
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Susceptibility of methionine to oxidation is an important concern for chemical stability during the development of a monoclonal antibody (mAb) therapeutic. To minimize downstream risks, leading candidates are usually screened under forced oxidation conditions to identify oxidation-labile molecules. Here we report results of forced oxidation on a large set of in-house expressed and purified mAbs with variable region sequences corresponding to 121 clinical stage mAbs. These mAb samples were treated with 0.1% H2O2 for 24 hours before enzymatic cleavage below the hinge, followed by reduction of inter-chain disulfide bonds for the detection of the light chain, Fab portion of heavy chain (Fd) and Fc by liquid chromatography-mass spectrometry. This high-throughput, middle-down approach allows detection of oxidation site(s) at the resolution of 3 distinct segments. The experimental oxidation data correlates well with theoretical predictions based on the solvent-accessible surface area of the methionine side-chains within these segments. These results validate the use of upstream computational modeling to predict mAb oxidation susceptibility at the sequence level.
Introduction
Throughout manufacturing, storage, transportation, and administration, therapeutic monoclonal antibodies (mAbs) are subject to biophysical and biochemical stress from multiple sources, which may lead to their degradation via aggregation, fragmentation, and chemical modifications, such as oxidation, deamidation, or isomerization. Among these undesirable degradation products, oxidation is one of the most commonly observed post-translational modifications. Oxidative stress in therapeutic mAbs may come from direct contact with oxygen, degradation of excipients in formulation, metal ion traces from production equipment or cell culture, exposure to light, vaporized sanitation agents, and cavitation.Citation1 MAb oxidation can lead to aggregate formation,Citation2 reduced biologic activity, shortened half-life,Citation3,4 and immunogenicity.Citation2 Although all 20 natural amino acids can in principle be oxidized,Citation5 intrinsic oxidation rates span 3 orders of magnitude. Residues containing sulfur and aromatic groups are the most labile to oxidation. In addition to the wide range of intrinsic reactivities, protein dynamics and conformation can further impact the measured reactivity of a given residue. This has been previously observed with reduced level of oxidation for methionine (Met) with either lower solvent exposure or higher protein conformational stability.Citation6-10
At the early stage of mAb discovery, assessing oxidation stability of therapeutic candidates to minimize downstream risks is critical. Early identification of oxidation-prone sites enables antibody engineering to eliminate oxidation liability of leading candidates while maintaining binding activity. Previous studies have mostly focused on the elucidation of oxidation sites on a small number of mAbs with higher resolution methods, such as tryptic peptide mapping (TPM). In this work, we present a combination of forced oxidation (24 hours) and enzymatic digestion (0.5 h) for a high throughput middle-down liquid chromatography-mass spectrometry (LCMS) approach (3 min/sample) with which we screened 121 samples with variable region sequences corresponding to clinical stage mAbs in 6 hours. This middle-down approach permits rapid analysis at segment resolution (LC, Fd, and Fc). Concurrent machine learning-based estimation of solvent-accessible surface area (SASA) for the methionine side-chain was found to correlate well with oxidation levels in Fd and LC species, suggesting high confidence in the predictions of the SASA model, and thus the prediction of oxidation propensity at the sequence level.
Results
One hundred and 21 mAbs at advanced clinical stages, with variable regions matching published sequences, were produced recombinantly in HEK293 cells.Citation11 All mAbs were expressed as IgG1 isotype, regardless of original drug isotype and purified by ProA. All the antibodies in this study have a kappa light-chain. These samples (∼1 mg/mL) were stressed by 0.1% H2O2 for 24 hours followed by quenching with 3-fold molar excess of methionine. The stressed mAbs were digested into Fab’2 and Fc by incubation with Ides enzyme at 37 ˚C for 30 minutes. Prior to LCMS, dithiothreitol (DTT) was added to reduce Fab’2 into Fd and LC segments.
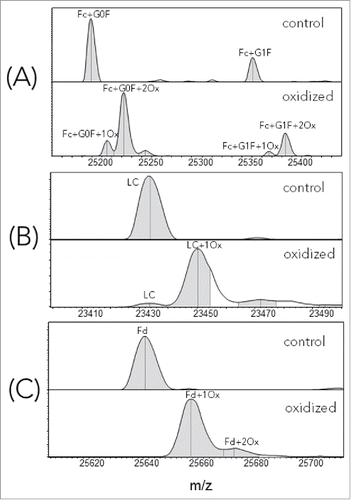
Representative LCMS spectra, corresponding to Fc, LC and Fd segments of control (unstressed) and oxidized infliximab, are shown in . To be concise, we used the US Adopted Name (USAN) for all samples made with IgG1 isotype and published variable region sequences. In , the peak corresponding to 2 Fc oxidation events was the dominant species, suggesting that at least 2 sites were oxidized under the current forced oxidation condition. This result is consistent with the observed oxidation in other experimental studiesCitation12 and with the known crystal structures of the Fc, where Met252 and Met428 side-chains have an average solvent exposure of 36% and 14%, respectively. The results of these calculations are presented in the Table S1 (Fc-SASA tab of the Excel file). Interestingly, for this particular mAb, both LC () and Fd () showed a single oxidation species as the dominant oxidation product.
Figure 1. Deconvoluted LCMS spectra of reduced, control and oxidized infliximab. (A) Fc; (B) LC; (C) Fd.
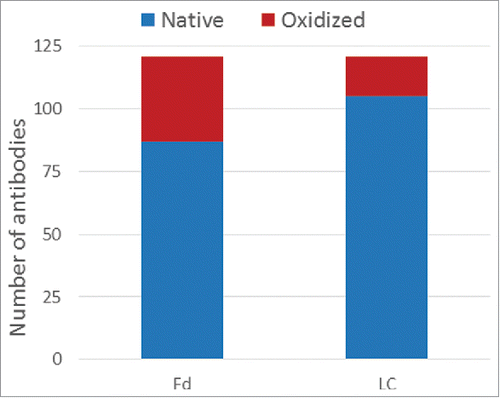
The consolidated results for the 121 mAbs are displayed in . To differentiate oxidation signal (+16 Da peak) from low level water adduct (+18 Da peak), a greater than 50% loss of the native (unmodified) species peak was used as a cutoff for assigning a true oxidation event. Motavizumab was considered as having an oxidation signal since the loss of its native species is 48%, which is close to the 50% threshold. For cases that passed this threshold, the number of oxidation events was assigned as the species with the dominant peaks. The experimental data are summarized in the Table S1 (Experimental tab of the Excel file). Of the 121 samples analyzed, 87 (72%) had 100% native Fd and 34 (28%) had one or more oxidation events within the Fd. For the LC, the corresponding numbers are 104 (86%) fully native and 17 (14%) with oxidation events. Overall, we observed more instances of oxidation in Fd than in LC within the current set of antibodies. These summary statistics are consistent with the higher incidence of methionine in the Fd (291) compared with the LC (124). To address concerns about expression host differences between transient HEK and stable mammalian cell lines, we obtained a total of 9 commercial drugs and performed the same oxidative stress. In every case, the same level of oxidation was observed for both LC and Fd (data not shown), suggesting that the oxidation propensity is encoded in the sequence and folding.
Figure 2. Consolidated forced oxidation experimental results for both Fd and LC.
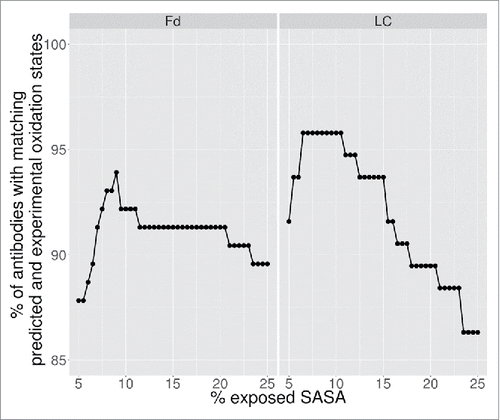
For the theoretical predictions, a methionine was considered oxidized if its predicted fractional SASA was greater than a cutoff threshold. Since all samples share the same kappa light chain constant domain and CH1 domain, which do not contain methionine, the theoretical predictions were focused exclusively on the variable regions. The cutoff value was optimized against the experimental data to maximize the number of antibodies with the correctly predicted oxidation state. The results of the optimization are shown in . A fractional SASA cutoff of 9% was found to exhibit the highest recapitulation of the experimentally assigned number of oxidation states. As seen from , the results were relatively insensitive to cutoff values in the range of 8–11%.
Figure 3. Determination of optimal predicted SASA cutoff.
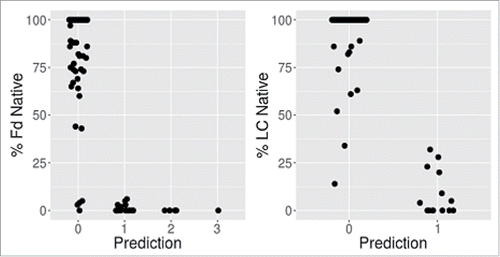
shows a comparison of the experimental and predicted number of oxidation states. Overall, we find significant correlation between predicted methionine side-chain SASA and oxidation propensity for both the Fd and the LC. shows the % native as a function of the number of predicted oxidation events using a 9% SASA cutoff. For both the Fd and LC, there are no false positives observed. A false positive case is defined as one where the calculation predicts an oxidation event that is not supported by experimental observation. There are, however, 7 and 4 apparent false negatives observed for the Fd and LC, respectively. Here the number of experimentally observed oxidation events exceeds the number predicted by the calculation. The 11 false negative cases are listed in . Some of these samples were further investigated by TPM to locate the site of oxidation at the single amino acid level.
Table 1. Comparison of experimental assignments versus computational predictions at 9% SASA cutoff.
Table 2. List of 11 false negatives in Fd and light chain.
Figure 4. High methionine SASA (> 9%) is indicative of HC (Fd) and LC oxidation event(s).
Three of the 4 false negatives for the LC, ponezumab, motavizumab and tovetumab, have a methionine at position 4. For 2 of these cases, ponezumab and motavizumab, which were investigated using peptide mapping, the oxidation of methionine at position 4 was confirmed (). The theoretical prediction of a SASA of 0–1% at this position is not consistent with the experimental data. In the entire data set, there are 61 instances of a LC with methionine at position 4 and no other methionine elsewhere. In this set, 57 cases have 100% native LC, 3 additional antibodies have % native LC above 70% but under 100%. The crystal structure of ponezumab (Protein Data Bank (PDB) ID: 3U0T) clearly shows that the methionine at FR1 in LC is buried. While the predictions are consistent with the majority of the data, the detection of oxidation for these cases indicates exceptions that could result from structural flexibility at the LC N-terminus that is not captured well by the modeling where the backbone is held fixed.Citation8 The fourth false negative consists of methionine at L48 in the LC-FR2 of gemtuzumab, which is rare in that all other antibodies in this set have an isoleucine at this position. We identified 9 PDB structures (1AD9, 1DBB, 2AEP, 2AJ3, 4XMP, 4ZYK, 5EA0, 5BZW, 5I9Q) containing a methionine at L48. The fractional SASA ranged from 0–0.6% over these experimental structures, thus suggesting the SASA prediction itself does not explain the failure to anticipate this oxidation event.
Table 3. Peptide mapping MS2 analysis of selected samples with false negative occurrences.
For all 4 HC cases investigated further by peptide mapping, the methionine near the end of H3 is likely oxidized, despite the fractional exposed SASA being ∼2%. The crystal structures for trastuzumab (PDB IDs: 1FVC, 1N8Z, 4HKZ, 4UB0) also show that the methionine toward the end of the H3 is buried. Within the 121 samples investigated, 22 have methionine at the same site displayed in ; despite being in the highly variable complementarity-determining region (CDR)H3, JH genes in both mouse and human encode for a methionine at this position, and thus explain the relatively high frequency of this feature. There are 7 cases with % native Fd <50%, 8 cases with 50% ≤ Fd < 100% and 7 cases with 100% native Fd. These data indicate that the factors affecting the oxidation of methionine at this position are not entirely captured by the current model for predicting SASA or by the static conformations in the crystal structures.
Table 4. Methionine near the end of H3.
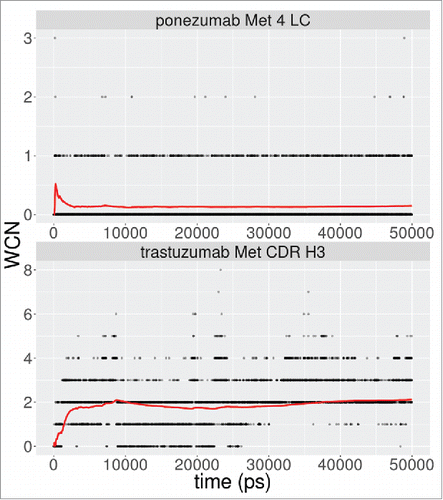
Molecular dynamics (MD) simulations were performed for 2 of the false negative cases, ponezumab and trastuzumab, to investigate whether explicit consideration of structural flexibility could explain the experimentally observed oxidation. Five-thousand snapshots over a 50 ns trajectory were analyzed to determine the 2-shell water coordination number (WCN)Citation7 and SASA for the sulfur atom, since these metrics have been shown to accurately capture oxidation propensity over a wide diversity of proteins.Citation8,13 The results are show in . For ponezumab, we did not observe any exposure of sulfur atom at L4. The average WCN over the entire trajectory was 0.15 waters. In the case of trastuzumab, for the methionine near the end of the CDR H3, we observed an average sulfur atom SASA of 0.005Å2 and an average WCN of 2.1 waters. While the sulfur atom is not exposed, this average of the WCN satisfies the condition of at least 2–3 waters needed for the oxidation of methionine,Citation8,13 which could indicate that conformational flexibility in solution enables oxidation at this position.
Figure 5. Water coordination number for methionine sulfur atom in a shell of radius 5.5Å. The red lines denote the running average over the simulation.
Discussion
Methionine oxidation in the Fc may lower the binding affinity of IgGs to Protein A and FcRn, leading to decreased recovery in process, and faster in vivo clearance.Citation12 Likewise, oxidation in the variable region may lead to decreased antigen binding activity or loss of function, as well as increasing heterogeneity. Selection of candidates stable to oxidation reduces downstream surprises and risks.
Sequence liability analysis, simply based on the presence or absence of methionine residues, does not always correlate with experimental results. Within the set of 121 antibodies investigated in this study, 115 and 95 have at least one methionine residue in the heavy chain or the light chain, respectively. Some of the methionine residues are not structurally exposed and do not add any risk of oxidation, under the conditions studied. In this work, all the exposed methionine in either LC or Fd, as predicted by the methionine side-chain SASA model, exhibited a significant propensity for oxidation under the forced oxidation condition. No false positive predictions were observed. This is useful for the early antibody discovery stage, where up to hundreds of candidates for each target require chemical stability assessment. Decreasing the time and cost in going from sequence to physical production of mAbs, and in subsequent stressing experiments, can add substantial value to a turnaround discovery cycle. It must be noted, however, that our study included several examples of false negative observations, where oxidation was observed experimentally without a corresponding prediction of an exposed methionine residue. The differences observed here could be due to expression host, process conditions, or the failure of the static crystal structure to capture the solution conformations that expose the methionine residue. The sequence-based machine learning model for SASA prediction model described here is based on the analysis of static crystal structures of 712 mAbs, and does not explicitly account for the dynamic conformation changes in solution. Accounting for structural flexibility using MD simulations has successfully captured relative oxidation propensitiesCitation8,13 better than using static SASA. For the 2 mAbs investigated in this study using MD, ponezumab and trastuzumab, the results were inconclusive. This observation emphasizes the importance of theoretical SASA prediction followed by high-throughput experimental verification to support early-stage discovery. We examined the correlation of oxidation propensity with biophysical characteristicsCitation11 for this set of mAbs, such as estimated isoelectric point, HEK expression titer, thermostability (Fab melting temperature) and hydrophobicity (hydrophobic interaction chromatography retention time), and found that all rank correlation coefficients were less than 0.2.
Materials and methods
The 121 antibodies were expressed in HEK293 cells. The VH and VL encoding gene fragments (Integrated DNA Technologies, Coralville, IA) were subcloned into heavy and light chain pcDNA 3.4+ vectors (ThermoFisher, Waltham MA). The variable region sequences are listed in the Table S1 (Sequence tab of the Excel file). All mAbs were expressed as IgG1 isotype. The corresponding vectors were co-transfected into HEK293 suspension cells. After 6 d of growth, the cell culture supernatant was harvested by centrifugation and passed over Protein A agarose (MabSelect SuRe™, GE Healthcare Life Sciences, Pittsburgh PA). The bound antibodies were then washed with phosphate-buffered saline and eluted with buffer (200 mM acetic acid / 50 mM NaCl pH3.5) into 1/8th volume 2 M HEPES, pH 8.0. The final products were buffer exchanged into 25 mM HEPES, 150 mM sodium chloride, and pH 7.3.
Thirty% H2O2 (H1009), DL methionine (64340), DTT (D0632), and HEPES (H3375) were purchased from Sigma. Sodium chloride (14734) was purchased from Hawkins. FabRICATOR (A0-FR1–050) was purchased from Genovis.
Commercial therapeutic mAb drugs were obtained from Myoderm.com.
Generation of oxidized mAbs
Samples were subject to 0.1% (w/w) H2O2 treatment of 24 hours with protection from light. Oxidation reaction was quenched by adding free methionine in solution at ∼4:1 (methionine:H2O2) molar ratio. Samples were stored at -80°C before further treatment or analysis.
Ides digestion
The oxidized mAb samples were digested using FabRICATOR (Genovis, 1 unit/µg IgG) at 37°C for 30 minutes. Following digestion, samples were stored at – 80°C until LCMS analysis.
LC-MS
The Ides-digested mAb samples were reduced by DTT, followed by middle-down LCMS analysis on a Bruker maXis4G mass spectrometer coupled with an Agilent 1100 HPLC (Agilent). A POROS R2 10 µm (2.1×30 mm) reversed phase column was used to remove salt in the samples. A fast LC flow at 2 mL/min allows the separation between sample and salt and elution of samples and regeneration of column to finish within a 2.1 min cycle. A T-junction is used to deliver only 0.15 mL/min sample flow into the mass spectrometer for sample analysis.
The Bruker maXis 4G mass spectrometer was run in positive ion mode with detection in the range of 750 to 2500 m/z. The remaining source parameters were set as follows; the capillary was set at 5500V, the Nebulizer at 4.0 Bar, dry gas at 4.0 l/min, and dry temp set at 200°C.
The MS spectra were analyzed using Bruker Data Analysis version 4.1 and the deconvolution was accomplished using maximum entropy deconvolution with a mass range of 20 to 30 kDa.
Tryptic peptide mapping
The TPM assay was performed as described previously except the digestion time and enzyme-to-protein ratio.Citation14 Briefly, 5 µL mAb samples at ∼1 µg/µL were mixed with 10 µL of the reduction buffer (8 M urea, 20 mM ammonium bicarbonate, 10 mM DTT, pH 7.8), and incubated at 70°C for 3 minutes. The reduced samples were then digested by adding 100 µL of 5 ng/µL trypsin solution (20 mM ammonium bicarbonate, pH 7.8) and incubating at 37°C for 20 minutes. The digestion was quenched by adding 4.8 µL of 20% trifluoroacetic acid. All tryptic peptides were eluted on an Agilent 1200 HPLC (Agilent) equipped with an Agilent AdvanceBio Peptide Mapping column (2.7µm, 2.1×150 mm). The gradient is 2–60% solvent B (0.1% formic acid at acetonitrile) in 30 minutes and the flow rate is 0.2 mL/min. Peptides separated on the column were detected by a UV detector and a Thermo Q Exactive mass spectrometer. Acquired MS data were analyzed using Thermo Biopharma Finder software followed by manual inspection.
Machine learning for prediction of methionine SASA
Input set of crystal structures
A curated set of 712 antibody crystal structures with kappa light chains was used in this study. The input to the curation process were the PDB structures from RCSBCitation15 as of November 2015. We parsed coordinate entries from the PDB and performed a sequence alignment of individual chains to human and mouse antibody variable (VH, Vκ) and junctional region (JH, Jκ) germline sequences obtained from the International ImMunoGeneTics Information System (IMGT) database.Citation16,17 Chains that returned alignments with at least 40 exact matches over the variable region and at least 6 exact matches in the junctional regions were retained, and the type (light or heavy) was assigned using the alignment with the highest identity over the variable region. Within a coordinate entry, pairs of light and heavy chains are assembled to generate the coordinates of the Fv (variable) region. If multiple light and heavy chain copies were present, the pair with the most number of light-heavy residue-residue contacts (using a 5 Å heavy-atom distance cutoff), subject to a minimum of 35 contacts, was designated as the representative Fv. The assignment of pairs was validated by cross-checking against the biologic unit information provided in the REMARK 350 section of the PDB header. Structures with missing backbone atoms were removed from further analysis. For structures containing identical CDR H3s, the highest resolution structure was retained in the final set. Except for CDR H3, the residues were re-numbered using the Kabat numbering scheme. Citation18 For CDR H3, we adopted a numbering scheme adapted from that proposed by IMGT.Citation17 YASARACitation19 was used to delete crystallographic waters and groups containing heteroatoms, as well as add any missing side-chains. Finally, all side-chains were optimized using steepest-descent minimization with the YASARA2 forcefield.
Generation of methionine mutations
Following the generation of the curated set of antibodies, a single methionine mutation was walked along the entirety of the heavy and light chains. The energy of the structure was minimized using the Dead-end-elimination (DEE) algorithmCitation20 on biconnected subgraphsCitation21 over a set of flexible side-chains that included the site of mutation and neighboring positions with a less than 5Å heavy-atom contact distance to the mutation site. Discrete rotamers were generated using the backbone-dependent Dunbrack rotamer libraryCitation22 in the following manner:
For the site of the mutation, rotamers with a probability greater than 0.1 times the highest probability rotamer were included. Nine variants were generated for each rotamer by a combinatorial perturbation of the χ1 and χ2 side-chain dihedrals by ± one standard deviation.
For the neighboring sites, the side-chain was kept at its native rotameric state. However, additional variants about this native state were generated in the same manner as for the mutation site by using the standard deviations from the closest rotamer in the rotamer library.
The OPLS-AACitation23 force-field was used to assign parameters and calculate energies for the van der Waals (VdW), Coulomb and side-chain torsional energies. A pairwise-decomposable Generalized-Born formulationCitation24 was used to calculate solvation energies with a solvent dielectric of 78.5 and solute dielectric of 4. A cutoff of 15 Å was used for the non-bonded energy calculation. Since the optimization was over a discrete space of side-chain conformations, the repulsive component of the VdW termCitation25 was scaled by 0.009.Citation26 The bond lengths, bend angles, improper torsions and backbone degrees of freedom were kept fixed.
In many cases, a mutation to methionine results in clashes that cannot be resolved by optimization over the side-chain degrees of freedom. This can potentially indicate that the structure is not an appropriate template for the new sequence containing the change to methionine. To reliably eliminate these cases, we performed an additional DEE optimization using the wild-type (WT) amino acid. Thus, for each position in each starting antibody structure, we generated 2 minimized structures; one with WT and one with the methionine mutant. Structures with methionine that showed more than one additional heavy-atom clash compared with the WT were discarded from further analysis. Two atoms were considered as clashing when the distance between them was less than 85% of the sum of their VdW radii.
An internal computational pipeline was developed in Python and C++ to perform the energy calculations and the DEE optimization.
Random forest regression
The SASA for methionine was calculated using the mutated and side-chain repacked structures prepared above. For each position i along the antibody sequence, a random forest regressor was trained to predict the SASA using the following model:where, SASAi is the fractional SASA calculated from structure containing the methionine mutation at position i, AAi is the original WT amino acid at the position, (CDR Lengths) is the set of lengths of the heavy- and light-chain CDRs and Neighbor AA to i are the amino acids at neighboring positions to i. A value of 164.67Å2 corresponding to the maximum exposed side-chain SASA of methionine,Citation28 was used to convert the absolute SASA to a fractional value. The amino acids in the model were encoded using an ordinal scale based on the ranks of the amino acid when ordered by increasing maximum exposed side-chain SASACitation28 from G < A < S < C < D < P < T < N < V < L < E < Q < H < I < M < K < F < Y < R < W.
To reduce the complexity of the model, we limited the set of neighbors in the model in the following manner. Using the 712 input structures, we calculated the probability that 2 positions were in contact using a Cβ-Cβ cutoff of 12 Å. The set of positions in contact with the target position with a probability exceeding 0.5 were used in the model equation.
The randomForestCitation29 package (version 4.6–12) in R 3.2.4 was used to fit the model with 2500 trees. All other parameters were set to their default values. The resulting forest was used to predict the SASA for methionine residues in the set of 121 antibodies. The results of the predictions are listed in the Table S1 (Predictions tab of the Excel file).
Molecular dynamics simulations
All-atom MD simulations for ponezumab (PDB ID: 3U0T) and trastuzumab (PDB ID: 4HKZ) Fab regions were performed using YASARA.Citation19 The AMBER03Citation30 forcefield with explicit water was used with long-range Coulomb interactions calculated using the Particle Mesh Ewald method. The simulations were run in an orthorhombic box with periodic boundary conditions for 50 ns at a temperature of 298 K and a pH of 7.4. The net charge on the system was neutralized by the addition of Na+ and Cl− ions. To minimize simulation artifacts due to long-range interactions, the cell dimensions were set to 50Å greater than the extent of the Fab molecule. There were ∼57500 waters and 160 pairs of Na+/Cl− ions in the simulation. The total number of atoms in the simulation were ∼180,000. Simulation snapshots were saved every 10 ps and used for subsequent analysis. Equilibration in the simulations was ascertained based on the Fv Cα RMSD from the starting structure (Fig. S1). For the methionine residues that were flagged as false negatives by the predicted SASA from the machine-learning method, the number of water molecules within a shell of 5.5Å,Citation8,13 and the SASA of the sulfur atom, were calculated over the last 40 ns of the simulation trajectory.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Data.zip
Download Zip (155.6 KB)Acknowledgments
All the antibodies characterized in this work were identified, produced and purified through the combined efforts of numerous people at Adimab, LLC, including those in the computational biology, molecular core and high throughput expression departments. We appreciate the critical review, discussion, guidance and support provided by Arvind Sivasubramanian, Kristin Rookey, William Roach, Michael Ruse, Eric Krauland, Dane Wittrup and Tillman Gerngross. We also thank the anonymous reviewers for comments and suggestions that resulted in an improved manuscript.
References
- Hawe A, Wiggenhorn M, van de Weert M, Garbe JH, Mahler HC, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci 2012; 101:895-913; PMID: 22083792; http://dx.doi.org/10.1002/jps.22812
- Torosantucci R, Schoneich C, Jiskoot W. Oxidation of therapeutic proteins and peptides: structural and biological consequences. Pharm Res 2014; 31:541-53; PMID: 24065593; http://dx.doi.org/10.1007/s11095-013-1199-9
- Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol 2011; 48:860-6; PMID: 21256596; http://dx.doi.org/10.1016/j.molimm.2010.12.009
- Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci 2009; 18:424-33; PMID: 19165723; http://dx.doi.org/10.1002/pro.45
- Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta 2005; 1703:93-109; PMID: 15680218; http://dx.doi.org/10.1016/j.bbapap.2004.08.007
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol 1971; 55:379-400; PMID: 5551392; http://dx.doi.org/10.1016/0022-2836(71)90324-X
- Chu JW, Yin J, Wang DI, Trout BL. Molecular dynamics simulations and oxidation rates of methionine residues of granulocyte colony-stimulating factor at different pH values. Biochemistry 2004; 43:1019-29; PMID: 14744147; http://dx.doi.org/10.1021/bi0356000
- Chennamsetty N, Quan Y, Nashine V, Sadineni V, Lyngberg O, Krystek S. Modeling the oxidation of methionine residues by peroxides in proteins. J Pharm Sci 2015; 104:1246-55; PMID: 25641333; http://dx.doi.org/10.1002/jps.24340
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 2014; 111;18601-6; PMID: 24324143; http://dx.doi.org/10.1073/pnas.1421779112
- Thirumangalathu R, Krishnan S, Bondarenko P, Speed-Ricci M, Randolph TW, Carpenter JF, Brems DN. Oxidation of methionine residues in recombinant human interleukin-1 receptor antagonist: Implications of conformational stability on protein oxidation kinetics. Biochemistry 2007; 46:6213-24; PMID: 17480058; http://dx.doi.org/10.1021/bi700321g
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A 2017; PMID:28096333; http://dx.doi.org/10.1073/pnas.1616408114
- Gao X, Ji JA, Veeravalli K, Wang YJ, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al. Effect of individual Fc methionine oxidation on FcRn binding: Met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J Pharm Sci 2015; 104:368-77; PMID: 25175600; http://dx.doi.org/10.1002/jps.24136
- Chu JW, Brooks BR, Trout BL. Oxidation of methionine residues in aqueous solutions: Free methionine and methionine in granulocyte colony-stimulating factor. J Am Chem Soc 2004; 126:16601-7; PMID: 15600366; http://dx.doi.org/10.1021/ja0467059
- Li X, Xu W, Wang Y, Zhao J, Liu YH, Richardson D, Li H, Shameem M, Yang X. High throughput peptide mapping method for analysis of site specific monoclonal antibody oxidation. J Chromatogr A 2016; 1460:51-60; PMID: 27432793; http://dx.doi.org/10.1016/j.chroma.2016.06.085
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000; 28:235-42; PMID: 10592235; http://dx.doi.org/10.1093/nar/28.1.235
- Lefranc MP. IMGT, the International ImMunoGeneTics Information System. Cold Spring Harb Protoc 2011; 2011:595-603; PMID: 21632786; http://dx.doi.org/10.1101/pdb.top115
- Lefranc MP, Pommi?? C, Kaas Q, Duprat E, Bosc N, Guiraudou D, Jean C, Ruiz M, Da Piédade I, Rouard M, et al. IMGT unique numbering for immunoglobulin and T cell receptor constant domains and Ig superfamily C-like domains. Dev Comp Immunol 2005; 29:185-203; PMID: 15572068; http://dx.doi.org/10.1016/j.dci.2004.07.003
- Al-Lazikani B, Lesk aM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927-48; PMID: 9367782; http://dx.doi.org/10.1006/jmbi.1997.1354
- Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009; 77 Suppl 9:114-22; PMID: 19768677; http://dx.doi.org/10.1002/prot.22570
- Pierce Na, Spriet Ja, Desmet J, Mayo SL. Conformational splitting: A more powerful criterion for dead-end elimination. Journal of Computational Chemistry 2000; 21:999-1009; http://dx.doi.org/10.1002/1096-987X(200008)21:11%3c999::AID-JCC9%3e3.0.CO;2-A
- Canutescu Aa, Shelenkov Aa, Dunbrack RL. A graph-theory algorithm for rapid protein side-chain prediction. Protein science : a publication of the Protein Society 2003; 12:2001-14; PMID: 12930999; http://dx.doi.org/10.1110/ps.03154503
- Dunbrack RL, Others. Rotamer libraries in the 21st century. Current opinion in structural biology 2002; 12:431-40; PMID: 12163064; http://dx.doi.org/10.1016/S0959-440X(02)00344-5
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides †. The Journal of Physical Chemistry B 2001; 105:6474-87; http://dx.doi.org/10.1021/jp003919d
- Pokala N, Handel TM. Energy functions for protein design I: efficient and accurate continuum electrostatics and solvation. Protein science : a publication of the Protein Society 2004; 13:925-36; PMID: 15010542; http://dx.doi.org/10.1110/ps.03486104
- Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein–Protein Docking with Simultaneous Optimization of Rigid-body Displacement and Side-chain Conformations. J Mol Biol 2003; 331:281-99; PMID: 12875852; http://dx.doi.org/10.1016/S0022-2836(03)00670-3
- Pierce B, Weng Z. ZRANK : Reranking Protein Docking Predictions With an Optimized Energy Function. Bioinformatics 2007; 1086:1078-86; PMID:17373710; http://dx.doi.org/10.1002/prot.21373
- Chennamsetty N, Quan Y, Nashine V, Sadineni V, Lyngberg O, Krystek S. Modeling the Oxidation of Methionine Residues by Peroxides in Proteins. J Pharm Sci 2015; 104:1246-55; PMID: 25641333; http://dx.doi.org/10.1002/jps.24340
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B 2010; 114:6614-24; PMID: 20411962; http://dx.doi.org/10.1021/jp911706q
- Liaw A, Wiener M. Classification and Regression by randomForest. R News 2002; 2:18-22.
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 2003; 24:1999-2012; PMID: 14531054; http://dx.doi.org/10.1002/jcc.10349