
ABSTRACT
Ch14.18 manufactured in Chinese hamster ovary (CHO) cells is currently being evaluated in clinical trials. Short-term infusion (STI) (8–20 h/day; 4–5 days) of 100 mg/m2 ch14.18/CHO (dinutiximab β) per cycle in combination with cytokines is standard treatment of neuroblastoma (NB) patients. As pain is a limiting factor, we investigated a novel delivery method by continuous long-term infusion (LTI) of 100 mg/m2 over 10 days. 53 NB patients were treated with 5–6 cycles of 6 × 106 IU/m2 subcutaneous interleukin-2 (d 1-5, 8-12), LTI of 100 mg/m2 ch14.18/CHO (d 8-18) and 160 mg/m2 oral 13-cis-retinoic acid (d 22-35). Human anti-chimeric antibody (HACA), antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity were determined. With LTI, we observed a maximum concentration of ch14.18/CHO (Cmax) of 12.56 ± 0.68 µg/ml and a terminal half-life time (t1/2 β) of 32.7 ± 16.2 d. The clearance values for LTI and STI of 0.54 ± 0.13 and 0.41 ± 0.29 L/d m2 and area under the serum concentration-time curve (AUC) values of 189.6 ± 41.4 and 284.8 ± 156.8 µg×d/ml, respectively, were not significantly different. Importantly, we detected ch14.18/CHO trough concentration of ≥ 1 µg/ml at time points preceding subsequent antibody infusions after cycle 1, allowing a persistent activation of antibody effector mechanisms over the entire treatment period of 6 months. HACA responses were observed in 10/53 (19%) patients, similar to STI (21%), indicating LTI had no effect on the immunogenicity of ch14.18/CHO. In conclusion, LTI of ch14.18/CHO induced effector mechanisms over the entire treatment period, and may therefore emerge as the preferred delivery method of anti-GD2 immunotherapy to NB patients.
Introduction
Disialoganglioside GD2 is ranked in the top 20% (12/75) of tumor-associated antigens in a priority list published by the National Cancer Institute.Citation1 It is highly expressed on neuroectodermal tumors such as neuroblastoma (NB), melanoma and Ewing sarcoma, but with low expression on normal tissues. Therefore, studies of passive immunotherapies based on administration of monoclonal Ab (mAb) against GD2 have been initiated and have shown promising results.Citation2,3 In NB treatment, the Children's Oncology Group (COG) reported the application of human/mouse chimeric Ab ch14.18 produced in SP2/0 cells.Citation4 In this randomized, open label Phase 3 clinical trial, treatment with short-term infusion (STI) of ch14.18 (4× 25 mg/m2/day, 8–20 h) in combination with cytokines resulted in increases of 20% and 11% in 2-year event-free and overall survival of high-risk NB patients, respectively.Citation4 In Europe, the International Society of Pediatric Oncology European Neuroblastoma Group (SIOPEN) commissioned a Good Manufacturing Practice (GMP) production of ch14.18 Ab in the most commonly used mammalian cell line for industrial production of recombinant protein therapeutics, Chinese hamster ovary (CHO) cells.Citation5 Such a major change in the Ab production process requires preclinical and clinical reevaluation, including its pharmacokinetics (PK) and pharmacodynamics (PD). For that reason, a Phase 1 bridging study was initiated to assess safety, PK and activity profiles of the recloned Ab ch14.18/CHO.Citation6 Analysis of 16 patients revealed that the toxicity profile, clinical activity and PK of ch14.18/CHO given as 8 h STI on five consecutive days (5 × 20 mg/m2) were comparable to ch14.18 produced in SP2/0 cells, allowing for approval of the use in randomized clinical trials.Citation6
One major obstacle associated with anti-GD2 Ab therapies is the induction of neuropathic pain,Citation4,6 which is an on-target side effect not observed with other human/mouse chimeric mAbs. In animal models, which approximate the pain associated with anti-GD2 Ab in humans in terms of timing and quality, GD2-specific binding to Aδ- and C pain fibers results in decreased mechanical stimulus thresholds.Citation7
We initiated a novel treatment schedule aiming at a reduction of toxicity. Instead of STI over five days (5 × 20 mg/m2/d, 8h infusion), 53 high-risk NB patients received the same cumulative dose of 100 mg/m2 ch14.18/CHO given as a continuous long-term infusion (LTI) over 10 days (10× 10 mg/m2/d). Similar to the reported randomized, open label Phase 3 clinical trial,Citation4 patients received ch14.18/CHO in combination with a cytokine (6× 106/d IU/m2 subcutaneous (s.c.) interleukin (IL)-2, d 1-5, followed by a combined application of IL-2 (d 8-12) with LTI of ch14.18/CHO (d 8-18)). A reduced toxicity profile of the new treatment protocol was indicated by significantly decreased morphine usage and low pain scores.Citation8
Here, we report immuno-pharmacological effects of ch14.18/CHO administered as a continuous infusion. For a comprehensive assessment of ch14.18/CHO PK, we optimized the two previously reported enzyme-linked immunosorbent assay (ELISA) methods based on an anti-idiotype Ab (anti-Id Ab) of ch14.18/CHO (ganglidiomab) to increase sensitivity and reliability of measurements.Citation9 The limit of detection (LOD) was further improved by the establishment and validation of a third ELISA method. Concentration-time curves of ch14.18/CHO in treated patients were used to calculate PK parameters using a two-compartmental model. We also describe the immune effector functions (antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC)) of ch14.18/CHO in patients. Finally, we analyzed the frequency of human anti-chimeric antibody (HACA) responses and correlated the findings with ch14.18/CHO concentration-time curves and patient-specific effector functions (ADCC and CDC).
In summary, we show Ab concentration-time curves translating into anti-NB immune responses over the entire treatment period of six months. These data demonstrate that ch14.18/CHO administered by LTI is an active treatment strategy for NB patients.
Results
Establishment of a ch14.18/CHO-ELISA with an improved limit of detection (“high sensitivity” ELISA)
We previously reported two ELISA methods for the detection of ch14.18/CHO in serum of NB patients with a detection range of 0.5–25 µg/ml.Citation9 To further improve the LOD, a third variant following a two-step procedure was established.
In the first step, the minimal optimal dilution factor was determined using the previously reported ELISA protocols (). Six serum samples containing known ch14.18/CHO concentrations were used to analyze standard curves at distinct sample dilution factors covering a concentration range of 0 – 1 µg/ml (1.0, 0.5, 0.25, 0.13, 0.06 and 0.03 μg/ml). Serum samples without Ab served as blanks. All samples were analyzed in triplicate. Standard samples were diluted 1:400, 1:200, 1:100 and 1:70 in PBS (pH 7.4) and analyzed (). The best linear fit of the standard curve was observed at a minimal dilution of 1:70 (R2 = 0.991). In contrast, higher dilutions resulted in a flat curve that could not be used for precise measurements.
Figure 1. Sensitivity and precision optimization of the ch14.18/CHO ELISA method. Sensitivity and precision of the ch14.18 ELISA method were improved following a two-step procedure (A and B). First, the optimal minimal sample dilution factor was evaluated using the reported ELISA protocol (A).Citation9 Standard samples (1.0, 0.5, 0.25, 0.13, 0.06, 0.03 and 0 μg/ml) were diluted 1:70 (closed triangles), 1:100 (open triangles), 1:200 (closed squares) and 1:400 (open squares) and analyzed by ELISA. Results are presented as mean OD of three replicates ± SD from two representative experiments. Second, the optimal minimal sample dilution factor of 1:70 was used to further improve the precision by distinct modifications of sample-, wash- and blocking-buffers as described in “Materials and Methods” (B). Standard samples (1.0, 0.5, 0.25, 0.13, 0.06, 0.03 and 0 μg/ml) were diluted 1:70 and analyzed by “high sensitivity” ELISA. Results are presented as mean OD of three replicates ± SD from two representative experiments. Optimization of the buffer composition resulted in an improved linear fit of the standard curve (R2 = 0.998, closed circles), compared to the previously reported ELISA protocol (R2 = 0.991, closed triangles). (C) LOD for the “high sensitivity” ELISA method was determined using 11 samples containing serum of a healthy donor without ch14.18/CHO (“zero analyte”) analyzed on the same plate. The solid line indicates the LOD of 58 ng/ml, calculated as follows:mean + 3× SD. When error bars are not visible they are covered by the symbol.
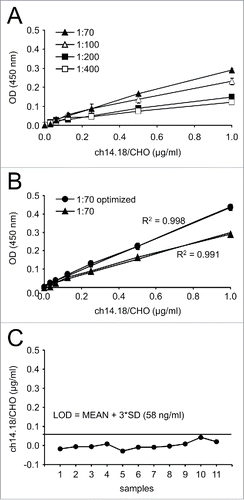
In the second step, we further optimized the ELISA precision by distinct modifications to the sample-, wash-, and blocking-buffer as described in the “Materials and Methods” section. These optimization steps were then evaluated at the optimal minimal dilution factor of 1:70. This resulted in an increased slope and an improved linear fit of the standard curve (R2 = 0.998) (, “1:70 optimized”). This optimized “high sensitivity” ELISA variant was used for further analysis of patient serum samples.
Determination of LOD
To evaluate the background caused by matrix interference, LOD for the optimized high sensitivity ELISA method was determined using 11 human serum samples of a healthy donor without ch14.18/CHO (“zero analyte”) diluted with sample buffer (1:70) and analyzed on the same plate. Analysis now revealed a LOD of 0.058 µg/ml (), showing an increased sensitivity by a factor of 9 compared to the previously published LOD (0.5 µg/ml).Citation9
ELISA validation
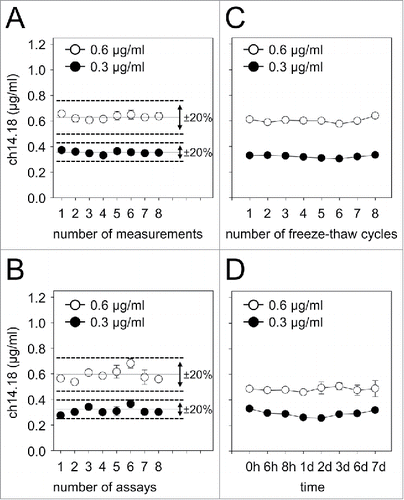
Coefficients of variation (CVs) of both within-assay and inter-assay test results were assessed according to international consensus recommendations for the bioanalytical method validation.Citation10 Experimental runs with CVs exceeding 20% were rejected from the analysis of PK parameters and subjected to reanalysis. Within-assay precision was determined by analysis of the CVs from eight samples containing known ch14.18/CHO concentrations in human serum (0.6 and 0.3 μg/ml) (). Each standard sample was analyzed in triplicate on the same plate, and the CVs were found to be <12% for both Ab concentrations, indicating a reproducible performance of the assay. Similarly, the inter-assay CVs were determined by changing operators on eight different days. This analysis revealed coefficients of 10% for 0.6 μg/ml and 13% for 0.3 μg/ml ch14.18/CHO in human serum (), clearly showing that results obtained were operator-independent and consistent over time.
Figure 2. Within- and inter-assay precision of ch14.18/CHO “high sensitivity” ELISA and stability of low ch14.18/CHO concentrations in serum. For reliable and reproducible detection of ch14.18/CHO concentrations with the “high sensitivity” ELISA, within- (A) and inter- (B) assay precision analyses were performed. Two samples containing known concentrations of ch14.18/CHO (0.6 μg/ml, open circles and 0.3 μg/ml, closed circles) were prepared in human serum and analyzed as described in the “Materials and Methods” section. CVs for both concentrations were calculated according to the formula:SD/mean × 100% and were found to be in the range of ± 20% (indicated by the dashed line). Mean values of the respective data sets are also indicated (solid line). To determine stability of low ch14.18/CHO concentrations in serum, samples of two defined concentrations (0.6 μg/ml (open circles) and 0.3 μg/ml (closed circles)) were prepared. Aliquots were subjected to either eight freeze-thaw cycles (C) or storage at RT for up to 168 h (7d) (D) and analyzed using the “high sensitivity” ch14.18/CHO-ELISA. When error bars are not visible they are covered by the symbol. Data represent mean ch14.18/CHO concentrations of at least three replicates ± SD from two representative experiments.
Stability of low ch14.18/CHO concentrations in serum samples
We expanded previously reported stability results of ch14.18/CHO in serum samples covering a concentration range of 4–17 µg/ml Citation9 by analysis of concentrations below 1.0 µg/ml. For this purpose, we analyzed serum samples containing two defined ch14.18/CHO concentrations (0.6 and 0.3 μg/ml). Samples were subjected to eight freeze-thaw cycles or stored at room temperature (RT) for up to 168 h (7d) followed by quantification with the “high sensitivity” ELISA. Neither repeated thawing () nor storage at RT for up to one week () affected ch14.18/CHO stability in serum samples. These data are important given that European NB trials are generally multi-center in nature and require reliable sample transfer to a central laboratory for analysis.
Procedure to evaluate ch14.18/CHO levels in patient serum
Run acceptance analysis
To accept or reject experimental runs, two quality-control (QC) samples of known ch14.18/CHO concentrations were prepared in human serum for each detection range (low sensitivity:7.0 and 20.0 μg/ml, intermediate sensitivity:1.0 and 2.0 μg/ml and high sensitivity:0.3 and 0.6 μg/ml). Respective QCs were included in each analysis followed by calculation of deviation of QCs from their respective nominal values. Experimental runs were excluded from the analysis of Ab PK, if deviation values of both QCs exceeded 20% from their respective nominal values.
Evaluation of ch14.18/CHO serum levels
Three established ELISA methods with low, intermediate and high sensitivity including respective QCs were used for detection of ch14.18/CHO in serum samples as described in “Materials and Methods” (). First, cycle one serum samples of 13/53 patients with complete sampling and per protocol treatment in our single center program based on LTI administration of 100 mg/m2 ch14.18/CHO over 10 days were analyzed (). Next, we reanalyzed serum samples of cycle one from 8/16 NB patients enrolled in a previously reported European Phase 1 clinical trial with STI of the same cumulative Ab dose of 100 mg/m2 over five days Citation6 using the same triple-ELISA strategy ().
Figure 3. Serum levels of ch14.18/CHO of cycle 1 in neuroblastoma patients. Samples collected from patients treated with (A) continuous infusion of ch14.18/CHO over 10 days (10× 10 mg/m2/d; LTI; 13/53) or (B) 8 h infusion over five days (5× 20 mg/m2/d; STI; 8/16) were evaluated using the triple-ELISA strategy as described in the Materials and Methods section. Ch14.18/CHO levels were analyzed prior to start (d 8), during (d 8-13 for STI and d 8-18 for LTI) and after the end of Ab infusion (d 14-40 for STI and d 19-43 for LTI). The intravenous administration of ch14.18/CHO is indicated as a solid line for LTI and as a dashed line for STI treatment regimen. Data represent concentration time curves of each patient (gray lines) and the mean (bold black line) for cycle 1.
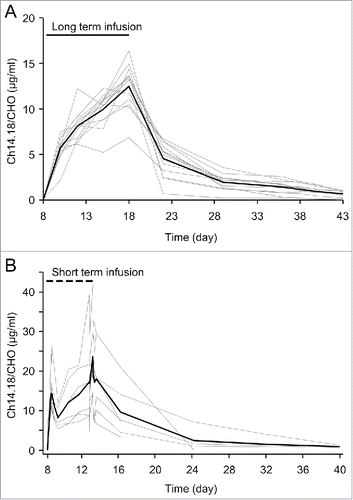
For LTI, we observed a continuous increase of ch14.18/CHO to a maximum of 12.56 ± 0.68 µg/ml at the last day of Ab application compared to a maximum of 21.46 ± 11.24 µg/ml for STI ().
Table 1. Cmax and AUC of ch14.18/CHO Ab given as LTI or STI in high-risk NB patients.
Analysis of population PK
Analysis was performed using ELISA data obtained from the first treatment cycle of Ab naïve patients from a total of 21 eligible patients treated with ch14.18/CHO following LTI- (n = 13/53) or STI (n = 8/16) treatment regimen.
The serum ch14.18/CHO concentration-time profiles for 100 mg/m2/doses administered as LTI and STI were biexponential and fitted by a two-compartment model. Two representative subjects of each treatment regimen are shown (patient LTI-1, LTI-2, STI-1 and STI-4), respectively ().
Figure 4. Ch14.18/CHO concentration-time profiles of LTI- and STI treated patients. Serum concentrations of ch14.18/CHO were determined with triple ELISA strategy and concentration-time fits were obtained with the two-compartment model. Results of two representative subjects of the LTI- (A) (left LTI-1, right LTI-2) and STI treatment regimen (B) (left STI-1, right STI-4) are shown. The population means and inter-individual variabilities (SD) of five pharmacokinetic parameters were estimated using a two-compartment model (CL, Vdss, MRT, t1/2 α, and t1/2 β; ) and the AUCs were calculated as AUC = dose/CL (). The clearance values obtained from the LTI and STI data were not statistically different (). Consequently, also no significant difference between the AUCs (189.6 ± 41.4 µg×d/ml for LTI and 284.8 ± 156.8 µg×d/ml STI) were found. Similarly, the terminal half-lives (t1/2 β) observed with STI and LTI were not different. Gender-specific and age related analysis of all parameters in patients treated by LTI or STI did not reveal significant differences.
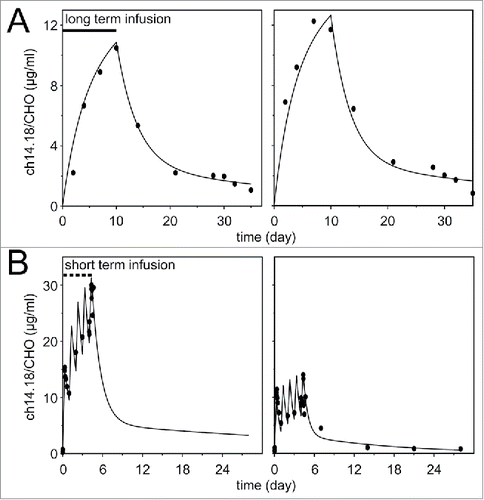
Table 2. Population mean values and inter-individual variability of PK parameters estimated from LTI and STI data using a two-compartment model.
Analysis of HACA and impact on PK
Since the induction of a HACA response may affect PK and effector functions of the therapeutic Ab, we analyzed patients treated with the LTI regimen for HACA development using the previously described ELISA method.Citation12 Samples were analyzed on d 8 (prior to the start of the subsequent Ab treatment cycle) in cycles 2, 3, 4 and 5 and compared to baseline (prior to first Ab administration; d 1, cycle 1; ). If HACA was detectable in any serum sample (LOD of HACA-ELISA 1.1 µg/ml), patients were defined as HACA-positive. We observed the development of HACA in 19% of analyzed patients (10/53). These data are in line with our previous report of HACA development in 21% patients treated with STI of ch14.18/CHO Citation6 and indicate a similar immunogenicity of ch14.18/CHO irrespective of the treatment regimen. For further analysis of HACA effects on ch14.18/CHO PK parameters we divided HACA-positive patients into two cohorts according to the level of HACA in analyzed serum samples: HACA-high responders (mean HACA value ≥ 10 µg/ml) and HACA-low responders (mean HACA value < 10 µg/ml). Within these two categories we observed four HACA-high responders (P1, P3, P5 and P9; 4/53) and six HACA-low responders (P2, P4, P6-8 and P10; 6/53).
Table 3. Analysis of HACA (µg/ml) in high-risk NB patients receiving LTI of ch14.18/CHO.
To investigate the effect of HACA response on ch14.18/CHO concentration-time curves over five treatment cycles, it is mandatory to have a sufficient sampling density per patient over the entire treatment period. Therefore, we only allowed patients to enter this analysis with <20% of missing samples. This criterion was applicable to 37/53 evaluable patients, which provide the basis for results shown in .
Figure 5. Effect of HACA on ch14.18/CHO concentration-time curves. Serum samples collected from 37/53 patients treated with ch14.18/CHO LTI were evaluated using the triple-ELISA strategy as described in “Materials and Methods” prior to start (d 1-8), during (d 8-18; indicated by the gray field) and after the end of Ab infusion (d 19-35) in every treatment cycle. Ch14.18/CHO levels in the circulation of HACA-negative patients (n = 30) (A) were compared to HACA-low (n = 4) (B) and HACA-high responders (n = 3) (C). Data are shown as mean values ± SEM of experiments performed in triplicate; t-test or Mann-Whitney Rank Sum test; §P < 0.05 vs. day 18, cycle 1; *P < 0.001 vs. baseline. When error bars are not visible they are covered by the symbol. Solid lines indicate the trend increase in Cmax over time and the 1 µg/ml ch14.18/CHO level. Numbers indicate the Cmax levels in cycles 1-5.
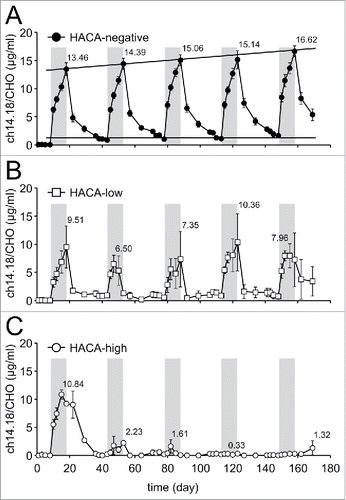
In this cohort of 37 patients, 30/37 patients were HACA-negative and seven patients were HACA-positive (four HACA-low- (4/37) and three HACA-high responders (3/37)). All HACA-negative patients (30/37) revealed increasing Cmax levels ranging from 13.5 ± 5.2 µg/ml (cycle 1) to 16.6 ± 4.1 µg/ml (cycle 5) (P < 0.05; cycle 1 vs. cycle 5), as well as increasing baseline concentrations at trough time points above an immunologically active Ab concentration level of 1 µg/ml 5 (P < 0.001; cycle 1 vs. cycle 2, 3, 4 and 5) (). These data indicate an active Ab concentration over the entire treatment period of six months. In contrast, development of HACA resulted in reduction of Ab levels in subsequent treatment cycles in HACA-low responders (cycles 2, 3 and 5) and HACA-high responders (cycles 3, 4 and 5) compared to HACA-negative patients; reduced and almost complete lack of administered ch14.18/CHO was observed in HACA-low- () and HACA-high responders (), respectively.
Determination of immune modulation and impact of HACA
We analyzed the induction of a GD2-specific ADCC () and CDC () response in patients treated by LTI with ch14.18/CHO (49 evaluable patients for ADCC (49/53; 41 HACA-negative patients (41/49), five HACA-low- (5/49) and three HACA-high responders (3/49)) and 53 patients for CDC (53/53; six HACA-low- (6/53) and four HACA-high responders (4/53)). For this, anti-NB killing activity of patient-specific effector cells and serum samples collected on day 8 in every cycle were analyzed using the calcein-AM based cytotoxicity assay.Citation14 Importantly, we could clearly demonstrate a strong increase of GD2-specific killing of NB cells in vitro mediated by ADCC (two-fold increase; ) and CDC (four-fold increase; ) in every treatment cycle on day 8 of Ab infusion (black columns) compared to baseline level (prior to Ab infusion; day 1, cycle 1) or to day 1 of the respective cycle (white column). Moreover, analysis of CDC activity in samples collected prior to subsequent ch14.18/CHO administrations (corresponding to ch14.18/CHO trough levels) revealed a steady increase over baseline CDC on day 1 of cycle 1 (, white columns), indicating a long-lasting activation of effector mechanisms over the entire treatment period. These data are in line with our ch14.18/CHO-ELISA results showing Ab concentrations above 1 µg/ml over the entire treatment period, sufficient for CDC induction. In contrast, ADCC activity at ch14.18/CHO trough time points were found to be comparable to baseline ADCC activity on day 1 of cycle 1 (, white columns) due to a lower sensitivity of the ADCC assay compared to the CDC assay.
Figure 6. Ch14.18/CHO-mediated ADCC and impact of HACA response. (A) Induction of GD2-specific ch14.18/CHO-mediated ADCC in 49 patients treated with the LTI regimen was determined in every cycle seven days after the start of Ab infusion (d 8) (black column) and compared to baseline ADCC (d 1) of the respective cycle (white column) using the calcein-AM-based cytotoxicity assay as described in “Materials and Methods.” Data are shown as mean values ± SEM of experiments performed in six replicates. Mann-Whitney Rank Sum test or one-way ANOVA, followed by appropriate post hoc comparison test; *P < 0.05 vs. baseline (prior to the first Ab infusion, d 1, cycle 1); §P < 0.05 vs. d 1 of the respective cycle. (B) The effect of HACA on ch14.18/CHO-mediated GD2-specific ADCC against NB cells was evaluated in 3/53 HACA-high responders (gray circles) and 5/53 HACA-low responders (white circles) and compared with 41/53 HACA-negative patients (black circles). The solid lines in black, white and gray indicate cycle-specific median values of ADCC activity in HACA-negative-, HACA-low- and HACA-high responders, respectively, and the solid thin line in black indicates baseline ADCC prior to the first Ab infusion (d 1, cycle 1). Data are shown as patient-specific ADCC (percentage values), Mann-Whitney Rank Sum test; *P < 0.05 vs. baseline. (C) The effect of HACA response on ADCC was determined at time points prior to subsequent treatment cycles (d 1 of cycles 2, 3, 4 and 5; ch14.18/CHO trough levels) and compared to baseline ADCC on day 1 of cycle 1 prior to the first Ab administration (). The solid line indicates baseline ADCC activity prior to the first Ab infusion (d 1, cycle 1). Data are shown as mean values ± SEM of experiments performed in six replicates. Differences between the groups were not significant, Mann-Whitney Rank Sum test or one-way ANOVA, followed by appropriate post hoc comparison test.
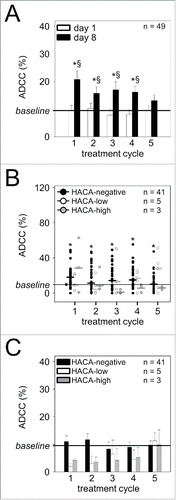
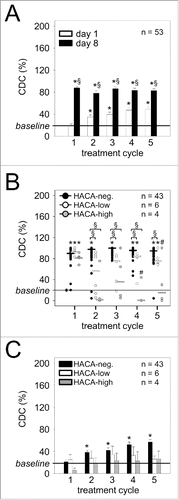
Figure 7. Ch14.18/CHO-mediated CDC and impact of HACA response. (A) Induction of a GD2-specific ch14.18/CHO-mediated CDC in 53/53 patients treated with the LTI regimen. CDC was determined in every cycle seven days after the start of Ab infusion (d 8) (black column) and compared to baseline (prior to the first Ab infusion, d 1, cycle 1) or day 1 of the respective cycle (white column) using the calcein-AM-based cytotoxicity assay as described in “Materials and Methods.” The solid line indicates baseline CDC activity prior to the first Ab infusion (d 1, cycle 1). Data are shown as mean values ± SEM of experiments performed in six replicates, Mann-Whitney Rank Sum test or one-way ANOVA, followed by appropriate post hoc comparison test; *P < 0.05 vs. baseline; §P < 0.05 vs. d 1 of the respective cycle. (B) The effect of HACA response on ch14.18/CHO-mediated GD2-specific CDC against NB cells was evaluated in serum samples of 4/53 HACA-high responders (gray circles) and 6/53 HACA-low responders (white circles) and compared with 43/53 HACA-negative patients (black circles). The solid lines in black, white and gray indicate cycle-specific median values of CDC activity in HACA-negative-, HACA-low- and HACA-high responders, respectively, and the solid thin line indicates baseline CDC prior to the first Ab infusion (d 1, cycle 1). Data are shown as CDC mean values ± SEM of experiments performed in six replicates, Mann-Whitney Rank Sum test or one-way ANOVA, followed by appropriate post hoc comparison test; *P < 0.05 vs. baseline; §P < 0.05 vs. HACA-negative patients of the respective cycle; #P < 0.05 vs. HACA-low responders of the respective cycle. (C) The effect of HACA response on CDC activity in patient serum was determined in HACA-positive patients at time points prior to subsequent treatment cycles (d 1 of cycles 2, 3, 4 and 5; ch14.18/CHO trough levels) and compared to HACA-negative patients and baseline CDC on day 1 of cycle 1 prior to the first Ab administration. Results show two cohorts of HACA-positive patients (HACA-low responders (white columns, n=6) and HACA-high responders (gray columns, n=4)) and HACA-negative patients (black columns, n=43). The solid line indicates baseline CDC activity prior to the first Ab infusion (d 1, cycle 1). Data are shown as mean values ± SEM of experiments performed in six replicates. Mann-Whitney Rank Sum test or one-way ANOVA, followed by appropriate post hoc comparison test; *P < 0.05 vs. baseline; #P < 0.05 vs. HACA-negative patients of the respective cycle.
To determine the effect of HACA development on the effector functions of ch14.18/CHO, we compared GD2-specific ADCC and CDC of HACA-negative patients with both cohorts of HACA-positive patients (). We also compared ADCC and CDC activity in patient samples at time points prior to subsequent treatment cycles (d 1, cycles 2, 3, 4 and 5) corresponding to the trough levels of ch14.18/CHO with baseline activity on day 1 of cycle 1 prior to the first Ab administration ( ).
As expected, in HACA-negative patients, ch14.18/CHO administration resulted in two-fold increased ADCC and four-fold increased CDC in every treatment cycle compared to the respective baseline ( ; black circles; HACA-neg.). In contrast, HACA-high- and HACA-low responders showed significantly reduced CDC on day 8 of Ab infusion compared to HACA-negative patients (). For ADCC, reductions of cytotoxicity levels in comparison to HACA-negative patients were not significant ().
Further, detailed analysis of HACA effects revealed significantly reduced CDC in cycle 4 and 5 (p = 0.015 and 0.050, respectively) and unchanged ADCC in cycles 2, 3, 4 and 5 in HACA-high responders compared to HACA-low responders (, gray and white circles, respectively). Within the six HACA-low responders, all patients revealed detectable ADCC and CDC at least once during therapy (, white circles). Conversely, we found a positive CDC response in the majority of the 10 HACA-positive patients (HACA-low and high responders combined). Specifically, in cycle 2 we found CDC levels over baseline in five of 10 HACA-positive patients. In cycle 3, six of nine HACA-positive patients showed a CDC response (one patient was not evaluable). In cycle 4, six of eight HACA-positive patients showed a CDC response (two missing samples) and in cycle 5, eight of 10 HACA-positive patients showed measurable CDC. Finally, we could even observe CDC in two of four HACA-high responders (, gray circles), indicating that a high HACA response does not necessarily translate into complete abrogation of ch14.18/CHO effector functions. This observation supports the contention that a HACA response does not necessarily lead to abrogation of immune effector functions of ch14.18/CHO.
Analysis of baseline values of ADCC and CDC in HACA-negative patients at trough time points prior to subsequent treatment cycles revealed a steady increase of CDC compared to day 1 of cycle 1 (, black columns), but unchanged levels of ADCC (, black columns). These data are in line with ch14.18/CHO Ab trough levels showing active Ab concentrations sufficient for CDC induction over the entire treatment period. These data again indicate a much higher sensitivity of the CDC assay to detect ch14.18/CHO effector functions compared to the ADCC as already pointed out for the entire cohort (). In contrast, ADCC and CDC analysis of HACA-positive patients within both HACA subgroups did not reveal a significant change of anti-tumor activity at ch14.18/CHO trough time points compared to baseline on day 1 of cycle 1 ( , white and gray columns).
Detection of anti-anti-Id Ab
Since the induction of a HACA response against ch14.18/CHO (Ab1) constitutes the first step in the formation of the anti-idiotypic network (Ab2), we aimed to identify an anti-anti-Id Ab (Ab3) response based on binding to the nominal antigen of Ab1, i.e., GD2. Therefore, we analyzed the serum samples from HACA-positive patients using a GD2 solid phase ELISA as previously described.Citation13 Only HACA-positive samples lacking a ch14.18/CHO signal in the high sensitivity ELISA were included to rule out cross reactions. We did not observe an increase in OD signals compared to the baseline sample (d 1, cycle 1; data not shown) using the GD2 solid phase ELISA for any evaluable HACA-positive patient. This negative finding was further confirmed by preincubation of serum samples with an excess of the anti-Id Ab ganglidiomab mimicking GD2, which did not influence the signal intensity observed in the GD2 ELISA (data not shown). Although LTI of ch14.18/CHO resulted in development of HACA in 19% of patients, we could not detect the induction of a GD2-specific anti-anti-Id Ab with this method.
Discussion
The treatment of high-risk NB patients with ch14.18 emerges as an important treatment option for this challenging malignancy.Citation2,3 For this purpose, dinutuximab (ch14.18/SP2/0) was approved for marketing (4×25 mg mg/m2 in combination with granulocyte-macrophage colony stimulating factor and IL-2) in the US in 2015. In order to ameliorate pain as the main on-target side effect of this treatment, we piloted a 10-day continuous LTI with ch14.18/CHO using the established cumulative dose of 100 mg/m2/cycle.Citation6 Here, we report population PK and PD with this novel method of ch14.18/CHO delivery. We first established an improved ELISA technique compared to our previous report,Citation9 following a three-step analysis procedure with an optimized limit of detection of 58 ng/ml ch14.18/CHO ().
With this method, we analyzed the effect of storage conditions on concentrations below 1 µg/ml of ch14.18/CHO in patient samples. According to our present data, patient samples could be stored at RT for seven days and subjected eight times to repeated thawing and freezing without significant influence on Ab concentration. These data are in line with our previous observations showing that neither thawing nor storage at RT for up to one week affected Ab stability in serum samples containing higher ch14.18/CHO concentrations (5, 11, 17 and 21 μg/ml).Citation9
Analysis of ch14.18/CHO concentration-time curves of 13/53 eligible LTI patients were used to calculate population PK parameters (), which did not show gender- or age-specific differences.
As we previously reported, the completion of a Phase 1 study with ch14.18/CHO delivered in the STI setting, Citation6 we used samples from these patients for reanalysis and comparison to LTI of ch14.18/CHO. This investigation revealed similar clearance parameters of LTI compared with STI of ch14.18/CHO.Citation6 As expected, we observed a continuous increase of ch14.18/CHO levels over the treatment period with maximum concentrations on the last day of Ab application followed by a decrease in Ab levels after the end of Ab infusion.
Clearance, t½ α and β, volume of distribution and mean residence time of ch14.18/CHO (dinutuximab β) were found to be comparable between both regimens.
However, when compared to published results for STI of ch14.18/SP2/0 (dinutuximab) (4 × 25 mg/m2/d = 100 mg/m2/cycle),Citation15 we found differences in parameters for drug exposure between ch14.18/CHO (dinutuximab β) and ch14.18/SP2/0 (dinutuximab).
For instance, AUC0-∞ and t½ β, with LTI of ch14.18/CHO (189.6 ± 41.4 µg*d/ml and 32.7 ± 16.2d, respectively) were higher compared to STI of ch14.18/SP2/0 (1.380 ± 398 μg h/ml = 57.50 ± 16.58 μg×d/ml and 6.9 ± 3.3d, respectively) (p < 0.001),Citation16 underlining an approximate 3-4 fold increased exposure of the same cumulative dose of ch14.18/CHO compared to ch14.18/SP2/0. Cmax values of 12.56 ± 0.68 µg/ml for ch14.18/CHO compared to 11.0 ± 3.1 µg/ml for ch14.18/Sp2/0 (p > 0.1) were not significantly different. The clearance values obtained from ch14.18/CHO applied by LTI and STI using the population-based model were about one-fourth of those reported.Citation15 It should be noted that, despite the problems in AUC tail extrapolation, the AUC values calculated by numerical integration are in reasonable agreement with those obtained by data fitting.
A second interesting observation made with the LTI treatment concept relates to ch14.18/CHO concentration-time curves in subsequent cycles (). Trough levels ≥ 1 µg/ml were found, which is established as an immunologically active ch14.18/CHO concentration,Citation5 also translating into effective CDC activity () at these time points. This trough level is also in sharp contrast to the report for STI of ch14.18/SP2/0 at 0.2 µg/ml.15 These findings are well in line with a greater exposure to ch14.18/CHO by LTI and demonstrate a persistent ch14.18/CHO mediated anti-NB effector function over the entire treatment period of six months.
As mAb-mediated immune effector functions are crucial for the anti-cancer activity of this class of molecules,Citation17 we analyzed ADCC and CDC activity in LTI patients on day 8 of each cycle, and compared results to pre-treatment baseline (d 1, cycle 1) and ch14.18/CHO trough levels prior to subsequent ch14.18/CHO infusions ( ). We demonstrated a four- and two-fold increase in CDC- and ADCC-activity, respectively, against LA-N-1 NB cells on day 8 compared to control time points, indicating that significant anti-NB effector function is engaged by LTI of ch14.18/CHO.
Based on the murine nature of the variable regions, treatment of patients with ch14.18/CHO can induce HACA affecting ch14.18/CHO PK and effector functions. On the other hand, HACA response was described to be associated with prolonged survival.Citation18 Therefore, we analyzed patients treated with the LTI regimen for HACA development using the previously described ELISA method.Citation12 We observed the induction of HACA in 10/53 (19%) of patients. These data are in line with our previous report showing HACA in 21% patients treated with STI of ch14.18/CHO Citation6 and in (3/14) 21% of patients treated with STI of ch14.18/SP2/0, Citation15 indicating similar immunogenicity of ch14.18/SP2/0 and ch14.18/CHO irrespective of the treatment regimen.
In order to evaluate the effect of HACA on PK, we separated patients into two cohorts designated HACA-high and HACA-low responders according to a cutoff HACA level of 10 µg/ml. HACA-high responders (≥10 µg/ml) revealed an almost complete absence of ch14.18/CHO in serum samples upon subsequent treatments. In contrast, HACA-low responders (<10 µg/ml serum HACA level) showed reduced, but detectable ch14.18/CHO levels compared to HACA-negative patients. Our data confirm known effects of HACA on concentration levels of therapeutic Ab in patient circulation.Citation18
Next, we assessed the effect of HACA on ADCC and CDC in both patient cohorts. As expected in HACA-negative patients, ch14.18/CHO administration resulted in two-fold increased ADCC and four-fold increased CDC compared to the respective baselines, confirming reported effector mechanisms of Ab-based therapy.Citation17 In contrast, development of HACA resulted in a reduction of both ADCC and CDC levels in subsequent cycles (). However, we could observe CDC in HACA-high responders compared to baseline, indicating that development of high titer HACA does not necessarily translate into complete abrogation of ch14.18/CHO effector functions. HACA-low responders showed a mixed pattern of ADCC and CDC with negative, reduced and normal level of cytotoxicity compared to HACA-negative patients. These observations support the fact that HACA induction does not necessarily result in abrogation of ch14.18/CHO-mediated immune effector functions. The effect of a HACA response on outcome is a subject for further analysis.
We also investigated the induction of an adaptive immune response against tumor-associated antigen GD2 via the anti-idiotypic network. Therefore, we analyzed the induction of GD2-specific Ab in HACA-positive patients using a GD2 solid phase ELISA as previously described.Citation13 We did not detect an anti-GD2-specific signal in any evaluable HACA-positive patient. This negative result was confirmed by preincubation of serum samples with excess of anti-Id Ab ganglidiomab mimicking GD2 which did not influence the signal intensity observed in the GD2 ELISA (data not shown). Although LTI of ch14.18/CHO resulted in development of HACA in 19% of patients, we could not detect the induction of a GD2-specific anti-anti-Id Ab with this method.
In summary, we show that LTI of ch14.18/CHO (dinutuximab β) results in higher exposure compared to STI of ch14.18/SP2/0 (dinutuximab), translating into induction of anti-NB immune responses over the entire treatment period of six months. These data provide an important baseline for the design of future clinical applications of ch14.18/CHO in high-risk NB patients.
Materials and methods
Ethic statement
This single center program complies with the Declaration of Helsinki. The bridging study was approved by the ethics committee and the competent national authority (Paul-Ehrlich Institute) (EudraCT Number:2005–001267–63). Patients or their parents or legal guardians gave written informed consent.
Patient characteristics
Patients analyzed in this report were treated with ch14.18/CHO either by LTI (10× 24 h; 10 mg/m2/d; 10d, 100 mg/m2/cycle) (53 patients) or by STI (5 × 8 h; 20 mg/m2/d; 5d, 100 mg/m2/cycle) (16 patients). Only patients with biopsy-proven high-risk NB were treated. Patients after first-line therapy had to have evaluable disease. Following second-line chemotherapy, patients were allowed to be treated without evidence of disease; previous treatment had to be discontinued three weeks prior to the start of ch14.18/CHO. Treatment with isotretinoin, growth factor or other immunomodulatory therapy needed to be completed at least seven days before treatment start. A performance score above 70% and a life expectancy of at least 12 weeks were required for inclusion.
The 53 patients treated with LTI received IL-2 for five days (6 × 106 IU/m2/d; s.c; d 1-5), followed by a combined application of IL-2 (6 × 106 IU/m2/d; s.c; d 8-12) with ch14.18/CHO (10 mg/m2/d; i.v. LTI; d 8-18) (100 mg/m2/cycle) and 13-cis-retinoic acid (p.o; d 22-35). At initial diagnosis, 46 patients had stage 4, four stage 3, one stage 2 and two stage 1 disease. Prior treatment included chemotherapy (53 patients), surgery (51 patients), radiotherapy (34 patients) and high-dose therapy (HDT) followed by peripheral blood stem cell rescue (PBSCR; 51 patients); 32 received meta-iodo-benzyl-guanidine (mIBG) therapy preceding HDT.
Age at diagnosis 4.41y (0.5-24.1), age at start of treatment 7.08y (1.9-25.5), International Neuroblastoma Staging System Stage 4:86.8%, International Neuroblastoma Risk Group stage M:92.5%, MYCN oncogene amplified:28.3%, 1p deletion or imbalance:42.1%. The median time from diagnosis to ch14.18/CHO therapy was 25 months (range 8-111 months). At treatment start, the performance scores were ≥90 (Lansky or Karnofsky).
The patient characteristics treated by STI of 100 mg/m2/cycle ch14.18/CHO (8 h, 20 mg/m2/d, 5d) were reported elsewhere.Citation6
Sampling time points and serum preparation
Blood of 53 patients treated with LTI of ch14.18/CHO was collected using BD Vacutainer plastic serum tubes (BD Biosciences, 366444) in every treatment cycle at the following time points:prior to Ab infusion (d 1, 3, 5, and 8), during Ab infusion (d 10, 12, 15 and 18) and after the end of Ab infusion (d 22, 29, 36, 38, 40, 43). Serum was prepared from clotted blood samples by centrifugation at 1,700 × g for 10 min at RT and stored in aliquots at −80°C.
Complete sampling and per protocol treatment in Ab naïve patients in cycle 1 was available for 13/53 patients. These patients were included in the analysis of PK parameters as described below. In order to evaluate ch14.18/CHO levels over five treatment cycles as well as the effect of HACA responses on the concentration-time curves of ch14.18/CHO, additional 24 patients with incomplete sampling were included in the analysis, increasing the number of evaluable patients from 13/53 to 37/53 (mean percentage for all missing samples did not exceed 20%). Ch14.18/CHO-mediated effector functions (ADCC and CDC) were evaluated based on patient sodium-heparin blood and serum samples (49 evaluable patients for ADCC and 53 patients for CDC) that were analyzed in every treatment cycle at baseline (prior to start of Ab infusion) and seven days after the start of Ab infusion. For comparison of PK of ch14.18/CHO LTI and PK previously reported for STI of ch14.18/CHO (5 × 8 h on consecutive days; 20 mg/m2/d; 100 mg/m2/cycle),Citation6 we reanalyzed serum samples. In this cohort, complete sampling of per protocol treated Ab naïve patients receiving 100 mg/m2 ch14.18/CHO in cycle 1 was available for 8/16 patients. In these patients, we used the method described below, which is different to the method used for the previous report.Citation6 Serum samples from NB patients treated by STI had to be collected at the following time points:prior to Ab infusion (d 7 and 8), during Ab infusion (d 9, 10, 11, 12 and 13) and after the end of Ab infusion (d 19, 26, 33 and 40). Additional time points on days 1 and 5 of STI were 0.5, 1, 2, 4 and 8 hours after end of infusion.
Evaluation of ch14.18/CHO levels in patient serum
Isolation of ganglidiomab from hybridoma supernatants
Specific detection of ch14.18/CHO was accomplished using the anti-Id Ab ganglidiomab as a capture Ab for the ELISA methods used. Ganglidiomab was isolated from hybridoma supernatants using a protein G-based technique as previously described.Citation9
ELISA detection strategy
For evaluation of ch14.18/CHO PK in serum of NB patients, we optimized our previously reported ELISA methods (detection range:0.5-25 µg/ml 9) and established a third ELISA method with an improved limit of detection (LOD) at 0.058 µg/ml.
Analysis of unknown patient serum samples followed a three-step procedure. Firstly, the “low sensitivity” ELISA method (detection range 3.0 – 25 µg/ml) was applied. Secondly, samples containing lower ch14.18/CHO levels than 3 μg/ml were subjected to reanalysis with a more sensitive ELISA method (detection range: 0.5 – 3.1 µg/ml), referred to as “intermediate sensitivity” ELISA. Thirdly, samples with ch14.18/CHO concentrations below 0.5 µg/ml were reanalyzed with a newly established “high sensitivity” ELISA method (detection range:0.058–1.0 µg/ml).
The reliability of the previously reported ELISA methods was improved by changing the blocking reagent from 1% (w/v) bovine serum albumin (BSA) in phosphate-buffered saline (PBS) (pH 7.4) to 5% (w/v) Blotting-Grade Blocker (Bio-Rad, 1706404) in PBS (pH 7.4). We also adapted the wash buffer by reducing the concentration of Tween-20 from 0.10% (v/v) to 0.05% (v/v) in PBS (pH 7.4), and changed the sample buffer from PBS (pH 7.4) to PBS (pH 7.4) supplemented with 2% (w/v) Blotting-Grade Blocker and 0.05% (v/v) Tween-20. All other ELISA procedures remained unchanged.Citation9
Establishment of the new “high sensitivity” ELISA (detection range:0.058 – 1.0 µg/ml)
In order to improve the sensitivity for reliable analysis of ch14.18/CHO concentrations below 1 µg/ml, we investigated the effect of a reduction of the sample dilution factor. For this purpose, we prepared six standard samples (1.00, 0.50, 0.25, 0.13, 0.06 and 0.03 μg/ml ch14.18/CHO) in triplicate, using human serum as a sample matrix (PAA, C11-002) and aliquots were stored at −80°C for single use. Standard aliquots without ch14.18/CHO were used as “0” μg/ml standard sample.
Further optimization followed a two-step procedure. First, we performed ELISA procedures as previously described Citation9 and determined the optimal minimal dilution factor required to obtain a linear standard curve. In the second step, we used the optimal dilution factor resulting from step one to further refine the LOD by modifications of the sample-, blocking- and wash-buffers as described below.
Step one:Distinct dilutions of standard samples in PBS (pH 7.4; PAA, H15-002) were analyzed (1:400, 1:200, 1:100 and 1:70) () as follows:First, 96-well immunoplates (PAA, PAA32296X PS 96 F) were coated with 250 ng per well ganglidiomab (100 μl per well; 0.1 M carbonate/hydrogen carbonate buffer, pH 9.6, 1 h, +37°C). After three wash steps with PBS (pH 7.4) supplemented with 0.1% (v/v) Tween-20 (Applichem, A4974,0500), the wells were blocked with 1% (w/v) BSA (200 μl per well; Sigma Aldrich, A9647) in PBS (pH 7.4, 1 h, +37°C) and washed three times (200 μl per well; 0.1% (w/v) BSA in PBS, pH 7.4). Ch14.18/CHO standard samples were diluted in PBS (pH 7.4) and incubated overnight (100 μl per well; +4°C). After three wash steps (200 μl per well; 0.1% (v/v) Tween-20 in PBS, pH 7.4) ch14.18/CHO bound to ganglidiomab was detected with 100 μl per well horseradish peroxidase-(HRP)-conjugated goat anti-human IgG Ab used as secondary Ab (1:20,000, 1 h, 37°C; Sigma Aldrich, A0170). Plates were washed with 0.1% (v/v) Tween-20 in PBS (pH 7.4) three times followed by incubation with a substrate reagent (R&D Systems Inc.., DY999) according to manufacturer's guidelines. The reaction was stopped by addition of 2 N H2SO4 (50 μl) after 30 min. Absorption was analyzed at 450 nm in a plate reader (BioTek Instruments GmbH, SIAFRTD). Results indicated an optimal minimal dilution factor at 1:70 ().
Step two:After the optimal dilution factor (1:70) for standard samples was defined, we implemented optimizations to the ELISA method to further improve the precision and the LOD (). We supplemented the sample buffer (PBS, pH 7.4) with 2% (w/v) Blotting-Grade Blocker and 0.05% (v/v) Tween-20 and reduced the Tween-20 concentration in the wash buffer from 0.10% (v/v) to 0.05% (v/v) (200 μl per well; 0.05% (v/v) Tween-20 in PBS, pH 7.4). The blocking buffer was changed to 5% (w/v) Blotting-Grade Blocker in PBS (pH 7.4). PBS (pH 7.4) containing 2% (w/v) Blotting-Grade Blocker and 0.05% (v/v) Tween-20 was used for dilution of a secondary Ab (1:10,000; HRP-conjugated goat anti-human IgG Ab). Briefly, 96-well plates were coated with 250 ng per well ganglidiomab as described above. After three wash steps with wash buffer, 200 µl blocking buffer per well was added (1 h, +37°C) and then plates were washed three times. Ch14.18/CHO standards prepared in sample buffer (1.0, 0.5, 0.25, 0.13, 0.06 and 0.03 µg/ml Ab) and tailored quality controls (QCs) as well as patient serum samples were diluted 1:70 using sample buffer and incubated overnight at +4°C (100 μl per well). After three wash steps, plates were incubated for 1 h at +37°C with a secondary Ab (100 μl per well; HRP-conjugated goat anti-human IgG; Sigma Aldrich, A0170) diluted 1:10,000 in 2% (w/v) Blotting-Grade Blocker/0.05% (v/v) Tween-20/PBS (pH 7.4). Finally, detection of ch14.18/CHO in samples was performed as described above.
Calculation of limit of detection
To evaluate matrix interference, we determined the LOD for the ELISA method with the highest sensitivity. For this purpose, serum of a healthy donor without ch14.18/CHO (“zero analyte”) was diluted 1:70 with sample buffer (2% (w/v) Blotting-Grade Blocker/0.5% (v/v) Tween-20/PBS, pH 7.4). Then, 11 samples were analyzed on the same plate as described above and the LOD was calculated as follows:mean + 3× standard deviation (SD) ().
ELISA validation
To validate the measurements performed using the “high sensitivity” ELISA method, within-assay and inter-assay precision were calculated as described previously.Citation9 Furthermore, acceptance analysis was carried out for each analytical run. Briefly, to account for the variability of measurements on the same plate, within-assay precision was determined using within-assay coefficient of variation (CV). Eight triplicated samples containing two known concentrations of ch14.18/CHO (0.6 and 0.3 μg/ml) were prepared in human serum and analyzed according to the ELISA protocol as described above. The within-assay precision CV values were calculated as follows:SD of replicates divided by mean of replicates × 100%. To analyze the effect of operator-dependent variability of the assay as well as assay variability over time, inter-assay CVs were calculated. We developed a set of tailored QCs containing two defined concentrations of ch14.18/CHO (0.6 and 0.3 μg/ml) and evaluated QCs from eight independent experiments analyzed by different operators on different days. The inter-assay CVs were calculated for each ch14.18/CHO concentration in accordance to the formula described in the “within-assay precision” section. We calculated the CVs for all assays of each QC batch and plotted the results over time.
Run acceptance analysis
To accept or reject analytical runs, we used two QCs containing defined concentrations of ch14.18/CHO (0.6 and 0.3 μg/ml). Both QCs were included in each analytical run and the deviation of the QC results from their nominal values was calculated as an absolute value (modulus) using the formula: │ (1 − mean divided by nominal value) × 100% │, according to international guidelines for bioanalytical method validation.Citation10 If the differences between both QCs and their respective nominal values exceeded 20%, the run was rejected and data were not included in the analysis. Only runs in which the deviation of QC from the nominal value was less than 20% were accepted.
Stability of low ch14.18/CHO concentrations in serum samples
To evaluate the effects of shipping conditions (temperature and time) on stability of ch14.18/CHO in serum samples containing low Ab concentrations (≤ 1 µg/ml), we prepared two sets of serum samples containing less than 1 µg/ml Ab (0.6 and 0.3 μg/ml). Aliquots were subjected to a maximum of eight freeze-thaw cycles or stored at RT for up to 168 h and analyzed as described above.
Determination of ch14.18/CHO PK in patients
Of 53 patients treated with 100 mg/m2/cycle ch14.18 LTI, 13 patients were evaluable for PK analysis with a complete sample set in cycle one of Ab naïve patients. Of 16 patients treated with STI, 8 patients were evaluable. A population approach (nonlinear mixed-effects modeling) with maximum likelihood estimation via the EM algorithm (MLEM) implemented in the software ADAPT 5 Citation11 was used for PK data analysis. In estimating the parameters of the model, data from all experiments in one group were analyzed simultaneously (8 STI and 13 LTI patients). The MLEM program provides estimates of the population mean and inter-subject variability (clearance (CL), volume distribution at steady state (Vdss), mean residence time (MRT), half-life time (t1/2 α and t1/2 β) and AUC which was calculated as AUC = dose/CL) as well as of the individual subject parameters (conditional means). We assumed log-normally distributed model parameters and the measurement error has a standard deviation that is a linear function of the measured quantity. ‘Goodness of fit’ was assessed using the Akaike Information Criterion and by plotting the predicted versus the measured responses.
Human anti-chimeric Ab response
In order to analyze the development of HACA in patients treated with ch14.18/CHO, a validated ELISA allowing for the specific detection of anti-ch14.18/CHO Ab in patient serum was performed at six different time points (d 1, 8, 15, 18, 22, 29) in every cycle as previously reported.Citation12 Briefly, based on the one arm binding principle, ch14.18/CHO was used as a capture Ab and biotinylated ch14.18/CHO as a detection Ab. 1% (w/v) BSA/PBS (pH 7.4) buffer served as a blocking reagent. Anti-Id mAb ganglidiomab specifically binding ch14.18/CHO Citation13 was used as a standard in a concentration range up to 25 µg/ml. Two QCs containing 5 and 15 µg/ml ganglidiomab were used for validation. After wash steps, a high sensitivity NeutrAvidin-HRP conjugate and a substrate reagent (R&D Systems Inc.., DY999) were added to detect HACA. The reaction was stopped by addition of 2 N H2SO4 and the absorption at 450 nm was determined with a plate reader. In the case of HACA serum levels exceeding 25 μg/ml, patient samples were prediluted in PBS (pH 7.4) and the respective dilution factor was considered for calculation of the final concentration.
Analysis of ADCC and CDC
To evaluate ADCC and CDC effector functions of administered ch14.18/CHO, both patient-specific effector cells and serum collected on the same day were analyzed at the following time points:prior to start of therapy on day 1 in cycle 1 (baseline) as well as in every Ab treatment cycle on day 8 after start of Ab infusion. Cytotoxicity was analyzed using a calcein-acetoxymethyl ester (AM)-based cytotoxicity assay as previously described.Citation14 Briefly, for ADCC, patient leukocytes and heat-inactivated serum (12.5% final concentration) collected on the same day were incubated with 5,000 calcein-AM-labeled human GD2-positive NB cells LA-N-1 for 4 h (effector-to-target cell ratio:40:1). For CDC, serum without effector cells (12.5% final concentration) was incubated with 5,000 calcein-AM-labeled LA-N-1 cells for 4 h. Heat-inactivated serum served as a negative control. Finally, supernatants of each well were transferred to black 96-well plates for determination of fluorescence at 495 nm excitation and 515 nm emission wavelengths using a multimode microplate reader. Patient samples as well as samples for spontaneous (target cells only) and maximum release (target cells disrupted using an ultrasonic homogenizer) were analyzed in at least six replicates. Cytotoxicity in percent mediated by ADCC was calculated according to the formula:(experimental release - spontaneous release)/(maximum release - spontaneous release) x 100%. For CDC the cytotoxicity was calculated according to the formula:(experimental release - negative control release)/(maximum release - negative control release) × 100 %.
Detection of anti-anti-idiotype antibodies
To determine a possible anti-anti-Id response in HACA-positive patients, we applied a previously reported GD2-solid phase ELISA.Citation9,13 Only serum samples collected during Ab infusion and between Ab treatment cycles in cycles 2, 3, 4 and 5 of HACA-positive patients that were found to be negative for ch14.18/CHO were subjected to the analysis.
Briefly, 96-well micro titer plates were coated for 1 h at 50°C with 50 ng/well GD2 (Sigma Aldrich, G0776) as a capture antigen prepared in 99.9% methanol. After evaporation of methanol, wells were blocked for 1 h at +37°C with 1% (w/v) BSA in PBS (pH 7.4) and washed three times (0.1% (w/v) BSA in PBS (pH 7.4)). Serum samples (100 µl/well) from HACA-positive patients diluted 1:50 with PBS (pH 7.4) were added in triplicate. Plates were incubated for 2 h at +37°C and washed five times (0.1% (w/v) BSA in PBS, pH 7.4). Binding of anti-anti-Id Ab to GD2 was analyzed by incubation with HRP-conjugated goat anti-human IgG Ab used as a secondary Ab, developed with a substrate reagent (R&D Systems Inc.., DY999) according to the manufacturer's guidelines. A GD2 metabolite ganglioside GD1b served as a negative control. Additionally, binding of anti-anti-Id Ab to GD2 was inhibited by preincubation of serum samples with an excess of ganglidiomab used as a surrogate of GD2 (final concentration 10 µg/ml).
Statistics
After testing for normality and equal variance across groups, differences between groups were assessed using either the Students t-test or one-way ANOVA test followed by appropriate post hoc comparison. A P level of < 0.05 was considered significant. All data are presented as mean ± SD or mean ± SEM. Analysis was performed using the software SigmaStat (Jandel, San Rafael, CA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge the cooperating group of pediatric oncologists of the SIOPEN-R-NET for the production of ch14.18/CHO Ab carried out by Polymun Scientific, Vienna, Austria, which was financed by charities throughout Europe. We also thank Sandra Pasewald, Manuela Brueser and Theodor Koepp (University Medicine Greifswald, Pediatric Hematology and Oncology, Greifswald, Germany) for excellent technical assistance. Financial support was provided by the Hector-Stiftung, Germany, University Medicine Greifswald, Germany and Deutsche Kinderkrebsstiftung, Germany. Apeiron Biologics, Vienna, Austria appropriated further funding.
Reference List
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens:a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323-37; PMID:19723653; http://dx.doi.org/10.1158/1078-0432.CCR-09-0737
- Modak S, Cheung NK. Disialoganglioside directed immunotherapy of neuroblastoma. Cancer Invest 2007; 25:67-77; PMID:17364560; http://dx.doi.org/10.1080/07357900601130763
- Yang RK, Sondel PM. Anti-GD2 Strategy in the Treatment of Neuroblastoma. Drugs Future 2010; 35:665; PMID:21037966; http://dx.doi.org/10.1358/dof.2010.035.08.1513490
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010; 363:1324-34; PMID:20879881; http://dx.doi.org/10.1056/NEJMoa0911123
- Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, Lewis G, Ladenstein R, Lode HN. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol 2005; 42:1311-9; PMID:15950727; http://dx.doi.org/10.1016/j.molimm.2004.12.018
- Ladenstein R, Weixler S, Baykan B, Bleeke M, Kunert R, Katinger D, Pribill I, Glander P, Bauer S, Pistoia V, et al. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients:a SIOPEN Phase 1 study. MAbs 2013; 5:801-9; PMID:23924804; http://dx.doi.org/10.4161/mabs.25215
- Xiao WH, Yu AL, Sorkin LS. Electrophysiological characteristics of primary afferent fibers after systemic administration of anti-GD2 ganglioside antibody. Pain 1997; 69:145-51; PMID:9060025; http://dx.doi.org/10.1016/S0304-3959(96)03280-0
- Lode HN, Jensen C, Endres S, Pill L, Siebert N, Kietz S, et al. Immune activation and clinical responses following long-term infusion of anti-GD2 antibody ch14.18/CHO in combination with interleukin-2 in high-risk neuroblastoma patients. J Clin Oncol 2015; 32:5 13-6-.
- Siebert N, Seidel D, Eger C, Brackrock D, Reker D, Schmidt M, Lode HN. Validated detection of anti-GD2 antibody ch14.18/CHO in serum of neuroblastoma patients using anti-idiotype antibody ganglidiomab. J Immunol Methods 2013; 398-399:51-9; PMID:24055592; http://dx.doi.org/10.1016/j.jim.2013.09.008
- DeSilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, Khan M, Tacey R, Hill H, Celniker A. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res 2003; 20:1885-900; PMID:14661937; http://dx.doi.org/10.1023/B:PHAM.0000003390.51761.3d
- D'Argenio D, Schumitzky A, Wang X. ADAPT 5 User's Guide:Pharmacokinetic/Pharmacodynamic Systems Analysis Software 2009. Biomedical Simulations Resource Los Angeles, CA
- Siebert N, Eger C, Seidel D, Juttner M, Lode HN. Validated detection of human anti-chimeric immune responses in serum of neuroblastoma patients treated with ch14.18/CHO. J Immunol Methods 2014; 407:108-15; PMID:24727144; http://dx.doi.org/10.1016/j.jim.2014.04.001
- Lode HN, Schmidt M, Seidel D, Huebener N, Brackrock D, Bleeke M, Reker D, Brandt S, Mueller HP, Helm C, et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD(2)-specific anti-neuroblastoma immune response. Cancer Immunol Immunother 2013; 62:999-1010; PMID:23591980; http://dx.doi.org/10.1007/s00262-013-1413-y
- Siebert N, Seidel D, Eger C, Juttner M, Lode HN. Functional bioassays for immune monitoring of high-risk neuroblastoma patients treated with ch14.18/CHO anti-GD2 antibody. PLoS One 2014; 9:e107692; PMID:25226154; http://dx.doi.org/10.1371/journal.pone.0107692
- Desai AV, Fox E, Smith LM, Lim AP, Maris JM, Balis FM. Pharmacokinetics of the chimeric anti-GD2 antibody, ch14.18, in children with high-risk neuroblastoma. Cancer Chemother Pharmacol 2014; 74:1047-55; PMID:25212536; http://dx.doi.org/10.1007/s00280-014-2575-9
- Uttenreuther-Fischer MM, Huang CS, Yu AL. Pharmacokinetics of human-mouse chimeric anti-GD2 mAb ch14.18 in a phase I trial in neuroblastoma patients. Cancer Immunol Immunother 1995; 41:331-8; PMID:8635190; http://dx.doi.org/10.1007/BF01526552
- Weiner LM, Surana R, Wang S. Monoclonal antibodies:versatile platforms for cancer immunotherapy. Nat Rev Immunol 2010; 10:317-27; PMID:20414205; http://dx.doi.org/10.1038/nri2744
- DeNardo GL, Bradt BM, Mirick GR, DeNardo S. Human antiglobulin response to foreign antibodies:therapeutic benefit? Cancer Immunol Immunother 2003; 52:309-16; PMID:12700946