
ABSTRACT
Of the inhibitory checkpoints in the immune system, programmed death 1 (PD-1) is one of the most promising targets for cancer immunotherapy. The anti-PD-1 antibodies currently approved for clinical use or under development bind to human PD-1 (hPD-1), but not murine PD-1. To facilitate studies in murine models, we developed a functional antibody against both human and murine PD-1, and compared the epitopes of such antibody to a counterpart that only bound to hPD-1. To quickly identify the epitopes of the 2 antibodies, we used alanine scanning and mammalian cell expression cassette. The epitope identification was based on PD-1-binding ELISA and supported by affinity ranking of surface plasmon resonance results. The hPD-1 epitopes of the 2 functional antibodies were also compared with the binding region on hPD-1 that is responsible for PD-L1 interaction. In silico modeling were conducted to explain the different binding modes of the 2 antibodies, suggesting a potential mechanism of the antibody cross-species binding.
Abbreviations
| PD-1 | = | programmed death-1 |
| PD-L1 | = | programmed death-ligand 1 |
| CDR | = | complementary-determining region |
| hPD-1 | = | human PD-1 |
| mPD-1 | = | murine PD-1 |
Introduction
Blockade of a specific pathway that shields tumor cells from immune response is promising in the treatment of multiple cancers, including advanced melanoma, ovarian cancer and advanced non-small cell lung cancer.Citation1-3 Programmed death-1 receptor (PD-1), an immune inhibitory receptor expressed on activated B cells, T cells and myeloid cells,Citation4 has critical roles in regulating stimulatory and inhibitory signals in the immune system.Citation5 PD-1 belongs to the CD28/CTLA-4 subfamily within the immunoglobulin super family. PD-1 ligands PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273) present not only on antigen presenting cells, but also on various types of tumor cells. As one of the immune-checkpoint proteins, PD-1 plays an important role in down-regulating T-cell activation and mediating the escape of tumor cells from immune surveillance by binding to its ligands PD-L1 and PD-L2 expressed on the surface of tumor cells.Citation6-8 Therefore, PD-1 is a target for immunotherapy.Citation9-12 To date, 2 anti-PD-1 antibodies (pembrolizumab and nivolumab) have been approved,Citation13, 14 and more are under clinical development.
Although those antibodies are useful in clinical treatment, so far no antibodies targeting human PD-1 (hPD-1) have been shown to cross-react to murine PD-1 (mPD-1). Prior to clinical studies, mouse models are often used to test the efficacy, pharmacokinetics and toxicity of drug candidates; therefore, an antibody that binds both hPD-1 and mPD-1 may simplify and accelerate preclinical studies of PD-1 antibody-based novel therapy, including combination therapy or bispecific antibodies.Citation15-17
Due to the relatively low homology between human and murine PD-1 sequence (62% in extracellular domain), developing a functional antibody cross-reactive to both human and murine PD-1 is especially challenging. However, because human PD-L1 can bind to both hPD-1 and mPD-1,Citation18, 19 we believed that the 2 PD-1 molcules share some structural similarities, and therefore it should be possible to develop a functional antibody binding to both hPD-1 and mPD-1. We immunized rats with hPD-1 and mPD-1, screened a large number of rat hybridoma clones, and successfully identified functional antibodies against both hPD-1 and mPD-1. One of the lead antibodies not only has high binding affinity on hPD-1 and mPD-1, but also shows functionality (significantly improved CD4+ T cell proliferation and interferon (IFN)-γ secretion in mixed lymphocyte reaction assay). It also prolonged survival time of tumor-bearing mice (data not shown). To our knowledge, this is the first report of a functional anti-PD-1 antibody with human and murine cross-species reactivity. Here, we report the epitope of this antibody and explore the mechanism of its cross-species binding activity.
Results
Characterization of antibodies
A series of monoclonal antibodies were generated by screening a large number of hybridoma clones after immunizing rats with hPD-1 and mPD-1 proteins. Among them, rat IgG2a antibody r16.88.9 (R9), which has a kappa light chain, was found to bind to both hPD-1 and mPD-1 (). Another rat IgG2b antibody r1.103.11 (R11), which has a lambda light chain, only bound to hPD-1 but not to mPD-1 (). The third hPD-1-binding rat antibody r11.148.10 (R10) was not functional in blocking the interaction of PD-1 and PD-L1, but it still bound to hPD-1 (). This antibody was chosen as a control antibody to be used to monitor the structure stability of PD-1 protein after alanine mutations. The binding activities of the 3 antibodies were confirmed via fluorescence activated cell sorting (FACS) using a hPD-1-positive cell line (). In the FACS assay, R10 was not capable of blocking hPD-1 binding to PD-L1, while R11 and R9 could block hPD-1 binding to hPD-L1 with IC50 at 13.14 and 2.07 nM, respectively.
Figure 1. Characterization of antibodies. (A) Cross-reactivity of anti-PD-1 antibodies to human/mouse PD-1. Each antibody was coated on 96-well plate overnight and incubated with hPD-1-His protein (filled shapes) and mPD-1-His protein (open shapes), and then HRP-conjugated anti-His antibody was added for detection. (B-D) Binding titration of the antibodies to hPD-1 expressed on CHO-S cells (B), competition of anti-PD-1 antibodies with hPD-L1 to hPD-1 expressed on CHO-S cells (C) and competition of anti-PD-1 antibody R9 with mPD-L1 to mPD-1 expressed on 293F cells were detected using FACS instrument as described in Materials and Methods. (E-F) Anti-PD-1 antibody R9 can enhance CD4+ T cell proliferation (E) and IFN-γ production (F) in a dose-dependent manner in mixed lymphocyte reaction assay as described in Materials and Methods. (unpaired, 2-tailed t test; * p< 0.05, ** p< 0.01, *** p< 0.001). (G-H) Comparison of R9, pembrolizumab (Pemb) and nivolumab (Nivo) in hPD-1 binding (G) and hPD-L1 competition (H) using FACS method as described in Materials and Methods.
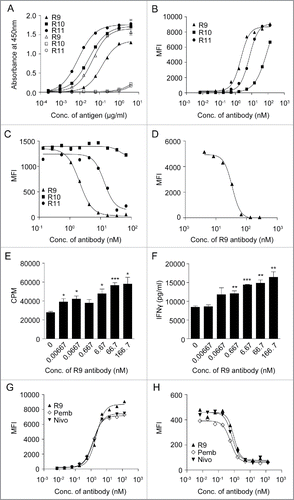
R9 could block hPD-1 binding to hPD-L1 with IC50 at 0.92 nM () and block mPD-1 binding to mPD-L1 with IC50 at 33.79 nM (). R9 also significantly improved CD4+ T cell proliferation and IFN-γ secretion in a dose-dependent manner in mixed lymphocyte reaction (MLR) assay ( and ).
Moreover, R9 showed comparable hPD-1 binding activity and hPD-L1 competition activity with pembrolizumab and nivolumab. In hPD-1 binding assay (), EC50 of R9 was 2.16 nM, which was close to pembrolizumab and nivolumab (EC50 at 1.32 nM and 1.50 nM, respectively). In hPD-L1 competition assay (), R9 was capable of blocking PD-1 binding to PD-L1 with IC50 at 0.92 nM, which was also very close to pembrolizumab and nivolumab (IC50 at 0.57 nM and 0.72 nM, respectively).
Epitope mapping
To identify the epitopes of R9, R11 and R10, we conducted an alanine scan on the extracellular domain of hPD-1, and examined how the mutations affected binding of the 3 antibodies. Codon of alanine residue on hPD-1 was mutated to glycine codon, and the codons of all other residues were mutated to alanine codon. The binding results were mainly based on enzyme-linked immunosorbent assay (ELISA) data and supported by surface plasmon resonance (SPR) data. The mutations on hPD-1 that significantly reduced antibody-binding (> 55%) are shown in Table S1 and Fig. S1. Analysis of all these residues on the hPD-1 crystal structures (PDB code 3RRQ and 4ZQK) revealed that some of the residues (e.g., Val144, Leu142, Val110, Met108, Cys123; see Table S1 for complete list) were fully buried in the protein. These residues are likely critical to maintain PD-1 structure rather than directly contacting antibodies. The observed binding reduction probably resulted from the instability or even collapse of the hPD-1 structure after alanine substitutions. This was evidenced by the fact that these mutations also affected PD-1 binding of the control antibody R10, which did not compete with PD-L1, and thus likely bound to a different location on hPD-1. These PD-1 residues were therefore not considered part of the epitopes of the antibodies. The final determined epitope residues are listed in . There were 9 positions for R11 and R9, and 10 residues for the control antibody R10. To understand how the discovered antibodies blocked hPD-L1, the hot spots residues interacting with hPD-L1Citation18 were also identified ().
Table 1. List of all identified hot spot residues on hPD-1. The first 2 columns are the hot spots for PD-L1 binding. Data were obtained from the latest paper of hPD-1/hPD-L1 co-crystal structure.Citation18 The rest are the hot spot residues binding antibodies, identified in alanine scanning experiments (Cutoff: binding fold change < 0.55). All the identified residues are listed in the sequence order (the original ranking can be found in Table S1).
It is noteworthy that some alanine-substitutions caused local structural perturbations that may not have been significant enough to affect the binding of R10. Therefore, use of the control antibody may not filter out all the false hot-spot residues. For example, analyzing the hPD-1 crystal structure revealed that residues Pro35 and Phe82 were partially buried in the protein, but the nuclear magnetic resonance (NMR) structures showed that they could be exposed sometimes. Since no further experiments were conducted to elucidate these details, we kept the data in as they were.
From the data in , it was quite clear that the 2 functional antibodies R11 and R9 shared a very similar epitope, whereas the nonfunctional antibody R10 had a different epitope. The epitope residues shared by R9 and R11 composed similar residues from C, C′, and G strands, as well as the FG loop. The only notable differences were residues Asp85 and Gln133, which were identified to be important to R11 binding, but not to R9. Checking the positions of these 2 residues on the PD-1 structures revealed that Gln133 was located in the G strand, which was structurally quite conserved in both human and murine PD-1 proteins, while Asp85 was located in the region where mPD-1 held a β-sheet, but hPD-1 had a flexible loop.Citation18, 20 The interactions associated with Asp85 could potentially be a more important obstacle that prevented the antibody R11 from being species cross-reactive.
The binding sites of hPD-L1 were expected to be close to the epitopes of R11 and R9, as its interactions to hPD-1 could be effectively blocked by the 2 antibodies. To evaluate this possibility, all residues shown in were therefore mapped on the hPD-1 crystal structure for better visualization (). Again, 2 functional antibodies R11 and R9 shared a similar epitope ( and ), except residue Asp85 (shown as a red dot) on the C'D loop of hPD-1 (corresponding to the C″ strand of mPD-1). Overall, the binding sites of hPD-L1 concentrated in the middle of C, F, G strands, the regions partially overlapped with the epitopes of 2 functional antibodies (). Antibody R11 and R9 blocked the binding of hPD-1 to hPD-L1 likely due to steric hinderance. In contrast, the data indicated that the control antibody R10 bound to a completely different location, not involving the PD-L1 binding sites at all ().
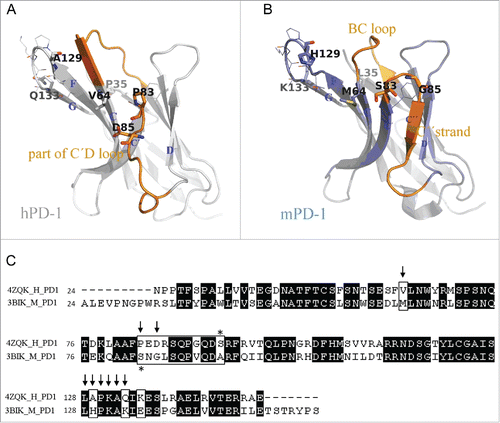
Figure 2. Hot spot residues mapped on hPD-1 structure. (A) hPD-L1 binding site. Data were obtained from the literature.Citation18 (B-D) Binding sites of antibody R11, R9 and the control antibody R10, respectively. Data were from . Colors in the pictures are included to help distinguish differences between epitopes. The structure of hPD-1 was taken from the crystal structure (PDB code 4ZQK), whose missing loops (Asp85-Asp92) were remodeled by adopting initial conformations from the NMR structure (PDB code 2M2D). The back β-strands are A′, B, E, D, A and G′. The front sheets are labeled in G, F, C and C′. The C″ strand observed in mPD-1 lost its secondary structure in hPD-1.Citation18, 20
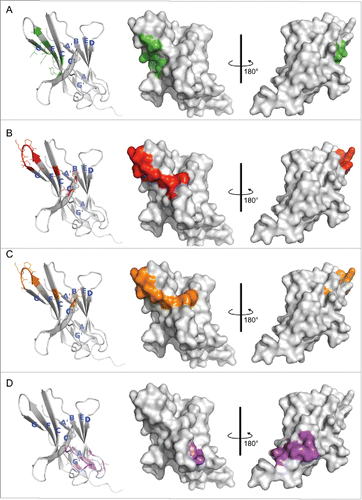
Data in and were consistent with the findings from the competition assay in , and clearly explained the reason why R11 and R9 could effectively block PD-L1 binding, and R10 cannot. Asp85, which is important to R11 binding, but not to R9, could potentially be the key residue that affects their cross-reactivity to mPD-1.
Kinetic analysis of macromolecular interaction using SPR biosensors
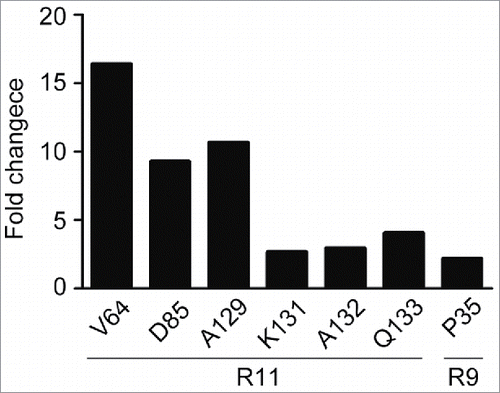
To further confirm the epitopes identified by ELISA, we measured the off-rate constant Kd of hPD-1 mutants to antibody R11 and R9 using SPR technology. Although SPR was less sensitive than ELISA, the obtained Kd values could be used for ranking antibody affinity. All data were normalized to the positive control (several different alanine codons were point-substituted for alanine residues on hPD-1) and the cutoff was set at 2-fold higher than the positive control. Six hot spot residues, Val64, Ala129, Asp85, Gln133, Ala132, Lys131 on hPD-1 were identified for R11 and one residue Pro35 for R9 (). All of the hot spot residues found by SPR were consistent with the epitopes detected by ELISA.
Figure 3. Kinetic analysis of macromolecular interaction. The off-rate constant K(d) of PD-1 mutants to R11 and R9 antibodies were measured using surface plasmon resonance biosensors. Fold changes were calculated based on the average value of control mutants. The fold changes of 6 residues to R11 and one residue to R9 exceeded the cutoff (2-fold).
Modeling the interaction of antibody and hPD-1 protein
As the epitopes of R9 and R11 were very similar, to explain the difference of the 2 antibodies in cross-species binding, we used in silico modeling to predict their binding mode and to shed light on the possible mechanism. Antibody Fv homology modeling and antibody-antigen docking were conducted to search their possible binding poses on the available hPD-1 structure. Complex models were selected and ranked based on comprehensive evaluations of computational docking scores, as well as experimental mutagenesis data. showed the final selected binding poses of 2 studied antibodies.
Figure 4. Putative binding modes of R11 (A) and R9 (B) to hPD-1, conducted by in silico modeling. The sticks on hPD-1 structures are the hot-spot residues listed in . The lines show the neighboring environment near the epitope residues. VH and VL frameworks were colored in green and cyan, respectively. Heavy chain CDRs were colored in salmon and light chain CDRs were colored in wheat. Computational docking results suggested that the Arg100A on CDR H3 of antibody R11 formed a salt bridge (yellow dashes) with Asp85 on hPD-1.
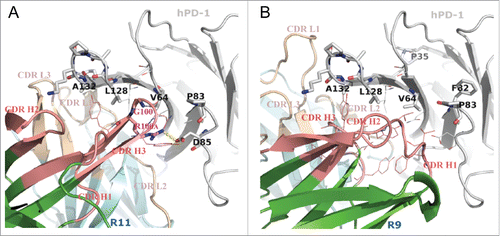
Since the epitopes of the 2 antibodies were very similar, it was not surprising that their binding modes shared some common features. Computational docking suggested that both antibodies used their heavy chains to bind to the hot-spot residues on C, C′ strand and C'D loop (C″ strands on mPD-1 correspondingly), i.e., residue Val64, Pro83 and Asp85. The consecutive epitope residues Leu128-Gln133 on the FG loop and G strand behaved like a linear epitope, buried in the middle of VL-VH interface in both cases.
Despite some common ground, the binding details were proposed to be quite different, which could help explain why they behaved differently in cross-reacting to mPD-1. Antibody R11 has a relatively long complementary-determining region (CDR) H3 loop (13 residues in Kabat numbering). Besides burying the FG loop, the docking model () suggested that it might also directly bind to Val64 and Asp85 of hPD-1 by means of 2 residues on the tip of the loop, which were Gly100 and its neighbor Arg100A. Because Gly100 had no side-chain atoms and was small enough, the tip of CDR H3 could insert into a hydrophobic cleft on hPD-1 made by Val64 and Leu128 without much resistance. This special posture in turn anchored and stabilized the CDR H3 loop, and thus provided a suitable distance for residue Arg100A to form a salt bridge with Asp85 in the hPD-1 C'D loop. Asp85 is located in the region where human and murine PD-1 show substantial structural deviations (see discussion below). Since antibody R11 could not cross-react to murine PD-1, the existence of this salt bridge might be critical in affecting the cross-reactivity capability of the antibody.
In contrast, antibody R9 has a very short CDR H3 loop (only 6 residues, Kabat numbering), and was predicted to bind to the hydrophobic residues on the F and G strands (). In addition, the kappa light chain of R9 has a CDR L1 that adopts a much extended canonical structure. The special structure of CDR L1 and H3, combined with the geometries of other CDRs, created a relatively wide and deep groove at VL-VH interface. The FG loop of hPD-1 might be buried in the groove and wrapped around by the CDRs with good structural complementarity. Unlike R11s CDR H3, the conformation and length of R9s CDR H3, once binding to F and G strands, could not contact the C'D loop of the antigen. The docking model suggested that CDR H1 was more likely to directly bind to the C'D region of hPD-1. Residue Asp85 on hPD-1 might form hydrogen bonds with the side chain or backbone atoms of some residues in CDR H1, but no strong interactions like salt bridges were observed in the model. This hypothesis was supported by the result of alanine mutation at Asp85, in which only moderate antibody affinity loss was observed for antibody R9 (Table S1). Antibody R9 was known to exhibit species cross-reactivity. Compared with R11, which had strong interactions to C'D loop of hPD-1, R9s unique cross-reactivity might originate from its binding mainly on the FG loop of the antigen.
The influences of hot spots to antibody cross-reactivity capability
Inspired by docking models, we further investigated the contributions of different parts of the epitope to the cross-reactivity behaviors of the antibodies. We mutated hot spot residues in hPD-1 to their corresponding amino acids in mPD-1, and measured how the antibodies responded to these changes.
The sequences and structures of PD-1 proteins of 2 species are shown in . The hPD-1 hot-spot residues (to R11 binding) that we identified, as well as the corresponding mPD-1 amino acids, are highlighted. Some of the hot-spot residues existed in both human and murine antigens (Leu128, Pro130, Lys131, Ala132), suggesting that they were less likely to affect antibody cross-reactivity capability. Others showed some obvious differences in sequences: Val64-Met64, Pro83-Cys83, Asp85-Gly85, Ala129-His129 and Gln133-Lys133. The positions of these residues were roughly located in 3 regions on PD-1 structure: C strand, C'D loop (including C′ strand) and FG loop (including G strand). We decided to mutate each of 3 hPD-1 segments to the corresponding mPD-1 sequences.
Figure 5. Comparison between human and murine PD-1 structures. (A) Crystal structure of hPD-1 (PDB code 4ZQK). The missing loop (Asp85-Asp92) were remolded. Sticks and lines on the structure are the marks of the identified hot-spot residues in charge of antibody R11 and R9 binding (). (B) Crystal structure of mPD-1 (PDB code 3BIK). Corresponding to the positions of hot-spot residues on hPD-1, sticks and lines on mPD-1 are the amino acids having different and same residue types, respectively. The structural differences between 2 proteins were marked in orange. (C) Sequence alignment of human and murine PD-1 structures. Arrows are the hot-spot residues identified for R11 binding. Marked box highlighted the amino acid differences in mPD-1.
Val64 is the only hot-spot residue in the C strand. The mutant of the first segment (M1) was therefore chosen as V64M. The second segment C'D loop exhibited big structural differences in human and murine PD-1. We superimposed 2 structures and selected the second mutant (M2) 83PEDRSQPGQDC93 to 83CNGLSQPVQDA93 based on the segments that did not overlap well. The third segment (M3) is the entire FG loop comprising Ala129 and Gln133. Considering Lys135-Glu135 was the only neighboring position showing an apparent difference, we included this position in the third mutant (A129H, Q133K, K135E), in case it might affect antibody cross-reactivity. The binding activities of these mutants to both antibodies were evaluated, and the results are displayed in .
Figure 6. The influences of some epitope hot spots to the capability of antibody cross-reactivity. The hot spots located on hPD-1 were changed to the corresponding mPD-1 amino acids and the binding activity of (A) R11 and (B) R9 were tested by ELISA. M1: V64M; M2: 83PEDRSQPGQDC93 to 83CNGLSQPVQDA93; M3: A129H, Q133K, K135E. 2 μg/ml of each antibody was coated at 96-well plate overnight and incubated with (A) 10 ng/ml and (B) 150 ng/ml mutants, then HRP-anti-His antibody were added for detection. The relative binding changes of the PD-1 mutants were normalized to the original binding of antibody and hPD-1 (unpaired, 2-tailed t test; * p< 0.05, ** p< 0.01, *** p< 0.001).
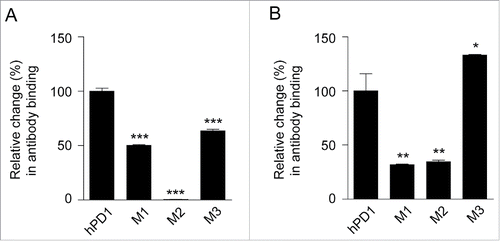
For R11, all M1-M3 mutants of hPD-1 reduced antibody binding, to the extent of 50.4%, 0.5% and 63.6%, respectively (). As expected, M2 mutant almost abolished the binding of PD-1 to the R11 antibody, which indicated the sequence and structure differences of C'D loop (including Asp85) clearly dominated the binding specificity of R11 to hPD-1. In contrast, antibody R9 responded to the 3 mutants in a very different way. M1 and M2 still reduced R9 binding to the level of 31.8% and 34.5%; however, mutant M3 exhibited obvious improvement in the affinity, with the relative binding changes to 133.2% (). This indicated that when R9 binds to mPD-1, the binding loss of other regions was compensated by the increase of better M3 binding. This result was consistent with the hypothesis derived from the docking model, in which that FG loop and G strand were predicated to be the most important segment for R9 binding.
Discussion
Although therapeutic antibodies are developed for human use, in the preclinical stage the efficacy and safety of the molecules need to be evaluated in animal models, which typically involve testing in rodents. Different approaches may be used to test therapeutic candidates. For example, therapeutic antibody candidates can be tested in transgenic mice carrying the corresponding human target, or a surrogate antibody that only binds to a murine target can be used. However, the most straightforward and cost-effective approach is to test an antibody that is cross-reactive to both human and murine targets in a mouse model.
The cross-reactivity of an antibody depends on the structures of 2 antigen homologs and the specific area where the antibody binds. Human and mouse PD-1 proteins share only 62% sequence identity in their extracellular domains, but the main structural differences lie only in the BC loop and the C'D loop of hPD-1 (or the BC loop and C″ strand of mPD-1), as shown in . NMR structures further confirm that hPD-1 contains a very flexible BC loop and has a complete structure loss in the C″ strand region.Citation20 Avoiding these 2 regions, but binding to structurally conserved areas on PD-1, could potentially allow a binding molecule to cross-react to both hPD-1 and mPD-1. This is how human PD-L1 binds to both human and murine PD-1 molcules, as revealed by their complex co-crystal structures (Fig. S2). Guided by this structural knowledge, we hypothesized that antibody R9 might have adopted the same interaction mechanism as hPD-L1, while R11 probably had significant interactions with the structure-variant regions, and therefore only binds to hPD-1. Although no critical hot spot was detected in the BC loop, alanine scanning experiments indeed identified Asp85 in the C'D loop of hPD-1 (corresponding to the C″ strand in mPD-1) as a critical residue for R11 binding, but not for that of R9. This seemed to support the conclusion that R9 did not contact the structural variant C'D loop much, causing it to be insensitive to the structural differences and cross-react with mPD-1. To further illustrate this possibility, we modeled these antibodies and their docking on PD-1.
Computational docking, conducted under the guidance of alanine scanning data, suggested that both antibodies should inevitably interact with the C'D loop of hPD-1 because of the epitope locations and antibody size compared with the PD-1 protein. R11 seemed to mainly bind C'D loop of hPD-1, as a strong salt bridge between CDR H3 and antigen C'D loop was observed (). Antibody R9 was also observed to contact the C'D loop using its CDR H1, but no salt bridge was found. Additionally, R9 might be capable of burying the antigen FG loop due to R9s deep groove at VL-VH interface (). The models suggest that the antibody's binding affinity could be contributed by multiple components.
Inspired by the hypothesis from docking models, we further tested the relative contribution of each epitope component to antibody binding. Alanine scanning told us how antibodies responded to alanine mutation on the antigen, which was informative to locate the epitope,Citation21 but did not provide answers about how the antibody responded and accommodated the murine amino acids at the corresponding location when cross-reacting to mPD-1. Three segments, C′ strand (Val64), C'D loop (83PEDRSQPGQDC93), and FG loop (Ala129, Gln133, Lys135) were mutated to their corresponding murine sequences, and antibody responses to these mutant were evaluated (). The results showed that the binding affinity of antibody R11 was indeed dominated by the interactions with C'D loop on the antigen. Antibody R9, however, exhibited a unique binding behavior. The human/murine chimeric sequence and structural differences in C′ strand and C'D loop obviously suppressed R9 binding, but the mutations in FG loop actually improved binding. Clearly, antibody R9 preferred the mPD-1 FG sequence. This set of data indicated that antibody R9 did not bypass the structural variant region on PD-1 to cross-react to both species. Instead, it actually lost certain binding on C′ strand and C'D loop when binding to mPD-1, but this was compensated by more favorable interactions with mPD-1s FG loop.
The delicate hPD-1/mPD-1 cross-reactivity mechanism revealed here highlights the challenge in generating human/murine PD-1 cross-reactive antibodies. Indeed, antibody R9 was generated by screening thousands of hybridoma clones (data not shown). Considering the importance and broad application of PD-1 antibodies in treating various cancers, we hope the reported antibodies, as well as their binding mechanism, will spark new ideas of potential rational design to achieve cross-reactivity and facilitate preclinical testing of antibody drug candidates.
Materials and methods
Generation of PD-1 expression cassettes
Human and murine PD-1 sequences were obtained from the Swiss-Prot database (Q15116 and Q02242, respectively). For each residue of the human PD-1 extracellular domain (ECD), point amino acid substitutions were made using 2 sequential PCR steps. A pcDNA3.3-hPD-1_ECD.His plasmid that encodes ECD of human PD-1 and a C-terminal His-tag was used as template, and a set of mutagenic primers with a melting temperature (Tm) more than 75°C and 10–15 bases of template-complementary sequence on both sides of desired point mutation codon were used for the first PCR step using the QuikChange lightning multi-site-directed mutagenesis kit (Agilent technologies, 210513), according to the manufacturer's instructions. Dpn I endonuclease was used to digest the parental template after mutant strand synthesis reaction. In the second PCR step, a pair of primers (pCMV: 5′-GCGTTGACATTGATTATTGAC; pTK: 5′-GGAAGAAAGCGAAAGGAGC) were designed for the amplification of the linear DNA expression cassette, which was composed of a CMV promoter, an extracellular domain (ECD) of PD-1, a His-tag and a herpes simplex virus thymidine kinase (TK) polyadenylation. Cassettes were purified using AxyPrep™ PCR clean-up kit (Axygen Biosciences, AP-PCR-250).
Different PD-1 expression cassettes were generated by using an overlap extension polymerase chain reaction (OE-PCR) techniqueCitation21, 22 and were extracted from agarose gels using NucleoSpin™ Gel and PCR Clean-up Kit (Macherey & Nagel, 740609.250).
Expression and quantification of PD-1 mutants
The FreeStyle™ 293 expression system (Life Technologies, K900001) was used for the expression of PD-1 mutants. Briefly, suspension-cultured HEK293F cells (Life Technologies, R79007) in 24-well plates (106 cell/ml/well) were transfected with a mixture of 1–2 μg cassette and 2 μL 293fectin™ reagent (Life Technologies, 12347019), and conditioned media were harvested 4∼5 d after transient transfection. PD-1 mutant protein in conditioned media was quantified by competition ELISA. In brief, gradient-diluted hPD-1_ECD.His protein as standard (range from 4000 ng/ml to 2 ng/ml) and diluted supernatant of each mutant were added to 250 ng/ml hPD-1_ECD.His protein pre-coated plates. Ten minutes later, 0.2 μg/ml of horseradish peroxidase (HRP)-conjugated anti-His antibody (Rockland Immunochemical, 200–303–382) was added to plates. The final reaction was developed using TMB substrate and absorbance was measured at 450 nm after termination with 2 M HCL. The concentration of each mutant in conditioned media was calculated from the standard curve.
ELISA binding assay
2 μg/ml of each antibody was coated on 96-well plate overnight for ELISA binding assay. After interacting with PD-1 mutant protein or with gradient diluted commercial hPD-1_ECD.His (Sino Biological, 10377-H08H) and mPD-1_ECD.His (Sino Biological, 50124-M08H), 0.2 μg/ml of HRP-conjugated anti-His antibody (Rockland Immunochemical, 200–303–382) was added as detection antibody. The absorbance was read at 450 nm as described above. The binding of silent alanine substitutions served as the positive control group and was used for normalization of fold changes.
FACS analysis
For antibody binding titration test, hPD-1 positive CHO-S cells (2× 105/well) were incubated with diluted anti-PD-1 antibodies, including pembrolizumab (purchased from Merck & Co., Inc.,) and nivolumab (synthesized based on 5C4 in US. Patent No. 9084776B2) at 4 °C for 1 h. FITC-conjugated goat anti-rat antibody (Jackson ImmunoResearch, 112–095–008) or FITC-conjugated goat anti-human antibody (Jackson ImmunoResearch, 109–095–008) was used as detection antibody. For hPD-1/hPD-L1 competition test, hPD-1 positive CHO-S cells (2× 105/well) were incubated with diluted antibodies and 5 μg/ml hPD-L1.ECD.mFc at 4 °C for 1 h. FITC-conjugated goat anti-mouse Fc (anti-mFc) antibody (Abcam, ab98716) was used as detection antibody. For mPD-1/mPD-L1 competition test, mPD-1 positive 293F cells (2× 105/well) were incubated with diluted R9 antibody and 5 μg/ml mPD-L1.ECD.mFc at 4 °C for 1 h, FITC-conjugated goat anti-mFc antibody (Abcam, ab98716) was used as detection antibody. After incubating with detection antibody at 4 °C in the dark for 1 h, cells were washed and then resuspended in PBS containing 1% BSA for subsequent flow cytometric analysis. The mean fluorescence intensity (MFI) values were detected using FACS instrument (BD Biosciences, BD FACSCanto™ II).
MLR assay
Primary dendritic cells (DCs) were mixed with 1× 105 CD4+ T cells at a ratio of 1:10 in the presence or absence of antibody, and were incubated at 37 °C, 5% CO2 for 5 d. The effect of antibody on CD4+ T cells proliferation was measured as the incorporation of 3H-thymidine (PerkinElmer, NET027001MC) into the proliferating cells in counts/min (CPM), and human IFN-γ secretion was measured by ELISA using matched antibody pairs (Thermo Fisher Scientific, M700A and M701B) according to the manufacturer's protocol.
SPR biosensor analysis
The off-rate constants K(d) of hPD-1 mutants to anti-PD-1 antibodies were measured based on SPR technology implemented in ProteOn™ XPR36 protein interaction analysis system (Bio-Rad Laboratories, 1760100). The GLC sensor chip (Bio-Rad Laboratories, 1765011) was activated and anti-PD-1 antibody in NaAc buffer was covalently immobilized on the chip. The chip was deactivated and washed until the baseline stabilized, then PD-1 mutants in conditioned media were injected into channels in parallel at a flow rate of 50 μL/min for an association phase of 300 seconds, followed by 600 seconds dissociation. Response units were monitored using the ProteOn imaging system and data analyses were performed with the ProteOn Manager™ software (Bio-Rad Laboratories, 1760210). Fold changes were calculated based on the average value of control mutants.
Antibody homology modeling
The Fv homology models of R11 and R9 antibodies were constructed based on their amino acid sequences, using the “Model Antibody” module in Discovery Studio. Both light and heavy chain were first annotated in Kabat numbering scheme, with CDR loops and framework regions all identified. The sequence of each segment (either CDR or framework) was then individually searched in the antibody structure database curated from PDB to find its closest structural templates. All searching hits were ranked by the sequence similarity to the queries. The VL-VH orientation templates were obtained in a similar way, but via searching the entire Fv sequences. Only those structures with high resolution (< 2.8Å) and low B-factor (< 50) were selected as candidates for further investigations. If certain conformational variability exists among the top searching hits (e.g., templates of CDR H3 loop), the MODELER program in Discovery Studio was used to model the structure via effectively combining the information from multiple templates. A rough antibody Fv structure was created after assembling all modeled structural segment into VL and VH domains, and superimposing them on the top of the orientation template. Further energy optimization of the entire Fv protein was then conducted, and the pose with the lowest energy was chosen to be the final antibody model.
Human PD-1 structures
PD-1 is a transmembrane glycoprotein composed of an Ig V-type extracellular domain, a transmembrane helix, and a cytoplasmic tail. The extracellular domain is responsible for binding partner recognition and immune checkpoint control, and therefore is extremely important. The crystal structures of hPD-1 extracellular domain, in both unbound and bound conditions to its ligand hPD-L1, are publicly available (PDB code 3RRQ and 4ZQK, respectively). However, both structures are missing the coordinates of important residues Asp85-Asp92. This is the region where mPD-1 holds a β-strand labeled as C.″ The failure of solving the crystal structure in this area is a strong indication that it might be a very flexible loop instead of a stable β-sheet. By inspecting the NMR structures of hPD-1 (PDB code 2M2D), it is further confirmed that this region is essentially a featureless loop and possesses significant flexibility.Citation20 To apply the crystal structures in the following modeling and analysis, the missing loop was built using the MODELER program in Discovery Studio, which borrowed and combined all loop conformations from NMR data. The final loop conformation was selected based on energy ranking. For consistency of terminology and convenience in comparing the human/murine PD-1 structures, we continued to use C″ to label this special loop (Asp85-Asp92).
Antibody-antigen docking
Computational protein-protein docking program ZDOCK, integrated in Discovery Studio, was used to predict the binding mode of discovered functional antibodies (R11 and R9) to hPD-1. Keeping the position of antigen fixed as a receptor, the antibody models were translated and rotated around the receptor in a rigid-body manner to search possible binding poses. Inspecting the structures of hPD-1 in PD-L1 bound and unbound conditions (PDB code 4ZQK and 3RRQ, respectively) revealed obvious conformational changes, especially in the region of loop CC′ (Met70-Asp77).Citation18 Since it is quite difficult to predict which state the antigen prefers to adopt when antibody binds, both conformations were taken into account in docking experiments. In addition, by knowing that CDR loops on antibody and hot spot residues on antigen (identified in alanine scanning experiments) are roughly the binding sites for each other, to reduce the searching space and relieve the scoring pressure, rigid-body docking was performed with loose constraints to ensure proper antibody-antigen contact. Furthermore, additional filtering process was conducted to filter decoys in which the pose of antibody had no overlap with PD-L1. The survived binding poses were refined and rescored using CHARMm-based optimization procedure (RDOCK). The most reasonable binding pose was selected after comprehensive evaluations of RDOCK scores and experimental mutagenesis information. The control antibody R10 and its binding behaviors were not modeled because it binds to a very different location and cannot block PD-L1.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Data.doc
Download MS Word (929.5 KB)Acknowledgments
We thank Baotian Yang, Xinhua Zhang (Biologics Discovery, WuXi Biologics, Shanghai, China) for providing cell lines and antibodies, and thank Hui Meng, Hong Wang, Chenghu Wang (Biologics Discovery, WuXi Biologics, Shanghai, China) for all their technical assistances.
References
- Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O'Day SJ, Hoos A, Humphrey R, Berman DM, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013; 1291:1-13; PMID:23772560; http://dx.doi.org/10.1111/nyas.12180
- Chester C, Dorigo O, Berek JS, Kohrt H. Immunotherapeutic approaches to ovarian cancer treatment. J Immunother Cancer 2015; 3:7; PMID:25806106; http://dx.doi.org/10.1186/s40425-015-0051-7
- Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med 2005; 3:9; PMID:15723705; http://dx.doi.org/10.1186/1479-5876-3-9
- Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol 2013; 6:74; PMID:24283718; http://dx.doi.org/10.1186/1756-8722-6-74
- Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007; 19:813-24; PMID:17606980; http://dx.doi.org/10.1093/intimm/dxm057
- Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, Siemens DR, Koti M, Craig AW, Graham CH. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget 2016; 7:10557-67; PMID:26859684; http://dx.doi.org/10.18632/oncotarget.7235
- Dong H, Chen L. B7-H1 pathway and its role in the evasion of tumor immunity. J Mol Med 2003; 81:281-7; PMID:12721664; http://dx.doi.org/10.1007/s00109-003-0430-2
- Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 2005; 54:307-14; PMID:15599732; http://dx.doi.org/10.1007/s00262-004-0593-x
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Ann Rev Immunol 2008; 26:677-704; PMID:18173375; http://dx.doi.org/10.1146/annurev.immunol.26.021607.090331
- McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med 2013; 2:662-73; PMID:24403232; http://dx.doi.org/10.1002/cam4.106
- Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med 2012; 366:2517-9; PMID:22658126; http://dx.doi.org/10.1056/NEJMe1205943
- Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, Konishi I. PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol 2016; 21:462-73; PMID:26899259; http://dx.doi.org/10.1007/s10147-016-0959-z
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348:56-61; PMID:25838373; http://dx.doi.org/10.1126/science.aaa8172
- Santabarbara G, Maione P, Rossi A, Palazzolo G, Gridelli C. Novel immunotherapy in the treatment of advanced non-small cell lung cancer. Exp Rev Clin Pharmacol 2016; 9:1571-81; PMID:27623999; http://dx.doi.org/10.1080/17512433.2016.1236681
- Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: Mouse tumor models, In vitro functional studies, and cynomolgus macaque toxicology. PloS One 2016; 11:e0161779; PMID:27610613; http://dx.doi.org/10.1371/journal.pone.0161779
- Spain L, Larkin J. Combination immune checkpoint blockade with ipilimumab and nivolumab in the management of advanced melanoma. Exp Opin Biol Ther 2016; 16:389-96; PMID:26750801; http://dx.doi.org/10.1517/14712598.2016.1141195
- Chen S, Li J, Li Q, Wang Z. Bispecific antibodies in cancer immunotherapy. Hum Vaccines Immunother 2016; 1-10; PMID:27249163; http://dx.doi.org/10.1080/21645515.2016.1141845
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015; 23:2341-8; PMID:26602187; http://dx.doi.org/10.1016/j.str.2015.09.010
- Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A 2008; 105:3011-6; PMID:18287011; http://dx.doi.org/10.1073/pnas.0712278105
- Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, Huo J, Yu C, Evans EJ, Leslie AJ, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem 2013; 288:11771-85; PMID:23417675; http://dx.doi.org/10.1074/jbc.M112.448126
- Wu PW, Li J, Kodangattil SR, Luxenberg DP, Bennett F, Martino M, Collins M, Dunussi-Joannopoulos K, Gill DS, Wolfman NM, et al. IL-22R, IL-10R2, and IL-22BP binding sites are topologically juxtaposed on adjacent and overlapping surfaces of IL-22. J Mol Biol 2008; 382:1168-83; PMID:18675824; http://dx.doi.org/10.1016/j.jmb.2008.07.046
- Waneskog M, Bjerling P. Multi-fragment site-directed mutagenic overlap extension polymerase chain reaction as a competitive alternative to the enzymatic assembly method. Anal Biochem 2014; 444:32-7; PMID:24084380; http://dx.doi.org/10.1016/j.ab.2013.09.021