
ABSTRACT
Secukinumab is a human monoclonal antibody that selectively targets interleukin-17A and has been demonstrated to be highly efficacious in the treatment of moderate to severe plaque psoriasis, starting at early time points, with a sustained effect and a favorable safety profile. Biotherapeutics—including monoclonal antibodies (mAbs)—can be immunogenic, leading to formation of anti-drug antibodies (ADAs) that can result in unwanted effects, including hypersensitivity reactions or compromised therapeutic efficacy. To gain insight into possible explanations for the clinically observed low immunogenicity of secukinumab, we evaluated its immunogenicity potential by applying 2 different in vitro assays: T-cell activation and major histocompatibility complex–associated peptide proteomics (MAPPs). For both assays, monocyte-derived dendritic cells (DCs) from healthy donors were exposed in vitro to biotherapeutic proteins. DCs naturally process proteins and present the derived peptides in the context of human leukocyte antigen (HLA)-class II. HLA-DR–associated biotherapeutic-derived peptides, representing potential T–cell epitopes, were identified in the MAPPs assay. In the T-cell assay, autologous CD4+ T cells were co-cultured with secukinumab-exposed DCs and T-cell activation was measured by proliferation and interleukin-2 secretion. In the MAPPs analysis and T-cell activation assays, secukinumab consistently showed relatively low numbers of potential T-cell epitopes and low T-cell response rates, respectively, comparable to other biotherapeutics with known low clinical immunogenicity. In contrast, biotherapeutics with elevated clinical immunogenicity rates showed increased numbers of potential T-cell epitopes and increased T-cell response rates in T-cell activation assays, indicating an approximate correlation between in vitro assay results and clinical immunogenicity incidence.
Abbreviations
| ADA | = | anti-drug antibody |
| APC | = | antigen-presenting cell |
| BSA | = | bovine serum albumin |
| CDR | = | complementarity-determining region |
| cpm | = | counts per minute |
| DCs | = | dendritic cell |
| ELISpot | = | enzyme-linked immunospot |
| HLA | = | human leukocyte antigen |
| IgG | = | immunoglobulin G |
| IL | = | interleukin |
| KLH | = | keyhole limpet hemocyanin |
| mAb | = | monoclonal antibody |
| MAPPs | = | major histocompatibility complex–associated peptide proteomics |
| PBMC | = | peripheral blood mononuclear cell |
| PBS | = | phosphate-buffered saline |
| PHA | = | phytohemagglutinin |
| SI | = | stimulation index |
| spw | = | spots per well |
Introduction
The proinflammatory cytokine interleukin (IL)-17A, produced by the Th17 subset of CD4+ T-helper cells, as well as other innate and adaptive immune cells, plays a pivotal role in the pathophysiology of psoriasis and other immune-mediated diseases.Citation1-6 Secukinumab, a human monoclonal antibody (mAb) that selectively targets IL-17A, has been demonstrated to be highly efficacious in the treatment of moderate to severe plaque psoriasis, starting at early time points, with a sustained effect and a favorable safety profile in Phase 3 studies.Citation7-11 Secukinumab also demonstrated efficacy in Phase 3 studies of subjects with psoriatic arthritis and ankylosing spondylitis.Citation12-16 The safety profile of secukinumab is favorable, with the majority of adverse events reported as mild or moderate in severity.Citation7-16
All biotherapeutics, including mAbs, are immunogenic to varying degrees in individual patients, and the percentage of individuals developing anti-drug antibodies (ADAs) can vary widely, depending on the nature of the target (i.e., internalizing cell surface receptor on antigen-presenting cells [APCs]),Citation17 as well as a range of factors that are not yet fully elucidated.Citation18-22 Factors known to influence biotherapeutic immunogenicity include intrinsic factors such as structural homology with respect to human amino acid sequencesCitation23 and posttranslational modifications.Citation24 In general, eliminating foreign amino acid sequences from antibodies reduces the frequency of ADAs and hypersensitivity reactions,Citation23 and human antibodies are believed to present lower immunogenicity potential compared with chimeric constructs.Citation22,25 However, even human antibodies (e.g., adalimumab), as well as other human proteins (e.g., interferons, erythropoietin), may demonstrate significant levels of immunogenicity for 2 reasons: 1) complementarity-determining regions (CDRs) of immunoglobulin G (IgG) molecules, which are highly diverse in terms of amino acid composition, have potential to raise an immune response,Citation26,27 and 2) tolerance to self-sequences can be broken. Product-specific attributes, including extrinsic factors such as dosing frequency, dose amount, administration route, and formulation (impurities, host cell proteins, tendency to aggregate), all influence immunogenicity.Citation22,28–30 In addition, patient- and treatment-specific factors, such as concomitant drug usage (e.g., for immunosuppression), as well as genetic and disease background of the patient population, have an effect on the immunogenic potential of a biotherapeutic compound,Citation22 which makes it difficult to compare immunogenicity rates between different clinical studies and between different biotherapeutics.
Factors unrelated to the compound that can affect the clinical immunogenicity responses of the different mAbs, such as patient-related or treatment-related factors, were not evaluated in this study. Instead, we focused on evaluation of the immunogenic potential of the biotherapeutics themselves in in vitro assays. For this purpose, we obtained approved comparator biotherapeutics, from a licensed pharmacy, and the marketed formulation of secukinumab as representatives of drug products applied to patients.
Production of high-affinity IgG isotype ADAs after treatment with biotherapeutics requires activation of the adaptive T-cell–dependent immune response, which involves endosomal cleavage of the biotherapeutic protein within a professional APC.Citation31,32 Linear peptide fragments derived from the biotherapeutic are then presented by APCs in the context of the individual's human leukocyte antigen (HLA) class II molecules.Citation31,33 T cells recognizing the peptide fragment as foreign, by virtue of T-cell receptors, initiate a CD4+ T-helper cell–mediated response, eventually leading to the release of cytokines and synthesis of ADAs by B cells.Citation34,35 HLA class II molecules are α/β heterodimers encoded by the highly polymorphic genes HLA-DR, HLA-DP, and HLA-DQ.Citation36,37 However, T-cell responses against allergens are rarely associated with HLA-DP and HLA-DQ, Citation38,39 and HLA-DP and –DQ are believed to play a minor role in the context of ADAs for reasons that are not fully understood.Citation40 We therefore focused mainly on HLA-DR-associated responses within our studies.
Immunogenicity of biologic therapies for psoriasis has been associated with production of ADAs that may affect drug pharmacokinetics, diminish treatment responses, or cause adverse reactions.Citation41-44 In Phase 3 clinical studies in subjects with moderate to severe psoriasis, secukinumab has demonstrated minimal immunogenicity up to Week 12 (primary endpoint), ranging from 0% to 0.41%.Citation7-11 Furthermore, in pooled analysis of these 6 different Phase 3 studies, secukinumab maintained a low rate (0.4%) of treatment-emergent ADAs up to Week 52, with longer term studies out to 2 years continuing to confirm this low rate (0.5%).Citation10 [Blauvelt A et al. Secukinumab treatment maintains efficacy in moderate to severe plaque psoriasis through second year of treatment: A randomized extension of the ERASURE and FIXTURE studies. Oral presentation at: 73rd Meeting of the American Academy of Dermatology; San Francisco, California, USA; March 2015.]
This study investigated the immunogenic potential of secukinumab and other biotherapeutics used to treat psoriasis by applying 2 different in vitro assays: a T-cell activation assay and a major histocompatibility complex–associated peptide proteomics (MAPPs) assay. T-cell activation was used to investigate whether CD4+ T cells are activated by APCs loaded with biotherapeutics. This assay encompasses several aspects of the activation of humoral immune responses, including antigen processing and presentation by professional APCs, as well as the induced T-cell responses.Citation45 The immunogenic potential of 5 mAbs (adalimumab, infliximab, rituximab, ustekinumab, and secukinumab) and 1 Fc-fusion protein (etanercept) was assessed by quantifying T-cell responses in terms of proliferation and IL-2 secretion in APC/T-cell cultures from 50 healthy and drug-naïve donors with a broad range of HLA class II haplotypes. Since the T-cell assay, as performed in this study, did not provide any information on the epitope sequences leading to the observed T-cell responses, MAPPs was independently applied to identify the sequence of biotherapeutic-derived HLA class II-associated peptides, which represent potential T-cell epitopes. The MAPPs assay evaluated differences in antigen presentation for the 5 different mAbs across 30 healthy donors, all of whom differed from the donors used in the T-cell assay. Both the MAPPs and T-cell assay results were compared with the clinically observed immunogenicity of secukinumab and the other biotherapeutics.
Results
T-cell assay
T-cell responses, following exposure to monocyte-derived dendritic cells (DCs) loaded with test biotherapeutics, were assessed by measuring 2 markers in parallel: CD4+ T-cell proliferation via [3H]-thymidine incorporation assay and IL-2 secretion via enzyme-linked immunospot (ELISpot) assay. The ELISpot is among the most sensitive methods for detecting T-cell responses to biotherapeutics.Citation45 Although there is generally a good correlation between IL-2 production and proliferation after CD4+ T cells have been activated, differences can sometimes occur. These differences can be attributed to the kinetics of T-cell responses in culture where transient proliferation responses can potentially be missed, particularly if the proliferation response occurs during the very early stages of the autologous T-cell culture (i.e., before Day 7). Alternatively, differences can be due to activation of specific T-cell subsets that undergo limited proliferation. Since the IL-2 ELISpot assay comprises a membrane pre-coated with capture antibody that binds secreted cytokine during the entire incubation time, both early and late responses will be detected. Proliferation and IL-2 ELISpot assays have therefore been interpreted independently, with differences and similarities highlighted between the respective assay data. The overall correlation between proliferation and IL-2 ELISpot assays was high in this study (89% for responses to the control antigen keyhole limpet hemocyanin [KLH]; Table S1), and donors responding to the tested biotherapeutics were thus defined as those mounting a positive response to each sample in both IL-2 ELISpot and proliferation assays. The marketed formulation of secukinumab and 5 approved biotherapeutics (adalimumab, infliximab, rituximab, ustekinumab, and etanercept), which were obtained from a licensed pharmacy, were individually assessed for immunogenic potential using DCs and CD4+ T cells from a cohort of 50 HLA-typed healthy donors.
T-cell responses to secukinumab and to comparator biotherapeutics with low clinical immunogenicity rates are in the same range
The frequencies of positive responses for T-cell proliferation and IL-2 ELISpot assays in the 50 blood donor samples are shown in . All donors produced a positive T-cell response against phytohemagglutinin (PHA) in IL-2 ELISpot assays, indicating the functionality of cells in culture (data not shown). In the T-cell proliferation assay, secukinumab showed overall a very compact response distribution, with 46 of 50 donors showing a response below and 4 donors only slightly above the threshold (stimulation index [SI] ≥1.9, P < 0.05). Likewise, few donors showed responses above the response threshold for etanercept (4 donors) and ustekinumab (3 donors). Infliximab, adalimumab, and rituximab, in contrast, showed more variable distributions of their responses, with 11, 10, and 7 donors, respectively, having responses above the response threshold ( and Table S1). Similarly, in the IL-2 ELISpot assay, secukinumab showed a compact response distribution, with only 4 donors deviating such that their SI was above the response threshold. The same distribution was true for ustekinumab and etanercept, for which 4 and 5 donors, respectively, showed IL-2 ELISpot signals above the threshold. Rituximab, in contrast, showed a highly variable response distribution, with 6 donors above the response threshold. Infliximab and adalimumab showed distinct subpopulations of the donor set with responses above the response threshold (10 and 8 donors, respectively) ( and Table S1).
Figure 1. Secukinumab exhibits low levels of T-cell responses in a low number of donors. Scatter plot showing the proliferation stimulation index (SI) data (A) and interleukin (IL)-2 enzyme-linked immunospot (ELISpot) SI data (B) from 50 donors incubated with the 6 test samples and humanized A33. Horizontal black lines represent mean values and red-dashed line indicates statistically significant SI ≥1 .9. Summary of T-cell proliferation SI and IL-2 ELISpot SI data for all values where SI ≥1 .9 and P<0.05 (C).
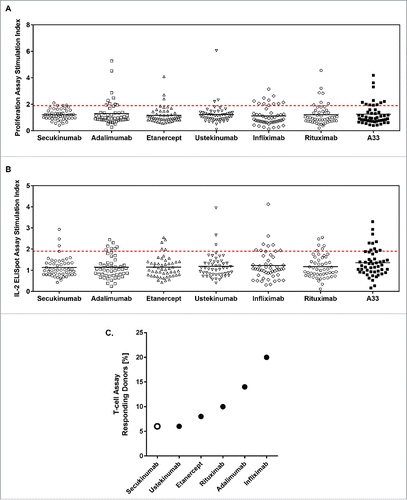
Overall, the percentage of donors mounting a positive T-cell response in both the IL-2 ELISpot and proliferation assays ranged from a high of 20% (infliximab) to 14% (adalimumab), 10% (rituximab), 8% (etanercept), and a low of 6% (for both secukinumab and ustekinumab) ( and Table S1).
MAPPs
Using the MAPPs assay, naturally presented HLA-DR–associated peptides were identified directly from 30 healthy donors' monocyte-derived DCs exposed to test biotherapeutics.Citation28,46–48 HLA-DR–associated peptides originate from a variety of proteins, which are naturally present in the endolysosomal cellular compartment, as well as from the test biotherapeutic.Citation28 Peptides can originate from different protein domains and typically occur as multiple length variants. Peptides sharing the same HLA-DR binding core build a “cluster” (Fig. S1), which represents a sequence region that may potentially, but not necessarily, be recognized as a T-cell epitope.
Clusters can partially overlap with respect to their amino acid sequence, but can be distinguished from one another by sufficiently different HLA binding properties such that different clusters are considered to be an additional distinct opportunity for recognition as a T-cell epitope. Depending on binding properties of the 2 HLA-DR alleles of an individual, donors can differ considerably regarding their pattern of presented clusters.
In the MAPPs analysis, antigen presentation is quantitated by 2 methods that characterize the content of potential T-cell epitopes for a test biotherapeutic. These methods are: 1) counting the number of all clusters for the whole molecule, taking repeated detections of each cluster in multiple donors into account (total clusters); and 2) counting the number of different clusters identified in a donor set for the whole molecule (see Fig. S1 for detailed explanation).
Different secukinumab preparations show a highly consistent cluster pattern by MAPPs
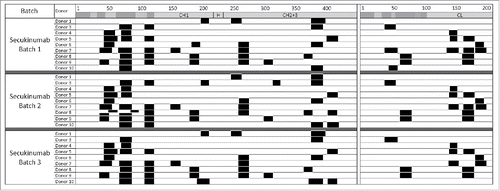
The MAPPs analysis performed with 3 different secukinumab batches and tested on monocyte-derived DCs from 9 healthy donors resulted in highly similar cluster patterns across secukinumab preparations, indicating the consistent quality of secukinumab protein (). Overall, a low number of different clusters were obtained for the secukinumab heavy (14 clusters) and light (6 clusters) chains.
Figure 2. Consistent cluster maps were generated across secukinumab preparations. Major histocompatibility complex–associated peptide proteomics cluster map of human leukocyte antigen-DR–associated peptides produced by 1.5 × 106 dendritic cells from 9 different donors exposed to 3 different secukinumab preparations. Clusters are indicated as black boxes, Complementarity-determining regions are indicated as shaded areas along the sequence of constant domains in heavy chain (CH; left) and light chain (CL; right) above each monoclonal antibody. H, hinge region.
Secukinumab contains low number of potential CD4+ T cell epitopes
The MAPPs cluster patterns of secukinumab were compared with patterns from a panel of therapeutic antibodies (adalimumab, infliximab, rituximab, and ustekinumab) in marketed formulations (). Etanercept was not included in the MAPPs analysis because, as a fusion protein containing endogenous tumor necrosis factor-receptor protein sequences, and lacking CH1 and CL constant domains, it would not be an adequate structural comparator to antibody molecules. The MAPPs assay was performed on 30 donors, in 3 independent sets of 10 donors each. One of the sets was measured using 50% more cells compared with the other 2 sets to increase sensitivity (). For comparison of the 3 donor sets, the numbers of different and total clusters in each dataset were normalized against the respective values for adalimumab as an arbitrary reference (). To obtain 1 single response value for MAPPs, which integrates the number of potential CD4+ T-cell epitopes, as well as their frequency in the donor set, the normalized numbers of different and total clusters were averaged (). In all 3 sets, secukinumab (42.9%, 60.5%, 51.4% relative response) and ustekinumab (46.5%, 61.0%, 47.7% relative response) ranked low in the average normalized cluster response. Infliximab (62.6%, 80.0%, 59.0% relative response) and rituximab (74.1%, 66.1%, 59.5% relative response) showed higher average normalized cluster responses, and the ranking order between the 2 molcules varied, which is likely due to the low number of donors tested in each set. Adalimumab consistently showed the highest response in all 3 donor sets (set as 100%; all data normalized to adalimumab). The total number of clusters was analyzed using a generalized linear model with Poisson errors and a log link. Adalimumab showed significantly higher numbers of total clusters compared with all other antibodies (Table S4). Differences in cluster numbers among the other biotherapeutics were observed although these differences did not reach statistical significance, probably due to the limited number of donors in each set.
Figure 3. Cluster maps vary across different therapeutic antibodies. Major histocompatibility complex–associated peptide proteomics cluster map of human leukocyte antigen-DR–associated peptides produced by 1.5 × 106 dendritic cells from 10 different donors exposed separately to 5 different therapeutic antibodies is shown. Clusters are indicated as black boxes, Complementarity-determining regions are indicated as shaded areas along the sequence of heavy chain (CH; left) and light chain (CL; right) above each monoclonal antibody. H, hinge region.

Figure 4. Secukinumab contains low numbers of different and total clusters. For comparison of the 3 independent sets (indicated by different symbols) of 10 donors each, different and total clusters for each biotherapeutic were quantified and normalized to adalimumab (100%) as an arbitrary reference. Average normalized cluster responses (C) were obtained from the normalized values of different (A) and total (B) clusters, averaging both values. The horizontal line indicates the mean of the 3 individual values. MAPPs, major histocompatibility complex–associated peptide proteomics.
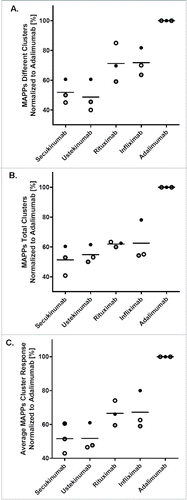
Despite the relatively low number of donors tested in each set, overall all 3 donor sets produced similar results (). The cluster patterns of the tested antibodies along the heavy and light chains showed obvious similarities in certain sequence regions such as the constant domains of the human IgG1 heavy chain (). In contrast, marked differences were observed in other regions such as in the variable domains. This is consistent with therapeutic antibodies showing high sequence diversity in variable domains, in particular in the CDRs.
MAPPs average normalized cluster response and T-cell responses show the same trend
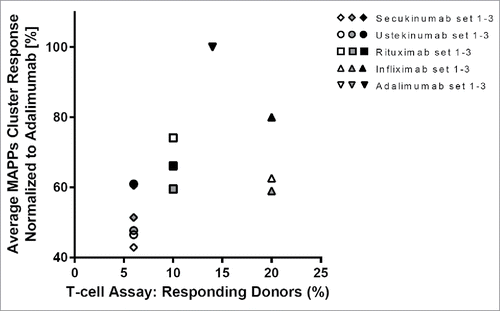
Comparison of the relative immunogenic potentials of the tested biotherapeutics, in the MAPPs assay and the T-cell assay (), indicates that results were generally comparable between these 2 in vitro assays (). Secukinumab and ustekinumab showed low immunogenicity potential in both assays, while rituximab, infliximab, and adalimumab consistently ranked higher in both assays, although the rank order of the 2 latter molecules was reversed between the 2 assays.
Figure 5. Consistent results from major histocompatibility complex–associated peptide proteomics (MAPPs) and T-cell assays. T-cell assay responses for each biotherapeutic from a total of 50 donors were plotted vs MAPPs average normalized cluster responses from a total of 30 donors.
Discussion
Secukinumab is a human IgG1κ mAb that selectively inhibits human IL-17A and has been associated with low immunogenicity (0.4% treatment-emergent ADA rate) in Phase 3 studies up to 52 weeks in subjects with moderate to severe plaque psoriasis. In general, multiple factors that are not clearly defined or understood contribute to biotherapeutic immunogenicity,Citation18-20,24,25,34 but these factors are known to include product-specific attributes, formulation, and tendency for aggregation.Citation28-30 Biotherapeutics currently used to treat psoriasis—such as adalimumab and infliximab—are reported to have higher immunogenicity compared with secukinumab in clinical trials (). However, such indirect comparisons of published immunogenicity prevalence in clinical trials might not account for differences in the time frame over which immunogenicity is observed, differences across clinical studies, and differences in ADA testing, including sampling time points, detection methodology, and reporting. For most biotherapeutics, these differences lead to high variability of reported clinical immunogenicity rates, which complicates not only the evaluation of the “true” immunogenicity of a biotherapeutic, but also the correlation of in vitro assay data with clinical immunogenicity. Harmonization of terms and concepts related to immunogenicity testing would facilitate interpretation and comparison of reported immunogenicity data.Citation49,50
Table 1. Reported Frequency of Anti-Drug Antibodies in Clinical Trials of Selected Biotherapeutics.
The main challenge in preclinical assessment of the immunogenicity potential of a biotherapeutic, using in vitro assays, is that responses against mAbs are much weaker, in general, compared with responses against pathogens, allergens, or vaccines. Weak responses to mAbs can be explained by a low precursor frequency of reactive T-cells, owing to negative T-cell selection processes, which specifically recognize endogenous antibodies. The high sequence similarity between mAbs and endogenous antibodies underlies the weak immune response against mAbs. The finding that patients often develop ADAs only several months after initial drug exposure further illustrates weak immune responses against mAbs.Citation51-52
In this study, T-cell and MAPPs assays were used to examine the immunogenicity potential of biotherapeutic preparations based on their ability to induce a T-cell–mediated immune response. The 2 assays reflect different aspects of the process of ADA development that occurs in patients. The in vitro T-cell assay addresses T-cell responses to whole protein biotherapeutics after processing and presentation by APCs,Citation45,53 whereas the MAPPs assay investigates the ability of biotherapeutics to be processed and presented by individual HLA-DR alleles on APCs (potential T-cell epitopes). Both assays were conducted with cells derived from healthy, drug-naïve blood donors for the following reasons: (1) drug-naïve healthy donors likely reflect the patient population better than patients who have been treated with biologics and already developed an ADA response, (2) it is infeasible to obtain the required number of cells from enough drug-naïve patients, (3) the aim of this study was to evaluate the assays in a setup independent of disease indication and the biotherapeutics assessed in this study are approved for several indications, each with different co-medications that can affect ADA responses, and (4) use of cells derived from healthy blood donors for in vitro studies is the current practice in this field.Citation35,54,55
The applied T-cell assay, conducted at the contract research organization Antitope, an Abzena company, separately measured T-cell proliferation by [3H]-thymidine incorporation and production of IL-2 by ELISpot. Cells from a different set of 50 drug-naïve, healthy donors representing the distribution of HLA class II allotypes expressed in the North American and European populations (Figure S2 and Table S2) were used to evaluate T-cell responses after exposure to biotherapeutics. The ability of secukinumab to initiate a T-cell response was comparable to other marketed biotherapeutics of known low clinical immunogenicity such as ustekinumab and etanercept (). Biotherapeutics with clearly higher clinical immunogenicity in the real-world setting (infliximab, adalimumab, and rituximab) scored higher than secukinumab in these assays, although the overall response rate was still relatively low and the observed responses were weak.
The low number of T-cell precursors reacting against a mAb poses a challenge in naïve T cell assays due to limited time for cell division and signal amplification during the duration of the culture,Citation35 which has an effect on assay sensitivity. Moreover, due to a possible direct interference of the tested biotherapeutics with T-cell activation in more sensitive whole peripheral blood mononuclear cell (PBMC) assays, DCs were first loaded with biotherapeutic proteins in isolation and then washed to remove the protein excess before adding the autologous CD4+ T-cells. This type of DC/T-cell co-culture contains a DC:T-cell ratio that is not physiological, leading to relatively high background, thus making the assay less sensitive compared with a PBMC-based assay. Considering the abovementioned factors, it is expected and consistent with other published studiesCitation29,55 that in the applied in vitro T-cell assay, which measures naïve T-cell responses of low precursor frequency, the dynamic range of the method was limited and the number of responding donors was low. Differences in T-cell responses between the different biotherapeutics were observed, although statistically significant differences were not reached according to analysis of variance and t test. Importantly, the signal ranges in the in vitro T-cell assay are dependent on the signal-to-noise ratio and assay specifics. Therefore, the degree and frequency of responses between clinical immunogenicity and in vitro assays cannot be directly compared.
In contrast, the MAPPs assay characterizes naturally derived HLA-DR–associated peptides obtained from DCs exposed to biotherapeutic proteins.Citation28,46 Testing of 3 different clinical batches of secukinumab in the MAPPs assay yielded highly reproducible cluster patterns (), indicating consistent secukinumab quality. The cluster patterns of secukinumab and other marketed biotherapeutic antibodies showed similarities, in particular in the constant domains of heavy chain (CH1, CH2, CH3) and light chain (CL), consistent with the conserved sequence in these domains among antibodies. Interestingly, some differences in the cluster pattern also occurred in conserved regions, which may be due to product-specific attributes such as formulation, tendency for aggregation, and differential enzymatic processing based on the overall structure of the biotherapeutic and influenced by the variable regions and differences in glycosylation. As one would expect, marked differences in the cluster pattern were observed in the variable domains, in particular around the CDR regions, which is consistent with the high sequence diversity between different antibodies in the variable domains, especially in the hypervariable CDRs.
Although the identified clusters of peptides all represent potential T-cell epitopes, not every cluster identified may effectively induce a T-cell response and immune responses are often dominated by the recognition of a limited number of epitopes, a phenomenon known as immunodominance. Two factors contribute to this phenomenon:Citation56 1) a high relative abundance of epitopes, and 2) the precursor frequency for naïve T cells recognizing different peptide-major histocompatibility complex combinations. The overall extent of antigen presentation, as determined by MAPPs in combination with the actually observed T-cell responses, is relevant when assessing the potential of a protein to induce a CD4+ T-cell-driven immune response. Although 1 epitope can be sufficient to elicit a strong response, immune responses of different individuals can be dominated by different epitopes and a higher number of different presented clusters will thus increase the likelihood that at least 1 of these clusters will be recognized as a true T-cell epitope. In addition, clusters recognized as T-cell epitopes by many different subjects will increase the likelihood for a high incidence of clinical ADA responses.Citation28,34,35
Based on these considerations, the numbers of presented different clusters and total clusters, together representing the number of potential T-cell epitopes in a protein and their frequency in a cohort of donors, were selected as parameters for comparison to the observed clinical immunogenicity of the tested biotherapeutics. Surprisingly, despite the complexity of the immune response, across biotherapeutics these parameters generally agree with the degree of clinical immunogenicity observed in different patient groups treated with these biotherapeutics. The average value for different clusters and total clusters, from the same population of donor cells, was low for secukinumab and in the same range as other biotherapeutics known to exhibit low clinical immunogenicity, such as ustekinumab (). In contrast, the values for rituximab and infliximab were elevated, and adalimumab showed the highest response of all compounds. Taking the average normalized cluster response as an indication of the relative immunogenic potential of the test biotherapeutic, an approximate positive correlation with clinical immunogenicity for these agents was observed.
Over the past years, the MAPPs assay has been applied to identify self- and tumor-derived peptidesCitation46-48 to characterize the effect of post-translational modificationsCitation57 and protein aggregationCitation28 on antigen processing and presentation. Our findings indicate that this assay, together with a functional T-cell assay, may provide valuable information for preclinical assessment of the relative immunogenicity potential of different biotherapeutics.
Both the MAPPs and T-cell assays showed comparable results indicating low immunogenic potential, for secukinumab and ustekinumab. The observed inverted rank order for infliximab and adalimumab in T-cell vs MAPPs assays may be explained by the different aspects of initiating an immune response addressed by the 2 assays. First, the performed MAPPs analysis was limited to HLA-DR due to the peptide isolation setup using an anti-HLA-DR antibody and did not evaluate HLA-DQ or HLA-DP binding. Second, glycosylated peptides could not be evaluated via MAPPs due to the analytical setup. As the MAPPs assay identifies potential T-cell epitopes only, it cannot assess which clusters are actually able to stimulate CD4+ T cells such that some presented clusters may be more relevant to the immunogenic potential of a biotherapeutic than others. For instance, the chimeric antibody biotherapeutics showed clusters in non-human sequence regions that are more likely to induce a T-cell response since the respective T cells have not undergone negative selection in the donor. Since the MAPPs assay does not take the T-cell precursor frequencies into account, the immunogenicity potential of infliximab and rituximab may be underestimated in the MAPPs assay compared with the human mAbs adalimumab, secukinumab, and ustekinumab. This may explain the inverted correlation between the MAPPs and T-cell assays for adalimumab and infliximab. The T-cell assay overcomes these limitations and provides a second independent in vitro analysis. However, the T-cell assay applied here does not identify the sequences of the individual T-cell epitopes, and most importantly, a response in this assay relies on the presence of reactive CD4+ T-cells. In future research, it would therefore be interesting to perform the MAPPs and T-cell assays consecutively using the same donor material and testing the main clusters identified in MAPPs for their T-cell reactivity. However, due to the limitations of the assay (mainly the amount of cells needed for each assay), this is currently infeasible. Neither assay-readout has the potential to specifically measure regulatory T-cell responses (defining positive in vitro T-cell responses based on IL-2 secretion and proliferation excludes regulatory T-cell responses). Moreover, interactions between the biotherapeutic and soluble or membrane-bound targets on APCs may potentially influence the cytokine profile and antigen uptake, and have an effect on the antigen presentation and T-cell activation profiles.
Further, a direct translation of the in vitro response rates and clinical immunogenicity incidence is not possible since immunogenicity is influenced by many factors not addressed by the assays applied in this study, such as patient- (e.g., indication, pre- or co-medication, HLA and other genetic factors) and treatment-related factors (e.g., duration of study, dosing route and regimen, co-medication, drug holidays).
Another caveat for clinical validation of prediction methods, such as those applied in this study, is the high variability of the published clinical immunogenicity data. Reported immunogenicity incidence for biotherapeutics depends on patient- and treatment-related factors, as well as on the immunogenicity assay that was used to measure ADA responses in the patient. Moreover, assay specifics such as sensitivity, drug tolerance, and sampling time all contribute to the complexity. Therefore, a valid correlation between in vitro data and clinical immunogenicity on different marketed compounds may only be possible by generating clinical immunogenicity data with harmonization of assays and sampling in clinical trials. Extensive collaborative efforts, such as the ABIRISK project (www.abirisk.eu) of the European Innovative Medicines Initiative, in which the authors are active contributing participants, might achieve this harmonization goal.
While a linear correlation between in vitro assays and clinical immunogenicity incidence is not possible given all the above-mentioned limitations, we investigated whether an approximate correlation might exist. Based on the reported clinical immunogenicity, the tested biotherapeutics could be separated into 2 main categories: 1) a low immunogenicity group including secukinumab, ustekinumab, and etanercept, and 2) a high immunogenicity group, including rituximab, adalimumab, and infliximab (although also for the latter 3 biotherapeutics, low immunogenicity prevalence has been reported in a few studies) (). The T-cell and MAPPs assay results could be similarly categorized such that secukinumab and ustekinumab, as well as etanercept (T-cell assay only), show low signals, whereas adalimumab, rituximab, and infliximab show high signals in the assays (), indicating indeed an approximate correlation between in vitro assays and clinical immunogenicity incidence.
This study did not intend to investigate mechanisms of causation of clinical immunogenicity nor should the presented data be mistaken for a complete explanation of causation of clinical immunogenicity. Rather, our in vitro data can be seen to provide insight, limited to the parameters investigated by the assays we used, to explain why different antibodies exhibit different incidences of clinical ADA. Our data indicate that certain biotherapeutics, such as ustekinumab and secukinumab, of which fewer peptides were presented to the immune system and had a lower probability for T-cell responses, are associated with a lower clinical immunogenicity incidence. In contrast, other biotherapeutics, such as adalimumab and infliximab, of which more peptides were presented to the immune system and which exhibited increased probability for a T-cell response, are associated with a higher clinical immunogenicity incidence. This approximate correlation between in vitro and clinical results suggests this approach is a relevant indicator of product characteristics that contribute to clinical immunogenicity. It is conceivable that such in vitro assays might be useful as an early checkpoint in the development process of non-immunogenic biotherapeutics, with the understanding that these in vitro assays can provide insight into contributing factors, but will not necessarily predict future clinical immunogenicity.
In summary, while precise prediction of the clinical immunogenicity incidence is not possible at this stage, our results for secukinumab and a number of other marketed biotherapeutics suggest an approximate correlation between the in vitro antigen-presentation potential and T-cell responses, and observed clinical immunogenicity incidence. Further research and large collaborative efforts by big consortia such as ABIRISK, as well as future data from studies using biosimilars or biobetters, will likely shed more light on mechanisms leading to immunogenicity and will facilitate better understanding of the predictive power of assays used to assess immunogenicity potential, e.g., by comparing responses observed with cells derived from healthy volunteers and patients who have been treated with the biotherapeutics.
Materials and methods
T-cell assay
The T-cell assay was performed by AntitopeCitation44 PBMCs were isolated from healthy donor buffy coats (from blood drawn within 24 hours). PBMCs were isolated from buffy coats by Lymphoprep (Axis-shield [NYC1114547]) density centrifugation, and CD8+ T cells were depleted using CD8+ RosetteSep™ (StemCell Technologies Inc. [15663]). Donors were characterized by identifying HLA-DR and HLA-DQ haplotypes using the HISTO Spot SSO HLA typing method (MC Diagnostics). T-cell responses to control antigen (KLH; Sigma [H7017]) were determined.
A cohort of 50 healthy donors (different from those used for the MAPPS assay) was selected to best represent the number and frequency of HLA-DR and -DQ allotypes expressed in North American and European populations. Analysis of HLA-DR and -DQ allotypes expressed in cohort against those expressed in the North American and European populations revealed that coverage of >80% was achieved, and that all major HLA-DR and -DQ allotypes (individual allotypes with a frequency >5% expressed in this population) were well represented (Table S2 and Fig. S2).
Preparation of monocyte-derived DC and autologous CD4+ T cells
To prepare monocyte-derived DCs, PBMCs from each donor were revived in AIM-V® (Life Technologies [12055-083]) culture medium and monocytes isolated using Miltenyi Pan Monocyte Isolation Kits (Miltenyi Biotech [130-096-537]) and LS columns (Miltenyi Biotech [130-042-401]) according to manufacturer's instructions. Monocytes were resuspended in AIM-V® (Life Technologies) supplemented with 1000 U/mL IL-4 (Peprotech [200-04]) and 1000 U/mL granulocyte-macrophage colony-stimulating factor (Peprotech [300-03]) (“DC culture media”) to a density of 4 to 6 × 106 cells/mL, and then distributed in 24-well plates (2 mL final culture volume). Cells were fed on Day 2 by half-volume DC culture media change. On Day 3, the test samples or controls (KLH and humanized A33) were added in DC culture medium to the cells, making a final concentration of 0.3 µM for the test sample, humanized A33, and KLH. In addition, an equivalent volume of DC culture medium was added to the untreated control wells. DCs were incubated with test biotherapeutic for 24 hours, after which cells were washed twice and resuspended in DC culture medium containing 50 ng/mL tumor necrosis factor-α (Peprotech [300-01A]) to induce cellular maturation.
Cells were fed on Day 7 by a half-volume medium change before harvesting on Day 8. The harvested DCs were counted and viability assessed using trypan blue (Sigma [T8154]) dye exclusion. DCs were then γ-irradiated (4000 rads) before use in the proliferation and ELISpot assays. Autologous CD4+ T cells were isolated on Day 8 by negative selection from frozen PBMCs using CD4+ T Cell Isolation Kit II (Miltenyi Biotech [130-096-533]) according to the manufacturer's instructions. Viability of cultured cells, as assessed by trypan blue exclusion, was comparable across the donor samples (data not shown).
After counting and assessing cell viability, 1 × 105 CD4+ T cells were added to 1 × 104 DCs in 96-well round bottom plates (ratio of 10:1). All test samples were tested in sextuplet cultures. Cells were cultured for 7 days before the cultures were pulsed with 1.0 µCi [3H]-thymidine (Perkin Elmer® [NET027Z]) in 50 μL AIM V® medium and incubated for a further 6 hours before harvesting onto filter mats using a TomTec Mach III cell harvester. Counts per minute (cpm) for each well were determined by Meltilex™ (Perkin Elmer®; Meltilex A, melt on solid scintillator [1450-441]; double thickness, glass fiber mats [1450-521]) scintillation counting on a Microplate Beta Counter in parallax, low background counting.
ELISpot plates (Millipore [MSIPS4W10]) were pre-wetted and coated overnight with 100 µL/well IL-2 capture antibody (R&D Systems [SEL202]) in phosphate-buffered saline (PBS). Plates were then washed 3 times in PBS, incubated overnight in blocking buffer (1% bovine serum albumin [BSA] in PBS) and washed in AIM-V® medium. CD4+ T cells and DCs were added to each well as for the proliferation assay (ratio 10:1).
Each test sample was tested in sextuplicate cultures and, for each donor, negative control (AIM-V® medium alone), no cells control, and mitogen-positive control (PHA at 2.5 µg/mL used as an internal test for ELISpot function and cell viability; Sigma [L1668]) were also included on each plate. After a 7-day incubation period, ELISpot plates were developed by sequential washing in dH2O and PBS (x3), prior to the addition of 100 µL filtered, biotinylated detection antibody (R&D Systems [SEL202]) in PBS/1% BSA. Following incubation at 37°C for 1.5 hours, plates were further washed in PBS (x3), and 100 µL of filtered streptavidin-AP (R&D Systems [SEL002]) in PBS/1% BSA was added for 1.5 hours (incubation at room temperature). Streptavidin-AP was discarded and plates were washed in PBS (x4). One hundred microliters of 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium substrate (R&D Systems [SEL002]) were added to each well and incubated for 30 minutes at room temperature. Spot development was stopped by washing the wells and the backs of the wells 3 times with dH2O. Dried plates were scanned on an Immunoscan® Analyzer and spots per well (spw) were determined using Immunoscan® Version 5 software.
T-cell assay data analysis
For proliferation and IL-2 ELISpot assays, an empirical threshold of an SI ≥2 has been previously established whereby test samples inducing responses above this threshold are deemed positive. Extensive assay development and previous studies have shown that this is the minimum signal-to-noise threshold, allowing maximum sensitivity without detecting large numbers of false-positive responses or omitting subtle immunogenic events. In order to further increase sensitivity, responses with an SI ≥ 1.9 were considered positive in this study. Consequently, for both proliferation (n = 3) and IL-2 ELISpot (n = 6) data sets, positive responses were defined by statistical and empirical thresholds:
Significance (P <0.05) of the response by comparing cpm or spw of test wells against medium control wells (cpm >150, spw >3) using unpaired 2-sample student's t-test.
SI ≥ 1 .9, where SI = mean of test wells (cpm or spw) / baseline (cpm or spw). Data presented in this way are indicated as SI ≥1 .9 (P <0.05).
In addition, intra-assay variation was assessed by calculating the coefficient of variance and standard deviation of the raw data from replicate cultures.
Preparation of test samples
Seven therapeutic antibody samples (secukinumab batch 1, 150 mg/mL; ustekinumab, 90 mg/mL; adalimumab, 50 mg/mL; etanercept, 50 mg/mL; infliximab, 10 mg/mL; rituximab, 10 mg/mL; A33, 7.68 mg/mL) were supplied and stored according to instructions provided. Test samples were diluted in AIM-V® culture medium (Invitrogen) just before use and the final assay concentration was 0.3 µM. KLH was stored at −20°C as a 10-mg/mL stock solution in water. For the studies, an aliquot of KLH was thawed before immediately diluting to 3 µM in AIM-V® (final concentration 0.3 µM). PHA (Sigma) was used as a positive control and a 1-mg/mL stock was stored at −20°C before diluting to a final concentration of 2.5 µg/mL in cell cultures. An immunogenic benchmark clinical control, humanized anti-A33 antibody,Citation58 was included for comparison with the test samples. A33 was immunogenic in clinical studies, but since only very limited clinical data are available, this antibody is only considered as an assay control and not as a comparator molecule.Citation59 Secukinumab batch 1 is the preparation used in clinical studies and in all analyses presented in this study, except for the comparison with other secukinumab batches in the MAPPs analysis ().
MAPPs assay
The MAPPs assay was established and conducted internally at Novartis.
Generation of HLA-DR–specific beads for immunoprecipitation
Monoclonal antibodies specific for HLA-DRα were generated using the mouse hybridoma cell line L243Citation60 and were used to isolate HLA-DR–peptide complexes. Protein A–purified anti–HLA-DR antibody was immobilized on N-hydroxysuccinimide–activated beads (GE Healthcare Bio-Sciences AB [28-9811-57]) according to the manufacturer's protocol and stored containing 0.02% sodium azide. For confirmation of HLA-DR depletion efficiency of the L243-conjugated beads, cell lysates before and after immunoprecipitation with the beads were analyzed by Western Blotting using the non-overlapping HLA-DR–specific mAb 1B5 (Lifespan Biosciences [LS-B2858]).
Isolation of monocytes
PBMCs were isolated from human buffy coats sampled from 30 healthy donors (Blood Donation Center SRK beider Basel, Basel, Switzerland; Interregionale Blutspende SRK AG, Bern, Switzerland) different from those used in the T-cell assay. Since only HLA-DR-derived peptides were analyzed, donors were isotyped for HLA-DR only (Table S3). PBMC isolation was achieved by density gradient centrifugation using Leucosep tubes (Greiner Bio One [227290]) according to manufacturer's recommendations.
Isolated PBMCs were labeled with human anti-CD14 microbeads (Miltenyi Biotech [MILT130-050-201]; STEMCELL Technologies Inc. [STMC18058]) according to the manufacturer's protocol and cells were separated within a magnetic field.
Differentiation of monocytes to immature DCs
Monocytes were resuspended in warm medium (RPMI 1640 w/o Glutamine [Gibco Invitrogen, 31870-074], 10% fetal calf serum [Gibco Invitrogen, 161140071], 1% Glutamax [Gibco Invitrogen, 35050038], 1% non-essential amino acids [Gibco Invitrogen, 11140035], 1% sodium-pyruvate [Gibco Invitrogen, 11360039], 1% kanamycin [Gibco Invitrogen, 15160047]) containing 33 ng/mL granulocyte-macrophage colony-stimulating factor (Miltenyi Biotech [130-093-867]) and 3 ng/mL IL-4 (Miltenyi Biotech [130-093-922]) to a final cell concentration of 0.3 × 105 cells/mL, and differentiated to immature DCs in cell-culture dishes for 5 days at 37°C and 5% CO2.
Stimulation and loading of immature DCs
On Day 5 of cell culture, immature DCs were induced to maturation by adding lipopolysaccharide (1 µg/mL, Sigma [L5866-10MG]) and loaded with the biotherapeutic of interest. After incubation for 24 hours at 37°C and 5% CO2, DCs were harvested and washed in PBS. After removal of liquid, the cell pellets were frozen at −70°C.
Isolation of HLA-DR–associated peptides
For isolation of HLA-DR–associated peptides, DC pellets obtained from either 1 × 106 or 1.5 × 106 cells were lysed in hypotonic buffer containing 1% Triton X-100 (Roche Diagnostics GmbH [11332481001]) and protease inhibitor tablets (Roche Diagnostics GmbH [11836153001]) for 1 hour at 4°C on a horizontal shaker at 1100 RPM. After centrifugation, the lysate was incubated overnight at 4°C with L243-conjugated beads for immunoprecipitation. After washing with wash buffer (PBS containing 0.5% Zwittergent) and several wash steps with distilled water, peptides were eluted from HLA-DR molecules by adding 0.1% trifluoracetic acid (Fluka [40967]) at 37°C and lyophilized using an Eppendorf Concentrator 5301 (Eppendorf AG).
Analysis of HLA-DR–associated peptidesCitation28,47,Citation48
Lyophilized peptides were resuspended in hydrophilic buffer containing 5% acetonitrile and 1.1% formic acid, and injected onto a self-packed fused-silica C18 reversed-phase nano-high-performance liquid chromatography column. Multiple injections were not performed due to limited amounts of sample. Peptide identification was performed using liquid chromatography (Nano Capillary System, Dionex Corporation) on a reversed-phase column connected to a mass spectrometer (Q-Exactive, Thermo) via electrospray ionization (LC-ESI-MS/MS). Chromatographic separation was achieved using a 5%–80% acetonitrile gradient buffer (buffer 1: 97.4% water, 2.5% acetonitrile, 0.1% formic acid; buffer 2: 17.4% water, 82.5% acetonitrile, 0.1% formic acid). Peptides were chromatographically separated using a 75-min or 130-min gradient depending on the number of cells used for the generation of peptide eluates (1 million or 1.5 million cells, respectively). Peptides were identified using a database search approach using the SEQUEST algorithm. Peptides with a delta mass of <10 ppm to the expected mass, cross correlation values of XCorr >2.3 for doubly charged ions, >2.8 for triply charged ions, and >3.3 for quadruply charged ions, and a delta cross correlation >0.1 were considered as true hits. In mAb-treated samples, mAb-derived peptides were detected multiple times and in several length variants clustering in sequence regions, increasing confidence in correctness of peptide identification. In addition, donors sharing the same HLA-DR alleles (HLA-DR haplotypes in Table S3) showed common peptide clusters.
Lack of identification of mAb-derived peptides in negative control samples ruled out false positive identification of mAb-derived peptides in the respective samples derived from the same donor.
The total number of clusters was analyzed using a generalized linear model with Poisson errors and a log link. Each of the individual sets used samples from different donors and these sets were analyzed both separately and combined. The terms included in the model as fixed effect were donor and biotherapeutic. For the combined set model, additional terms for set and the interaction between set and biotherapeutic were included.
P-values in Table S4 were obtained from fitting 4 separate models to the data, 3 relating to the modeling of each separate set and 1 for the analysis of the 3 sets combined.
All procedures were in accordance with the Helsinki Declaration of 1975.
Disclosure of potential conflicts of interest
Anette Karle, Sebastian Spindeldreher, and Frank Kolbinger are full-time employees of and have stocks and/or stock options in Novartis.
Supplementary_Datas.zip
Download Zip (287.1 KB)Acknowledgments
The authors would like to thank the subjects and investigators who participated in the studies: Stephan Koepke and Sascha Gottlieb generated and compiled the MAPPs assay results. Philip Jarvis performed the statistical analysis.
BioScience Communications, New York, New York, provided editorial assistance, and was supported by Novartis Pharma AG.
Antitope, performed the T-cell assay experiments and was supported by Novartis Pharma AG.
References
- Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, Russell CB. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol 2013; 133:17-26; PMID:22673731; http://dx.doi.org/10.1038/jid.2012.194
- Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, Krueger JG. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 2008; 128:1207-11; PMID:18200064; http://dx.doi.org/10.1038/sj.jid.5701213
- Johansen C, Usher P, Kjellerup RB, Lundsgaard D, Iverson L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 2009; 160:319-24; PMID:19016708; http://dx.doi.org/10.1111/j.1365-2133.2008.08902.x
- Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villaneuva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 2011; 187:490-500; PMID:21606249; http://dx.doi.org/10.4049/jimmunol.1100123
- Res PC, Piskin G, deBoer OJ, van der Loos CM, Teeling P, Bos JD, Teunissen MB. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggest their involvement in the pathogenesis of psoriasis. PLoS One 2010; 5:e14108; PMID:21124836; http://dx.doi.org/10.1371/journal.pone.0014108
- Ortega C, Fernandez AS, Carrillo JM, Romero P, Molina IJ, Moreno JC, Santamaria M. IL-17-producing CD8+T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol 2009; 86:435-43; PMID:19487306; http://dx.doi.org/10.1189/JLB.0109046
- Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, et al. ERASURE Study Group; FIXTURE Study Group. Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med 2014; 371:326-38; PMID:25007392; http://dx.doi.org/10.1056/NEJMoa1314258
- Blauvelt A, Prinz JC, Gottlieb AB, Kingo K, Sofen H, Ruer-Mulard M, Singh V, Pathan R, Papavassilis C, Cooper S; FEATURE Study Group. Secukinumab administration by pre-filled syringe: efficacy, safety, and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol 2015; 172:484-93; PMID:25132411; http://dx.doi.org/10.1111/bjd.13348
- Paul C, Lacour JP, Tedremets L, Kreutzer K, Jazayeri S, Adams S, Guindon C, You R, Papavassilis C. JUNCTURE Study Group. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol 2015; 29:1082-90; PMID:25243910; http://dx.doi.org/10.1111/jdv.12751
- Mrowietz U, Leonardi CL, Girolomoni G, Toth D, Morita A, Balki SA, Szepietowski JC, Regnault P, Thurston H, Papavassilis C, SCULPTURE Study Group. Secukinumab retreatment-as-needed vs. fixed-interval maintenance regimen for moderate to severe plaque psoriasis: a randomized, double-blind, noninferiority trial (SCULPTURE). J Am Acad Dermatol 2015 Jul; 73(1):277–36.e1; http://dx.doi.org/10.1016/j.jaad.2015.04.011. Epub 2015 May 14.
- Thaçi D, Humeniuk J, Frambach Y, Bissonnette R, Goodman JJ, Shevade S, Gong Y, Papavassilis C. STATURE Study Group. Secukinumab in psoriasis: randomized, controlled phase 3 trial results assessing the potential to improve treatment response in partial responders (STATURE). Br J Dermatol 2015 Sep; 173(3):777–87; http://dx.doi.org/10.1111/bjd.13814. Epub 2015 May 16.
- Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewé R, Nash P, Pricop L, Yuan J, et al. Secukinumab interleukin-17A inhibition in patients with psoriatic arthritis. N Engl J Med 2015; 373:1329-39; PMID:26422723; http://dx.doi.org/10.1056/NEJMoa1412679
- McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, van der Heijde D, Landewé R, Conaghan PG, Gottlieb AB, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015; 386:1137-46; PMID:26135703; http://dx.doi.org/10.1016/S0140-6736(15)61134-5
- Baeten D, Braun J, Baraliakos X, Sieper J, Dougados M, Emery P, Deodhar AA, Porter B, Martin R, Mpofu S, et al. Secukinumab, a monoclonal antibody to interleukin-17A, significantly improves signs and symptoms of active ankylosing spondylitis: results of a 52-week phase 3 randomized placebo-controlled trial with intravenous loading and subcutaneous maintenance dosing. Arthritis Rheumatol 2014; 66:S360.
- Sieper J, Braun J, Baraliakos X, Baeten DL, Dougados M, Emery P, Deodhar AA, Porter B, Andersson M, Mpofu S, et al. Secukinumab, a monoclonal antibody to interleukin-17A, significantly improves signs and symptoms of active ankylosing spondylitis: results of a phase 3, randomized, placebo-controlled trial with subcutaneous loading and maintenance dosing. Arthritis Rheumatol 2014; 66:S232; http://dx.doi.org/10.1002/art.38604
- Mease PJ, McInnes I, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewé R, Nash P, Pricop L, Yuan J, et al. Secukinumab, a human anti–interleukin-17A monoclonal antibody, improves active psoriatic arthritis and inhibits radiographic progression: efficacy and safety data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study. Arthritis Rheumatol 2014; 66:S423.
- Xue L, Hickling T, Song R, Nowak J, Rup B. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. 2016 jan; 183(1):102–13; http://dx.doi.org/10.1111/cei.12711
- Purcell RT, Lockey RF. Immunologic responses to therapeutic biologic agents. J Invest Allergol Clin Immunol 2008; 18(5):335-42
- Leach MW, Rottman JB, Hock MB, Finco D, Rojko JL, Beyer JC. Immunogenicity/hypersensitivity of biologics. Toxicol Pathol 2014; 42:293-300; PMID:24240973; http://dx.doi.org/10.1177/0192623313510987
- Descotes J. Immunotoxicity of monoclonal antibodies. mAbs 2009; 1:104-10; PMID:20061816; http://dx.doi.org/10.4161/mabs.1.2.7909
- De Simone C, Amerio P, Amoruso G, Bardazzi F, Campanati A, Conti A, Gisondi P, Gualdi G, Guarneri C, Leoni L, et al. Immunogenicity of anti-TNFα therapy in psoriasis: a clinical issue. Expert Opin Biol Ther 2013; 13:1673-82; PMID:24107126; http://dx.doi.org/10.1517/14712598.2013.848194
- Jullien D, Prinz JC, Nestle FO. Immunogenicity of biotherapy used in psoriasis: the science behind the scenes. J Invest Dermatol 2014; 135:31-8; PMID:25120005; http://dx.doi.org/10.1038/jid.2014.295
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010; 9:767-74; PMID:20811384; http://dx.doi.org/10.1038/nrd3229
- De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trend Immunol 2007; 28:482-90; http://dx.doi.org/10.1016/j.it.2007.07.011
- Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity residue in the CDR regions. mAbs 2010; 2:256-65; PMID:20400861; http://dx.doi.org/10.4161/mabs.2.3.11641
- van Schouwenburg PA, Kruithof S, Votsmeier C, van Schie K, Hart MH, de Jong RN, van Buren EE, van Ham M, Aarden L, Wolbink G, et al. Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies. J Biol Chem 2014; 289:34482-8; PMID:25326381; http://dx.doi.org/10.1074/jbc.M114.615500
- Malucchi S, Bertolotto A. Clinical aspects of immunogenicity to biotherapeutics. In: van de Weert M, Moller EH, eds. Immunogenicity of Biopharmaceuticals. New York, NY: Springer Science-Business Media, LLC. 2008.
- Rombach-Riegraf V, Karle AC, Wolf B, Sorde L, Koepke S, Gottlieb S, Krieg J, Djidja MC, Baban A, Spindeldreher S, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS One 2014; 9:e86322; PMID:24466023; http://dx.doi.org/10.1371/journal.pone.0086322
- Joubert MK, Hokum M, Eakin C, Zhou L, Deshpande M, Baker MP, Goletz TJ, Kerwin BA, Chirmule N, Narhi LO, Jawa V. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem 2012; 287:25266-79; PMID:22584577; http://dx.doi.org/10.1074/jbc.M111.330902
- Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol 2014; 11:99-109; PMID:23919460; http://dx.doi.org/10.3109/1547691X.2013.821564
- Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther 2007; 7:405-18; PMID:17309332; http://dx.doi.org/10.1517/14712598.7.3.405
- Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol 2008; 8:607-18; PMID:18641646; http://dx.doi.org/10.1038/nri2368
- Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DA, Strominger JL. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 1992; 358:764-8; PMID:1380674; http://dx.doi.org/10.1038/358764a0
- Weber CA, Mehta PJ, Ardito M, Moise L, Martin B, De Groot AS. T cell epitope: friend or foe? Immunogenicity of biologics in context. Adv Drug Deliv Rev 2009; 30:965-76; http://dx.doi.org/10.1016/j.addr.2009.07.001
- Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol 2013; 149:534-55; PMID:24263283; http://dx.doi.org/10.1016/j.clim.2013.09.006
- Mehra NK. Histocompatibility antigens. Encyclopedia of Life Sciences 2001. Available at: http://web.udl.es/usuaris/e4650869/docencia/GenClin/content/recursos_classe_(pdf)/revisionsPDF/HLA.pdf. Accessed 18 June 2015.
- Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 2009; 54:15-39; PMID:19158813; http://dx.doi.org/10.1038/jhg.2008.5
- Oseroff C, Sidney J, Vita R, Tripple V, McKinney DM, Southwood S, Brodie TM, Sallusto F, Grey H, Alam R, et al. T cell responses to known allergen proteins are differently polarized and account for a variable fraction of total response to allergen extracts. J Immunol 2012; 189:1800-11; PMID:22786768; http://dx.doi.org/10.4049/jimmunol.1200850
- McKinney DM, Southwood S, Hinz D, Oseroff C, Arlehamn CS, Schulten V, Taplitz R, Broide D, Hanekom WA, Scriba TJ, et al. A strategy to determine HLA class II restriction broadly covering the DR, DP, and DQ allelic variants most commonly expressed in the general population. Immunogenetics 2013; 65:357-70; PMID:23392739; http://dx.doi.org/10.1007/s00251-013-0684-y
- De Groot AS, Tassone R, Cousens LP, Terry F, Martin R, Ardito M, Martin W. In silico prediction of HLA-DP and -DQ epitope content is poorly correlated with clinical immunogenicity of therapeutic proteins, manuscript in preparation, Abstract presented at the American Association of Pharmaceutical Scientists (AAPS) National Biotechnology Conference (NBC), San Diego, CA, USA. May 20-22, 2013. (http://www.epivax.com/wp-content/uploads/2013/04/EpiVax_AAPS_DPDQ_Poster_16May13_Final.pdf).
- Hsu L, Snodgrass BT, Armstrong AW. Antidrug antibodies in psoriasis: a systematic review. Br J Dermatol 2014; 170:261-73; PMID:24117166; http://dx.doi.org/10.1111/bjd.12654
- Bito T, Nishikawa R, Hatakeyama M, Kikusawa A, Kanki H, Nagai H, Sarayami Y, Ikeda T, Yoshizaki H, Seto H, et al. Influence of neutralizing antibodies to adalimumab and infliximab on the treatment of psoriasis. Br J Dermatol 2014; 170:922-9; PMID:24329764; http://dx.doi.org/10.1111/bjd.12791
- Carrascosa JM. Immunogenicity in biologic therapy: implications for dermatology. Actas Dermosifiliogr 2013; 104:471-9; PMID:23622932; http://dx.doi.org/10.1016/j.ad.2013.02.005
- Farhangian ME, Feldman SR. Immunogenicity of biologic treatments for psoriasis: therapeutic consequences and the potential value of concomitant methotrexate. Am J Clin Dermatol 2015 Aug; 16(4):285-94. http://dx.doi.org/ 10.1007/s40257–015–0131–y
- Anthony DD, Milkovich KA, Zhang W, Rodriguez B, Yonkers NL, Tary-Lehmann M, Lehmann PV. Dissecting the T cell response: proliferation assays vs. cytokine signatures by ELISPOT. Cells 2012; 1:127-40; PMID:24710419; http://dx.doi.org/10.3390/cells1020127
- Kropshofer H, Spindeldreher S, Röhn TA, Platania N, Grygar C, Daniel N, Wölpl A, Langen H, Horejsi V, Vogt AB. Tetraspan microdomains distinct from lipid rafts enrich select peptide-MHC class II complexes. Nat Immunol 2002; 3:61-8; PMID:11743588; http://dx.doi.org/10.1038/ni750
- Röhn TA, Reitz A, Paschen A, Nguyen XD, Schadendorf D, Voight AB, Kropshofer H. A novel strategy for the discovery of MHC class II-restricted tumor antigens: identification of a melanotransferrin helper T-cell epitope. Cancer Res 2005; 65:10068-78; PMID:16267033; http://dx.doi.org/10.1158/0008-5472.CAN-05-1973
- Kropshofer H, Singer T. Overview of cell-based tools for pre-clinical assessment of immunogenicity of biotherapeutics. J Immunotoxicol 2006; 3:131-6; PMID:18958693; http://dx.doi.org/10.1080/15476910600845625
- Rup B, Pallardy M, Sikkema D, Albert T, Allez M, Broet P, Carini C, Creeke P, Davidson J, De Vries N, et al. ABIRISK Consortium. Standardizing terms, definitions and concepts for describing and interpreting unwanted immunogenicity of biopharmaceuticals: recommendations of the innovative medicines initiative ABIRISK consortium. Clin Exp Immunol 2015 May 8; 181(3):385-400; http://dx.doi.org/10.1111/cei.12652. [Epub ahead of print].
- Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, Quarmby V, Richards S, Schneider CK, Subramanyam M, Swanson S, et al. American Association of Scientists. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J 2014; 16:658-73; PMID:24764037; http://dx.doi.org/10.1208/s12248-014-9599-2
- Tovey MG, Legrand J, Lallemand C. Overcoming immunogenicity associated with the use of biopharmaceuticals. Expert Rev Clin Pharmacol 2011; 4:623-631; PMID:22114889; http://dx.doi.org/10.1586/ecp.11.39
- Bendtzen K. Immunogenicity of Anti-TNF-α Biotherapies: I. Individualized medicine based on immunopharmacological evidence. Front Immunol 2015 Apr 8; 6:152; http://dx.doi.org/10.3389/fimmu.2015.00152
- Jaber A, Baker M. Assessment of the immunogenicity of different interferon β-1a formulations using ex vivo T-cell assays. J Pharm Biomed Anal 2007; 43:1256-61; PMID:17118612; http://dx.doi.org/10.1016/j.jpba.2006.10.023
- Sauerborn M. The Immunogenicity of Therapeutic Antibodies. Handbook of Therapeutic Antibodies: Wiley-VCH Verlag GmbH & Co. KGaA. 2014; pp. 665-680.
- Wullner D, Zhou L, Bramhall E, Kuck A, Goletz TJ, Swanson S, Chirmule N, Jawa V. Considerations for optimization and validation of an in vitro PBMC derived T cell assay for immunogenicity prediction of biotherapeutics. Clin Immunol 2010; 137: 5-14; PMID:20708973; http://dx.doi.org/10.1016/j.clim.2010.06.018
- Kim A, Sadegh-Nasseri S. Determinants of immunodominance for CD4 T cells. Curr Opin Immunol 2015; 34:9-15; PMID:25576665; http://dx.doi.org/10.1016/j.coi.2014.12.005
- Karle AC, Oostingh GJ, Mutschlechner S, Ferreira F, Lackner P, Bohle B, Fischer GF, Vogt AB, Duschl A. Nitration of the pollen allergen bet v 1.0101 enhances the presentation of bet v 1-derived peptides by HLA-DR on human dendritic cells. PLoS One 2012; 7(2):e31483; PMID:22348091; http://dx.doi.org/10.1371/journal.pone.0031483
- Scott AM, Lee FT, Jones R, Hopkins W, MacGregor D, Cebon JS, Hannah A, Chong G, Papenfuss A, Rigopoulos A, et al. A phase I trial of humanized monoclonal antibody A33 in patients with colorectal carcinoma: biodistribution, pharmacokinetics, and quantitative tumor uptake. Clin Cancer Res 2005; 11:4810-7; PMID:16000578; http://dx.doi.org/10.1158/1078-0432.CCR-04-2329
- Carrasquillo JA, Pandit-Taskar N, O'Donoghue JA, Humm JL, Zanzonico P, Smith-Jones PM, Divgi CR, Pryma DA, Ruan S, Kemeny NE et al. 124I-huA33 antibody PET of colorectal cancer. J Nucl Med 2011; 52:1173-80; PMID:21764796; http://dx.doi.org/10.2967/jnumed.110.086165
- Ting YT, Temme S, Koch N, McLellan AD. A new monoclonal antibody recognizing a linear determinant on the HL-DRα chain N-terminus. Hybridoma 2009; 28:423-9; PMID:20025501; http://dx.doi.org/10.1089/hyb.2009.0050
- Hsu L, Armstrong AW. Anti-drug antibodies in psoriasis: a critical evaluation of clinical significance and impact on treatment response. Expert Rev Clin Immunol 2013; 9:949-58; PMID:24128157; http://dx.doi.org/10.1586/1744666X.2013.836060
- Krieckaert CL, Bartelds GM, Lerns WF, Wolbink GJ. The effect of immunomodulators on the immunogenicity of TNF-blocking therapeutic monoclonal antibodies: a review. Arthritis Res Ther 2010; 12:217-22; PMID:21029481; http://dx.doi.org/10.1186/ar3147
- Mazilu D, Opris D, Gainaru C, Iliuta M, Apetrei N, Luca G, Borangiu A, Gudu T, Peltea A, Gorseanu L, et al. Monitoring drug and antidrug levels: a rational approach in rheumatoid arthritis patients treated with biologic agents who experience inadequate response while being on a stable biologic treatment. Biomed Res Int 2014:702701; http://dx.doi.org/10.1155/2014/702701
- de Vries MK, Wolbink GJ, Stapel SO, de Vrieze H, van Denderen JC, Dijkmans BA, Aarden LA, van der Horst-Bruinsma IE. Decreased clinical response to infliximab in ankylosing spondylitis is correlated with anti-infliximab formation. Ann Rheum Dis 2007; 66:1252-4; PMID:17472991; http://dx.doi.org/10.1136/ard.2007.072397
- Zisapel M, Zisman D, Madar-Balakirski N, Arad U, Padova H, Matz H, Maman-Sarvagyl H, Kaufman I, Paran D, Feld J, et al. Prevalence of TNF-α blocker immunogenicity in psoriatic arthritis. J Rheumatol 2015; 42:73-8; PMID:25399390
- Krieckaert CL, Jamnitski A, Nurmohamed MT, Kostense PJ, Boers M, Wolbink G. Comparison of long-term clinical outcome with etanercept treatment and adalimumab treatment of rheumatoid arthritis with respect to immunogenicity. Arthritis Rheum 2012; 64:3850-5; PMID:22933315; http://dx.doi.org/10.1002/art.34680
- de Vries MK, Brouwer E, van der Horst-Bruinsma IE, Spoorenberg A, van Denderen JC, Jamnitski A, Nurmohamed MT, Dijkmans BA, Aarden LA, Wolbink GJ. Decreased clinical response to adalimumab in ankylosing spondylitis is associated with antibody formation. Ann Rheum Dis 2009; 68:1787-8; PMID:19822712; http://dx.doi.org/10.1136/ard.2009.109702
- Paramarta JE, Baeten DL. Adalimumab serum levels and antidrug antibodies towards adalimumab in peripheral spondyloarthritis: no association with clinical response to treatment or with disease relapse upon treatment discontinuation. Arthritis Res Ther 2014; 16:R160; PMID:25074046; http://dx.doi.org/10.1186/ar4675
- van Kujik AW, de Groot M, Stapel SO, Dijkmans BA, Wolbink GJ, Tak PP. Relationship between the clinical response to adalimumab treatment and serum levels of adalimumab and anti-adalimumab antibodies in patients with psoriatic arthritis. Ann Rheum Dis 2010; 69:624-5; PMID:20223840; http://dx.doi.org/10.1136/ard.2009.108787
- van Vollenhoven RF, Emery P, Bingham CO 3rd, Keystone EC, Fleischmann R, Furst DE, Macey K, Sweetser M, Kelman A, Rao R. Longterm safety of patients receiving rituximab in rheumatoid arthritis clinical trials. J Rheumatol 2010; 37:558-67; PMID:20110520; http://dx.doi.org/10.3899/jrheum.090856
- Mok CC. Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther 2013; 4:87-100; http://dx.doi.org/10.2147/DDDT.S41645
- Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, Wang Y, Shen YK, Doyle DK, Mendelsohn AM, et al. PSUMMIT 2 Study Group. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumor necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis 2014; 73:990-9; PMID:24482301; http://dx.doi.org/10.1136/annrheumdis-2013-204655
- Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, et al. CERTIFI Study Group. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med 2012; 367:1519-28; PMID:23075178; http://dx.doi.org/10.1056/NEJMoa1203572
- Emi Aikawa N, de Carvalho JF, Artur Alemida Silva C, Bonfa E. Immunogenicity of anti-TNF-α agents in autoimmune diseases. Clin Rev Allergy Immunol 2010; 38:82-9; PMID:19565360; http://dx.doi.org/10.1007/s12016-009-8140-3
- Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, Aelion JA, Lee SH, Codding CE, Kellner H, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis 2013; 72:863-9; PMID:22730366; http://dx.doi.org/10.1136/annrheumdis-2012-201601