a chimeric recombinant monoclonal antibody by capillary electrophoresis with laser-induced fluorescence detection. Anal Chem 1999; 71:5185–92; PMID:10575965; http://dx.doi.org/10.1021/ac990376z
- Callewaert N, Van VH, Van HA, Laroy W, Delanghe J, Contreras R. Noninvasive diagnosis of liver cirrhosis using DNA sequencer-based total serum protein glycomics. Nat Med 2004; 10:429–34; PMID:15152612; http://dx.doi.org/10.1038/nm1006
- Gennaro LA, Salas-Solano O, Ma S. Capillary electrophoresis-mass spectrometry as a characterization tool for therapeutic proteins. Anal Biochem 2006; 355:249–58; PMID:16712766; http://dx.doi.org/10.1016/j.ab.2006.04.002
- Balaguer E, Demelbauer U, Pelzing M, Sanz-Nebot V, Barbosa J, Neususs C. Glycoform characterization of erythropoietin combining glycan and intact protein analysis by capillary electrophoresis - electrospray - time-of-flight mass spectrometry. Electrophoresis 2006; 27:2638–50; PMID:16817164; http://dx.doi.org/10.1002/elps.200600075
- Taichrib A, Pioch M, Neususs C. Multivariate statistics for the differentiation of erythropoietin preparations based on intact glycoforms determined by CE-MS. Anal Bioanal Chem 2012; 403:797–805; PMID:22430131; http://dx.doi.org/10.1007/s00216-012-5924-8
- Gennaro LA, Salas-Solano O. On-line CE-LIF-MS technology for the direct characterization of N-linked glycans from therapeutic antibodies. Anal Chem 2008; 80:3838–45; PMID:18426228http://dx.doi.org/10.1021/ac800152h
- Balaguer E, Neususs C. Glycoprotein characterization combining intact protein and glycan analysis by capillary electrophoresis-electrospray ionization-mass spectrometry. Anal Chem 2006; 78:5384–93; PMID:16878873; http://dx.doi.org/; http://dx.doi.org/10.1021/ac060376g
- Reusch D, Haberger M, Maier B, Maier M, Kloseck R, Zimmermann B, Hook M, Szabo Z, Tep S, Wegstein J, et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles–part 1: separation-based methods. MAbs 2015; 7:167–79; PMID:25524468; http://dx.doi.org/10.4161/19420862.2014.986000
- Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal Biochem 2007; 364:8–18; PMID:17362871; http://dx.doi.org/10.1016/j.ab.2007.01.023
- Jensen PH, Karlsson NG, Kolarich D, Packer NH. Structural analysis of N- and O-glycans released from glycoproteins. Nat Protoc 2012; 7:1299–310; PMID:22678433; http://dx.doi.org/10.1038/nprot.2012.063
- Harvey DJ. Fragmentation of negative ions from carbohydrates: part 1. Use of nitrate and other anionic adducts for the production of negative ion electrospray spectra from N-linked carbohydrates. J Am Soc Mass Spectrom 2005; 16:622–30; PMID:15862764; http://dx.doi.org/10.1016/j.jasms.2005.01.004
- Harvey DJ. Fragmentation of negative ions from carbohydrates: part 2. Fragmentation of high-mannose N-linked glycans. J Am Soc Mass Spectrom 2005; 16:631–46; PMID:15862765; http://dx.doi.org/10.1016/j.jasms.2005.01.005
- Harvey DJ. Fragmentation of negative ions from carbohydrates: part 3. Fragmentation of hybrid and complex N-linked glycans. J Am Soc Mass Spectrom 2005; 16:647–59; PMID:15862766; http://dx.doi.org/10.1016/j.jasms.2005.01.006
- Wormald MR, Petrescu AJ, Pao YL, Glithero A, Elliott T, Dwek RA. Conformational studies of oligosaccharides and glycopeptides: complementarity of NMR, X-ray crystallography, and molecular modelling. Chem Rev 2002; 102:371–86; PMID:11841247; http://dx.doi.org/10.1021/cr990368i
- Halbeek H. Structural Characterization of the Carbohydrate Moieties of Glycoproteins by High-Resolution (1)H-NMR Spectroscopy. Methods Mol Biol 1993; 17:115–48; PMID:21400136
- Yamaguchi Y, Nishima W, Re S, Sugita Y. Confident identification of isomeric N-glycan structures by combined ion mobility mass spectrometry and hydrophilic interaction liquid chromatography. Rapid Commun Mass Spectrom 2012; 26:2877–84; PMID:23136018; http://dx.doi.org/10.1002/rcm.6412
- Tangvoranuntakul P, Gagneux P, Diaz S, Bardor M, Varki N, Varki A, Muchmore E. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci U S A 2003; 100:12045–50; PMID:14523234; http://dx.doi.org/10.1073/pnas.2131556100
- Orvisky E, Stubblefield B, Long RT, Martin BM, Sidransky E, Krasnewich D. Phosphomannomutase activity in congenital disorders of glycosylation type Ia determined by direct analysis of the interconversion of mannose-1-phosphate to mannose-6-phosphate by high-pH anion-exchange chromatography with pulsed amperometric detection. Anal Biochem 2003; 317:12–8; PMID:12729595; http://dx.doi.org/10.1016/S0003-2697(03)00109-X
- Keck R, Nayak N, Lerner L, Raju S, Ma S, Schreitmueller T, Chamow S, Moorhouse K, Kotts C, Jones A. Characterization of a complex glycoprotein whose variable metabolic clearance in humans is dependent oN-terminal N-acetylglucosamine content. Biologicals 2008; 36:49–60; PMID:17728143; http://dx.doi.org/10.1016/j.biologicals.2007.05.004
- Rohrer JS, Thayer J, Weitzhandler M, Avdalovic N. Analysis of the N-acetylneuraminic acid and N-glycolylneuraminic acid contents of glycoproteins by high-pH anion-exchange chromatography with pulsed amperometric detection. Glycobiology 1998; 8:35–43; PMID:9451012; http://dx.doi.org/10.1093/glycob/8.1.35
- Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol 2010; 28:863–7; PMID:20657583; http://dx.doi.org/10.1038/nbt.1651
- Higgins E. Carbohydrate analysis throughout the development of a protein therapeutic. Glycoconj J 2010; 27:211–25; PMID:19888650; http://dx.doi.org/10.1007/s10719-009-9261-x
- Manzi A. Acid hydrolysis for release of monosaccharides. Curr Protoc Mol Biol 2001; 17:17.16.1-17.16.11; Chapter 17:Unit17
- Townsend RR, Hardy MR. Analysis of glycoprotein oligosaccharides using high-pH anion exchange chromatography. Glycobiology 1991; 1:139–47; PMID:1823157; http://dx.doi.org/10.1093/glycob/1.2.139
- Angeloni S, Ridet JL, Kusy N, Gao H, Crevoisier F, Guinchard S, Kochhar S, Sigrist H, Sprenger N. Glycoprofiling with micro-arrays of glycoconjugates and lectins. Glycobiology 2005; 15:31–41; PMID:15342550; http://dx.doi.org/10.1093/glycob/cwh143
- Kuno A, Uchiyama N, Koseki-Kuno S, Ebe Y, Takashima S, Yamada M, Hirabayashi J. Evanescent-field fluorescence-assisted lectin microarray: a new strategy for glycan profiling. Nat Methods 2005; 2:851–6; PMID:16278656; http://dx.doi.org/10.1038/nmeth803
- Pilobello KT, Krishnamoorthy L, Slawek D, Mahal LK. Development of a lectin microarray for the rapid analysis of protein glycopatterns. Chembiochem 2005; 6:985–9; PMID:15798991; http://dx.doi.org/10.1002/cbic.200400403
- Zheng T, Peelen D, Smith LM. Lectin arrays for profiling cell surface carbohydrate expression. J Am Chem Soc 2005; 127:9982–3; PMID:16011345; http://dx.doi.org/10.1021/ja0505550
- Dan X, Liu W, Ng TB. Development and Applications of Lectins as Biological Tools in Biomedical Research. Med Res Rev 2015; PMID:26290041
- Uchiyama N, Kuno A, Tateno H, Kubo Y, Mizuno M, Noguchi M, Hirabayashi J. Optimization of evanescent-field fluorescence-assisted lectin microarray for high-sensitivity detection of monovalent oligosaccharides and glycoproteins. Proteomics 2008; 8:3042–50; PMID:18615430; http://dx.doi.org/10.1002/pmic.200701114
- Hirabayashi J, Yamada M, Kuno A, Tateno H. Lectin microarrays: concept, principle and applications. Chem Soc Rev 2013; 42:4443–58; PMID:23443201; http://dx.doi.org/10.1039/c3cs35419a
- Ebe Y, Kuno A, Uchiyama N, Koseki-Kuno S, Yamada M, Sato T, Narimatsu H, Hirabayashi J. Application of lectin microarray to crude samples: differential glycan profiling of lec mutants. J Biochem 2006; 139:323–7; PMID:16567396; http://dx.doi.org/10.1093/jb/mvj070
- Tateno H, Uchiyama N, Kuno A, Togayachi A, Sato T, Narimatsu H, Hirabayashi J. A novel strategy for mammalian cell surface glycome profiling using lectin microarray. Glycobiology 2007; 17:1138–46; PMID:17693441; http://dx.doi.org/10.1093/glycob/cwm084
- Cook MC, Kaldas SJ, Muradia G, Rosu-Myles M, Kunkel JP. Comparison of orthogonal chromatographic and lectin-affinity microarray methods for glycan profiling of a therapeutic monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci 2015; 997:162–78; PMID:26114652; http://dx.doi.org/10.1016/j.jchromb.2015.05.035
- Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev 2012; 28:147–75; PMID:22616486; http://dx.doi.org/10.5661/bger-28-147