
ABSTRACT
Characterization of charge-based variants by mass spectrometry (MS) is required for the analytical development of a new biologic entity and its marketing approval by health authorities. However, standard peak-based data analysis approaches are time-consuming and biased toward the detection, identification, and quantification of main variants only. The aim of this study was to characterize in-depth acidic and basic species of a stressed IgG1 monoclonal antibody using comprehensive and unbiased MS data evaluation tools. Fractions collected from cation ion exchange (CEX) chromatography were analyzed as intact, after reduction of disulfide bridges, and after proteolytic cleavage using Lys-C. Data of both intact and reduced samples were evaluated consistently using a time-resolved deconvolution algorithm. Peptide mapping data were processed simultaneously, quantified and compared in a systematic manner for all MS signals and fractions. Differences observed between the fractions were then further characterized and assigned. Time-resolved deconvolution enhanced pattern visualization and data interpretation of main and minor modifications in 3-dimensional maps across CEX fractions. Relative quantification of all MS signals across CEX fractions before peptide assignment enabled the detection of fraction-specific chemical modifications at abundances below 1%. Acidic fractions were shown to be heterogeneous, containing antibody fragments, glycated as well as deamidated forms of the heavy and light chains. In contrast, the basic fractions contained mainly modifications of the C-terminus and pyroglutamate formation at the N-terminus of the heavy chain. Systematic data evaluation was performed to investigate multiple data sets and comprehensively extract main and minor differences between each CEX fraction in an unbiased manner.
Abbreviations
| AF | = | acidic fraction |
| BF | = | basic fraction |
| CEX | = | cation ion exchange chromatography |
| CHO | = | Chinese hamster ovary |
| ESI-MS | = | electrospray mass spectrometry |
| Fab | = | fragment antigen-binding |
| Fc | = | fragment crystallizable |
| HC | = | heavy chain |
| LC | = | light chain |
| LC-UV/ESI-MS | = | liquid chromatography – UV / electrospray mass spectrometry |
| mAb | = | monoclonal antibody |
| MS | = | mass spectrometry |
| m/z | = | mass-to-charge ratio |
| PTMs | = | post-translational modifications |
| UV | = | UV |
Introduction
Monoclonal antibodies (mAbs) and related glycoproteins belong to a fast-growing group of biopharmaceuticals.Citation1 Antibodies and related formats can trigger immune responses, which makes them ideal therapeutic candidates to target cancer and diseases with immunological background.Citation2 Additionally, it is well established that this class of large biomolecules exhibit higher specificity and longer clearance time than small molecule drugs.Citation3
Typically, these biomolecules are expressed at high titer in stable transfected mammalian cell lines like immortalized Chinese hamster ovary (CHO) cells.Citation4 Although the primary structure of the mAbs is unequivocally transcribed and translated from the coding DNA and mRNA sequences, respectively, the resulting drug substance may show some degree of microheterogeneity introduced during expression, purification, and storage.Citation5 Such heterogeneity is mostly linked to post-translational modifications (PTMs), which may include differences in the glycosylation pattern,Citation6 disulfide bridge scrambling,Citation7 N-terminal pyroglutamate formation,Citation8 C-terminal lysine processing,Citation9 oxidation of methionine (and less frequently of tryptophan, histidine, and tyrosine) residues,Citation10 deamidation and isomerization of asparagine and aspartic acid residues,Citation11 glycation of lysine residues,Citation12 and peptide backbone cleavage.Citation13 As some of these modifications might alter the product stability, alter pharmacokinetics as well as pharmacodynamics, reduce potency, and increase immunogenicity, in-depth PTM characterization and understanding of the product degradation pathwaysCitation14 is required during the technical development of a novel therapeutic glycoprotein. Such studies are useful because the level of microheterogeneity must be monitored and maintained during the life cycle of biopharmaceutical products. Since most of these modifications have an effect on the mAb surface charge, charge-sensitive analytical methods, such as cation ion exchange chromatography (CEX), are well suited and widely used to monitor charge variants.Citation15 Identification of these variants mainly requires the collection of CEX fractions and further characterization using orthogonal methods, such as mass spectrometry (MS).Citation16,17
Classical MS data analysis of charge variants at the intact mAb level requires the computation of average m/z signals for each MS peak observed. The different charge envelopes are then deconvoluted separately for all MS peaks through a manual or automated process. Information, e.g., mass and intensity, collected from the resulting deconvoluted average mass spectra are then compared for each MS peak detected in each CEX fraction. The same would be applied to the analysis of light (LC) and heavy (HC) chain charge variants after disulfide bridge reduction. Although this methodology is valid and widely used, mass shift determination may be impaired by multiple deconvolution processes and mAb variants may co-elute under the same MS peak. In many cases, efforts to recognize and quantify co-eluting variant signals may be hampered by average spectral noise and the major signal of the predominant species. Conversely, such a deconvolution process of a charge envelope is not performed for peptide mapping analyses, for which 2 main approaches can be undertaken. The first common approach is to overlay the ultraviolet (UV) or total ion chromatograms from the different CEX fractions, identify some trace variations, and evaluate the associated MS and MS/MS data. The main issue with this approach is that variants may be either hidden under major UV/MS peaks or in the background noise. The second widely used approach relies on a database search to assign peptide identifications to detected signals based on their MS and MS/MS data. A prerequisite for a reliable output is to have high quality MS/MS data for both intense and weaker ions. However, results are biased toward the modifications specified for the search, and any unexpected modification(s) of a peptide will not be detected. While several search engines allow multiple modifications to be scanned, the number of peptide misassignments, i.e., false positives, will automatically increase. Importantly, the relative quantification across multiple samples will be performed on identified peptides, whether misassigned or not, and will exclude species not assigned.
Here, we describe an approach for the unbiased and in-depth MS-based characterization of fractions collected from a CEX analysis of a stressed IgG1, at the intact, chain, and peptide levels. For the analyses of intact and reduced samples, we chose to perform time-resolved deconvolution for each CEX fraction in parallel instead of performing repeated MS peak-based average spectrum deconvolution. This approach has 3 main advantages: Firstly, it increases the accuracy in the determination of mass differences between multiple MS peaks in a single process. Secondly, the scan-after-scan deconvolution retains retention time information for all species, enhancing the detection of relevant lower abundant signals, separated in time from main species. Thirdly, the visualization of the deconvoluted data as a 3-dimensional map (mass vs. time vs. intensity) enables a fast and intuitive visual detection of differences and patterns in multiple samples. Applying this improved approach enabled the identification of degradation products like chain clips, half antibodies, free chains, as well as differences in the glycosylation profile and glycation level. At the peptide level, relative quantification was performed before assignment to successfully map low abundant changes in CEX fractions. All m/z values in each data set were detected in parallel in all CEX fractions, clustered with charge state calculation and quantified. Comparison of all clustered signals, i.e., putative unmodified and modified peptides, was performed across samples and further characterization was performed on signals enriched in investigated CEX fractions. This resulted in the identification of relevant differences in deamidation, glycosylation, fragmentation, glycation, and leader sequence extensions at low levels. The use of a 3-dimensional map (m/z vs. time vs. intensity) for characterization purposes is discussed.
Results
mAb fractionation on CEX
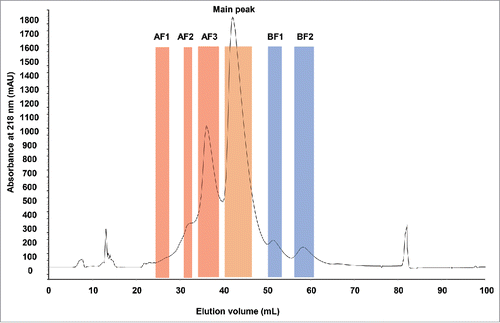
Analysis of the stressed mAb with analytical CEX resulted in the identification of acidic and basic variants (Fig. S1A). Six fractions were collected from a semi-preparative CEX column (), including 3 fractions corresponding to acidic variants, 2 fractions to basic variants and the main UV peak (main peak). Both basic fractions (BF1 and BF2) were produced from well-resolved but relatively small UV peaks. The acidic fraction AF3 represented a clear UV peak as well, whereas fractions AF1 and AF2 corresponded to relatively less resolved UV peaks. Collected fractions were reinjected on the analytical CEX column to check their respective purity (Fig. S1B).
Figure 1. CEX UV chromatogram of stressed monoclonal IgG1 antibody. The boundaries of the collected fractions are indicated by colored areas. AF: acidic fractions in red. BF: basic fractions in blue. Main peak is highlighted in orange. UV absorbance in mAU was measured at 218 nm. Elution volume is indicated in mL.
On-line LC-UV/ESI-MS analysis of the intact CEX fractions
Each CEX fraction was analyzed as received by online reversed-phase liquid chromatography – UV / electrospray mass spectrometry (LC-UV/ESI-MS). A “zoomed-in” overlay of the UV data are shown in . Several minor UV peaks were observed in addition to the intact mAb UV peak. Retention time shifts observed for some of the UV peaks across different CEX fractions were independent from the fraction position in the sample list and the chromatography reproducibility (median standard deviation of 0.01 min, c.f. Materials and Methods). This suggested that these shifts reflected the heterogeneity of the different charged variants of the intact mAb. For data evaluation, the underlying MS data were divided into 3 sections of ∼18 min, 19 min, and from 19.3 min to 21.1 min for the intact mAb MS peak. Time-resolved deconvolution was performed using Genedata Expressionist and its proprietary “harmonic suppression” algorithm. The result of the intact mAb MS peak is displayed in . Five signals could be observed at a retention time of ∼19.5 min with different intensities, representing the intact mAb with its different glycoforms. Displaying the time-resolved deconvoluted spectra of the intact mAb MS peak from all 6 fractions allowed visual identification of differences in each fraction (). The main peak and all 3 acidic fractions had a similar deconvoluted mass for the intact antibody, as shown with the white cross overlaid on the G0F/G0F form, whereas the deconvoluted mass of this form was lower in the basic fractions. The mass difference for BF1 was ∼18 Da, which could correspond to the more basic pE version of the molecule. In fraction BF2, this mass shift was ∼57 Da, which could correspond to the loss of glycine combined with prolinamide modification on the C-terminus of the HC.Citation18,19
Figure 2. Characterization of the CEX fractions by LC-UV/ESI-MS as intact. (A) Overlay of the reversed phase (RP)-UV chromatograms for the main peak, acidic (AF) and basic (BF) fractions at 214 nm. (B) Time-resolved deconvolution of the main RP peak for the CEX main peak. The horizontal axis displays the deconvoluted mass in kiloDalton (kDa) and the vertical axis the retention time in minutes (min). MS peak intensity is color-coded as shown on the left bar. Five major MS peaks are labeled as 1: G0F/G0, 2: G0F/G0F, 3: G0F/G1F, 4: G0F/G2F or G1F/G1F, 5: G1F/G2F. (C) Comparison of CEX fractions by time-resolved deconvolution, with the white cross positioned on the predominant G0F/G0F form. A shift in retention time and glycosylation relative abundance is observed for acidic fractions. Lower mass species are detected in basic fractions. (D) Glycosylation profile indicating the relative abundance (%) of glycosylations in acidic fractions and the main peak. The relative distribution was calculated for the glycoforms G0/G0F, G0F/G0F, G0F/G1F, 2G1F or G0F/G2F, and G1F/G2F. Larger glycosylations are enriched in acidic fractions. Maximum experimental variation: ± 0.2% (coefficient of variation < 3%). (E) Time-resolved deconvolution of the reversed phase early-eluting MS peaks in acidic fractions in comparison with the main peak. Fab fragment was identified in AF1. Fab-DK and Fab-DKT were enriched in AF2. Fab-DKTH was enriched in AF3 while none of these fragments were found in the main peak.
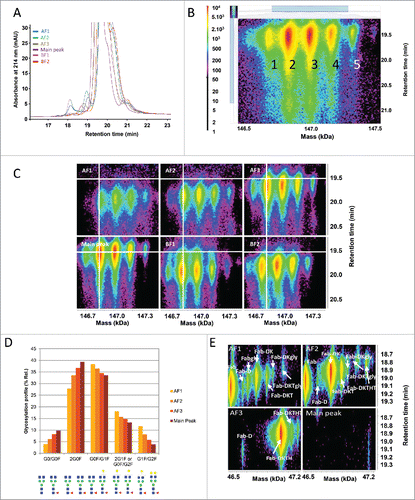
Besides C- and N-terminal modifications, the data revealed a change in the glycosylation profile. Fractions AF1 and AF2 showed less afucosylated glycan forms than the AF3 and the main peak. Additionally, the G0F/G0F form was no longer the main glycoform for both AF1 and AF2, which predominantly contained the G0F/G1F glycoform. This change was quantified by calculating the volume of the different glycoforms in the acidic fractions and the main peak (). Since these changes could not be explained by a change of the net charge of the molecule, it is hypothesized that the larger glycoforms led to a change in the 3-dimensional structure, which modified the charges available on the surface of the molecule.Citation20 Another possible explanation would be that an increased amount of glycated mAb, leading to the same mass shift, was recovered in the acidic fractions due to a loss of a free ϵ-amino group on lysines and a reduction in the net charge of the molecule.
The early eluting UV peaks in the LC-UV/ESI-MS analysis of the intact molecule are generally breakdown products of the whole antibody, either due to peptide bond cleavage or a reduction of disulfide bonds resulting in free LC or HC dimers (data not shown).Citation21 Signals corresponding to Fab fragments of the molecule eluted at ∼18.6 min, predominantly in the acidic fractions AF1, AF2, and AF3. The time-resolved deconvolution of this area is shown in , and displays different signals corresponding to the Fab fragment with varying number of additional amino acids on the HC. Fab was the major form in fraction AF1, whereas Fab-DK and Fab-DKT were identified in fraction AF2, and Fab-DKTH in fraction AF3. The distribution in the different acidic fractions is in good agreement with the chemical properties of the additional attached amino acids. The counterpart of the Fab fragments, the Fc, was detected in fractions AF1 and AF2 only (data not shown). This consistent observation suggests that either both Fc and Fab fragments exhibit less positive charges on their surface (as separate entities smaller than the intact antibody), or that cleavage occurred after fractionation. The hinge region is a known mAb degradation hotspot.Citation13,21
On-line LC-UV/ESI-MS analysis of the CEX fractions after reduction and carbamidomethylation
The aim of these analyses was to confirm the findings described above using a more resolutive sample preparation. This is demonstrated in the time-resolved deconvolution of the HC species separated by reversed phase chromatography (). The main peak contained signals expected for the HC without C-terminal lysine and with glycosylation G0, G0F, and G1F. The acidic fractions AF1 to AF3 showed highly comparable signals with masses corresponding to the HC with G0F and G1F.
Figure 3. Characterization of the HC species by LC-UV/ESI-MS. (A) Time-resolved deconvolution of the HC species separated by reversed phase chromatography for the different CEX fractions. HC with G0F, 0K and with G1F, 0K were the main signals observed in the acidic fractions (AF) and the main peak, whereas lower mass species potentially corresponding to HC with G0F, pE, 0K and HC with G0F, 0GK, are enriched in basic fractions BF1 and BF2, respectively. Masses are indicated in kiloDalton (kDa). (B) Comparison of the deconvoluted average mass spectrum and the time-resolved deconvolution. Each top panel corresponds to the average signal of the bottom time-resolved deconvolution for the main peak, BF1, and BF2, demonstrating that multiple MS peak-based deconvolution steps are required to analyze accurately each species within a single sample and across samples. MS peaks are numbered in top and bottom panels to ease comparison. Time-resolved deconvolution enables to derive the main retention time for each major and minor species, which is required to distinguish in-source artifacts from isobaric true variants.
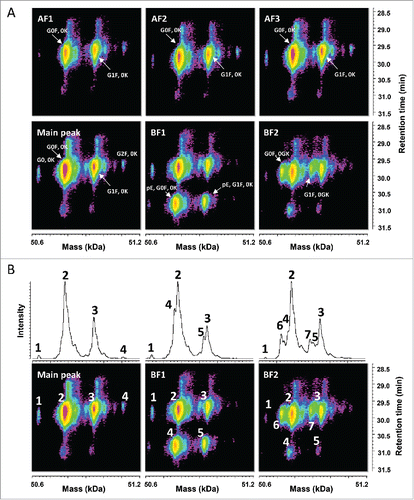
The basic fractions BF1 and BF2 showed additional signals with lower deconvoluted masses and eluting at different times, which could be assigned, based on the measured mass, to the HC without C-terminal lysine (0K), glycosylation G0F and G1F with pyroglutamate at the N-terminus in BF1, and to the HC without C-terminal GK (0GK), glycosylation G0F and G1F, probably with formation of prolinamide in BF2. shows a direct comparison between the result for time-resolved deconvolution and the classical averaged spectra deconvolution. In this latter case, separate MS peak-based spectrum deconvolutions would be required to accurately measure the mass of each species within a single sample but also across multiple fractions. The use of 3-dimensional heatmaps allows a faster and more intuitive comparison between different samples. Patterns can easily be recognized and relevant variants can be visually distinguished from isobaric in-source artifacts, co-eluting with the main species.
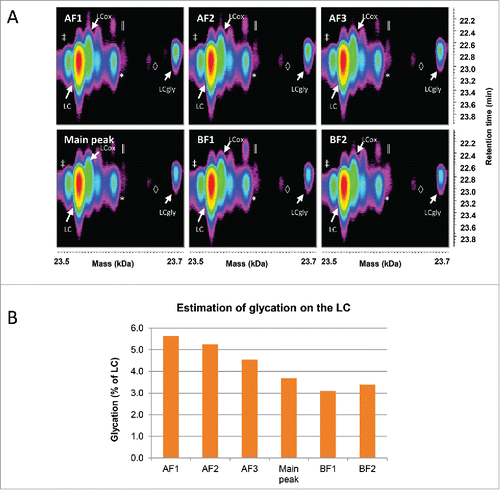
Time-resolved deconvolution of the LC enabled the detection of glycation across all fractions (), whose relative abundance was estimated in . This revealed that the amount of glycated LC was decreasing from the acidic fractions to the basic fractions. This is expected, since the glycation removes a positive charge on the LC due to its bonding to the side chain of a lysine residue. The overall amount of glycation in the sample was estimated at a relatively low level (below 6%), but this level dropped from the most acidic fraction to the most basic one by nearly 50%.
Figure 4. Characterization of the LC species by LC-UV/ESI-MS. (A) Time-resolved deconvolution of the LC species separated by reversed phase chromatography for the different CEX fractions. The most intense LC signal (red) corresponds to the unmodified LC, while the rightmost signal of each panel with the mass of LC+162 Da corresponds to the glycated LC (LCgly). LCox: oxidized LC, *: Guanidine adduct, ‡: In-source dehydration, ║: overalkylation, ◊: TFA adduct. (B) The relative abundance of the glycation on the LC was estimated as a percentage of the total volume of both glycated and unmodified LC species. The basic fractions contain less glycated LC than acidic fractions. Maximum experimental variation: ± 0.01% (coefficient of variation < 3.5%).
Peptide mapping analysis of reduced/carbamidomethylated Lys-C digest by LC-UV/ES-MS/MS
Each digested sample was analyzed twice in LC-UV/ESI-MS mode only to obtain reliable quantitative data, and, once in LC-UV/ESI-MS/MS mode, to obtain relevant sequence information. Expressionist was used to evaluate the peptide mapping data. First, chemical background noise such as streaking was removed and retention time alignment was performed. Then, each m/z signal was detected simultaneously in all data sets. In the next step, the detected MS peaks were grouped if they were related to the same isotopic cluster with a defined retention time, charge state, and a calculated monoisotopic mass. The various data processing steps are visualized in Fig. S2. A total of 2780 clusters, including multiple charge states for the same peptides, were detected. The volume, maximum intensity, and mass were systematically calculated for each cluster. We estimated that only 20–30% of these signals could be attributed to the mAb and its charged variants, the rest being due to chemical background ions, noise, adduct ions, in-source fragments, or matrix ions.
After assigning the intense MS peaks found at similar levels in all samples to peptide identifications, further investigation was performed for the signals that differed in cluster intensities in the different CEX fractions or which were only present in a subset of the fractions. The differences could be highlighted by displaying the data in a sample-to-sample scatter plot as shown in for fraction AF1 against the main peak. The volumes of most of the clusters (∼70%) was unchanged between AF1 fraction and the main peak, i.e., with a volume fold-change < 1.5. Clusters more abundant in AF1 than in the main peak are shown in red (≥ 1.5-fold-change). Technical replicates for these 2 fractions are shown in Fig. S3; the histogram distribution of all technical replicates in Fig. S4 shows that 95% of the data are comprised within the log2-transformed fold-change range of [-0.47; 0.47].
Figure 5. Characterization of peptide mapping signals. (A) Scatter plot of all clusters in the main peak and AF1. After chemical noise subtraction and background noise reduction, MS signals and associated isotopic distributions were grouped together in appropriate clusters defined by charge state, monoisotopic mass, and a retention time. Clusters consisting of only one m/z peak were filtered out from the cluster list before relative quantification across CEX fractions. Each cross corresponds to a single cluster. Red crosses highlight clusters enriched in AF1 fraction as compared with the main peak. Blue line: 1.5-fold difference, red line: 5-fold difference, green line: 10-fold difference. (B) Estimation of glycated LC peptide 1 (L-L1gly) compared with LC peptide 1 (L-L1) obtained after Lys-C cleavage. Abundance of L-L1, L-L1gly and its miscleaved peptide L-L1–2, L-L1–2 gly in the different fractions was monitored. Glycated peptides were more abundant in acidic fractions. The miscleaved peptide L-L1–2 was more abundant in acidic fractions, possibly due to a digestion artifact. (C) Three-dimensional map of the unglycosylated HC peptide containing Asn297 (brown cluster) and its deamidated form (blue cluster). Horizontal axis represents m/z and vertical axis shows the retention time in minutes (min). Intensity is increasing from white to dark blue. The deamidated form of the peptide is eluting first and is enriched in acidic fractions.
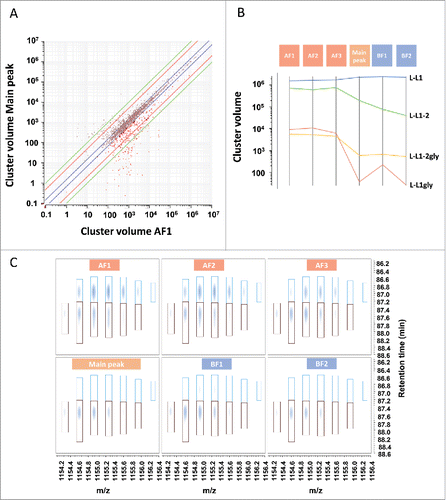
Further filtering was performed to select relevant signals (> 1000 in volume). Consequently, the number of interesting clusters was reduced to ∼10% of the overall number of clusters, and further characterization was performed only on these clusters. For example, glycation of the LC N-terminus was detected in 2 different peptides (one being a miscleaved peptide due to KP sequence). The 2 peptides confirmed the previous findings qualitatively and quantitatively (). The overall amount of glycated L-L1 peptide was ∼0.5% in acidic fractions, and at trace level in the main peak. The data also showed that the miscleaved peptide was much more pronounced in the acidic fractions. This phenomenon was observed for all other peptides containing a potential miscleavage site. Such differences between the fractions could not be explained with a different charge profile and are probably artifacts of the digestion.
Using this approach, it was possible to identify the deamidated non-glycosylated version of the HC peptide containing the consensus sequence for N-glycosylation. A similar amount of unglycosylated HC peptide was present in all fractions, although the amount of deamidated peptide was higher in the acidic fractions, as expected (). MS/MS spectral interpretation confirmed that Asn297 of the consensus glycosylation site was deamidated (data not shown). The overall amount of this peptide in comparison to the glycosylated version was estimated at a level below 1%.
Using these approaches it was possible to identify and monitor across fractions, in addition to the expected deamidation sites, glycation and pyroglutamate version, differences in the glycosylation pattern of consensus glycopeptides (sialic acid), clipping, C-terminal prolinamide, and leader sequence extension on the HC for which an automated MS/MS search would not retrieve a score, as compiled in . The glycosylation G2F was enriched in AF1 at cluster level, while only residual changes could be monitored for G0, G0F, and G1F at cluster level. Using 3 different charge states, a significant decrease in the relative percentage of G0F (among G0, G0F, G1F, and G2F) in AF1 compared with the main peak could be confirmed, as well as the significant increase in the relative percentage of G2F in AF1 (Fig. S5).
Table 1. HC modifications detected in acidic or basic fractions. Detected modifications are sorted according to their respective position in the sequence of the HC. Cluster identification numbers (ID), retention time (RT), mass-to-charge ratio (m/z), charge, mass, MS/MS consolidated score, and the logarithmic transformation (log2) of the fold changes in acidic (AF) and basic (BF) fractions in comparison with the main peak are shown. Positive log2(fraction/main peak ratio) values, corresponding to enrichment in acidic/basic fractions compared with the main peak are shown in red, while negative values, corresponding to a lower amount recovered in acidic/basic fractions for a specific modification are shown in blue. Color scale is graduated from dark blue for values ≤ −5 to dark red for values ≥ 5. Standard error of the mean (SEM) is shown for duplicate measurements after the log2 values. *: MS/MS not assigned due to unexpected modification. 0: Peptide identified by precursor mass only.
Discussion
MS-based identification and monitoring of PTMs is required to characterize in-depth the degradation pathways of biopharmaceuticals during technical development. Standard data analysis methodologies still remain biased toward the detection and identification of major proteoforms, sometimes in a time-consuming exercise, instead of comprehensively dissecting all data in an unbiased manner to allow fast and accurate elucidation of all species present in a set of multiple samples. We attempted to reduce this gap by interpreting 3-dimensional maps (e.g., mass vs. time vs. intensity) of charge variants of a stressed IgG1 at intact, chain, and peptide level, in comparison with standard peak-based approaches. CEX fractions were analyzed simultaneously and thoroughly at these 3 different levels. In our hands, Expressionist was able to cope with gigabytes of data in parallel, with results obtained in short periods (an hour to half a day), depending on the number of samples analyzed and processing parameters used. Speed of analysis was mainly dependent on the size of data files acquired, a vendor-specific attribute, which could be even reduced with shorter chromatographic methods (data not shown). Using such unbiased analysis, a signature of consistent PTMs specific to each CEX fraction could be determined across all analyses for this particular antibody. Glycosylations larger than G0F, including sialic acids, were identified in acidic fractions along with free LC, HC dimer, and Fab fragments. The basic fractions revealed N-terminal and C-terminal modifications of the HC, including pyroglutamate formation, clipping, and leader sequence extensions.
We were able to monitor glycosylation and glycation levels consistently across acidic and basic fractions in comparison to the main one. Glycation of the LC, which results in the same mass shift of +162 Da, was more pronounced in acidic fractions, as previously shown by others.Citation17,22,23 Glycation of recombinant mAbs has been shown to occur in the presence of reducing sugars during mammalian cell culture.Citation23 Low level charge variants present in different CEX fractions were quantified at peptide level below 1% for glycation and deamidation.
We outlined the benefits of 3-dimensional maps when a peak-based analysis would probably fail in enabling the fast detection of true species from isobaric artifactual signals created in the instrument source. By retaining valuable time information for each species, time-resolved deconvolution enabled the detection of lower abundant variants and the straightforward comparison of multiple samples consistently when the use of independent deconvolution steps typically slows down data analysis of a single sample and focuses on main variants only. Moreover, these latter results can be biased to the deconvolution process or the mass and time windows settings defined by the analyst. Changing from this tedious process has proven highly beneficial, especially for the qualitative evaluation and relative quantification of PTMs such as acidic and basic charge variants in multiple fractions from intact down to peptide level.
At the peptide level, signal comparison between fractions was performed for each clustered species without any bias toward search parameters and results. We believe this is a major improvement for the peptide-centric approach with regard to the discovery and further characterization of low abundance PTMs and sequence variants. The parallel data processing also facilitated the peptide identification for a cluster using MS/MS spectra acquired in one or more fractions. This allowed the successful monitoring of the precursor signal across fractions, including those in which no MS/MS could be triggered. The methodology presented here enabled the detection of major and minor differences between samples down to the trace level, which were then verified and assigned to peptide identifications. Therefore, such multilayer unbiased data analysis would enable comprehensive mapping of chemical and post-translational modifications that charge exchangers, whether used off-line or online with, for example, reversed phase liquid chromatography, can reveal in stressed biologics.Citation15,24 These microheterogeneities could be both tracked and further polished during technical development, while the monitoring of their levels during the life cycle of the product could become more accurate.
Materials and methods
Materials
For this study, a recombinant IgG1 mAb was used and manufactured at Novartis. To increase the amount of degradation products, the mAb was stored for 12 months at 25ºC before analysis.
Guanidine hydrochloride 8 M (24115) was purchased from Pierce, EDTA disodium salt, dehydrate (E5134–250G) was purchased from Sigma, 1 M Tris-hydrochloride pH 8 solution (Ultrapure, 15568–025) was purchased from Gibco, dithiothreitol (DTT; 43815–1G) was purchased from Sigma, iodoacetamide (Bioultra, I1149–5G) was purchased from Sigma, acetonitrile (LC-MS grade, 9821) was purchased from Biosolve, water (LC-MS grade, 9823–02) was purchased from J.T.Baker, isopropanol (LC-MS grade, 34965) from Fluka, trifluoroacetic acid (TFA) ampoules (MS grade, 28904) were purchased from Pierce and Lys-C (MS grade, 125–05061) was purchased from Wako.
Collection of CEX fractions before MS analysis
The fractions were collected on an Äkta Avant system (GE Healthcare) for scale up of the standard method run on HPLC. The scale factor was 160, and 9 runs were performed with 8 mg material injection each. The column used was a Propac WCX-10 (250 mm × 9 mm), operated at room temperature. The mobile phases were 25 mM Tris pH 7.8, 1% 250 mM NaCl as A and 25 mM Tris pH 7.8, 18% 250 mM NaCl as B. Total run time was 75 min, achieved with 4 column volumes by starting at 25% B to finish at 70% B. The column was then washed with 100% B. The collection was done in 15 mL tubes and followed by a buffer exchange in 10 mM histidine pH 6.00 with Millipore Centriprep – 15 (30 kDa cut off). The final concentration was determined with a NanoDrop1000 instrument (Thermo Scientific).
Analysis of intact mAb
Twenty µg of the collected fractions were directly injected on the column and analyzed by LC-UV/ESI-MS using an Agilent 1200 HPLC (Agilent) and a q-ToF Synapt G2-S (Waters Corporation) mass spectrometer. The mobile phases were 0.1% TFA in water as A, 0.09% TFA in 70:20:10 isopropanol:acetonitrile:water as B, and the flow rate was set to 0.2 mL/min. A polymeric reversed phase (PLRPS) column was used for the separation (2.1 × 150 mm, 3 µm, 300 Å) and was operated at 60°C. The UV traces were recorded at 214 and 280 nm. The separation was achieved over 40 min by starting at 35% B and maintaining it for 4 min, then ramping to 50% B in 24 min, then to 80% B in 1 min, and maintaining again for 5 min. Finally, one minute later, the composition was set to 35% B and maintained over 5 min to equilibrate the column for the next run. Median standard deviation for the retention time of the intact mAb peak was estimated at ± 0.01 min. The HPLC was directly coupled to the mass spectrometer. The instrument was operating in positive mode with a capillary voltage of 3 kV. The cone was set at 120 V and the source offset at 80 V. The source temperature was set at 120°C and the desolvation temperature at 500°C. Lockmass correction was applied every 20 scans from an average of 3 scans by infusion of 1 pmol/μL of Glu-1-Fibrinopeptide B.
Analysis of reduced/carbamidomethylated mAb
A hundred µg of each CEX fraction were first lyophilized to dryness. The lyophilizates were denatured in 150 µL of 6 M guanidine-HCl, 50 mM TrisHCl pH 8, 5 mM Na2EDTA, and incubated with 1.5 µL of 1 M DTT for reduction of disulfide bonds for 1 h at 37°C. Then, 3 µL of 1 M iodoacetamide were added for 1 h in the dark for the alkylation. One further µL of 1 M DTT was finally used to quench the alkylation.
Seven µg (10 µL) of these samples were injected on column and analyzed by LC-UV/ESI-MS using an Agilent 1200 HPLC (Agilent) and a q-ToF Synapt G2-S (Waters Corporation) mass spectrometer. The buffers A and B, the flow rate, the column, the column temperature, and the UV traces are the same as in the analysis method for the intact mAb. The separation was achieved over 54 min by starting at 32% B and maintaining it for 4 min, then ramping to 47% B in 37 min, then to 100% B in 1 min and maintaining again for 5 min. Finally, one minute later, the composition was set to 32% B and maintained over 6 min to equilibrate the column for the next run. Median standard deviation for the retention time of the LC and HC peaks was estimated below 0.02 min. The HPLC was directly coupled to the mass spectrometer. The instrument was operating in positive mode with a capillary voltage of 3 kV. The cone was set at 40 V and the source offset at 60 V. The source temperature was set at 120°C and the desolvation temperature at 500°C. Lockmass correction was applied every 20 scans from an average of 3 scans by infusion of 1 pmol/μL of Glu-1-Fibrinopeptide B.
Data evaluation for the intact and reduced/carbamidomethylated analysis
The MS data were directly imported into the Expressionist software package (Genedata) without any prior data conversion. An adapted and application specific workflow was built and used for chemical noise reduction, spectrum baseline subtraction, spectrum smoothing, retention time and m/z restriction, and finally time-resolved deconvolution. The scan-per-scan deconvolution was performed with the embedded Genedata algorithm with typical parameters: harmonic suppression deconvolution method with 0.5 Da step. After optimization of the workflow for the specific application, this process was run automatically. For estimations, we used volumes to ensure that data evaluation was performed using multiple data points. Volumes are defined as the integral of the intensity values inside the peak boundaries, i.e., intensity values × retention time window × mass width.
Analysis of peptide mapping of mAb
Two hundred µg of each CEX fraction were first lyophilized to dryness. The lyophilizates were denatured in 150 µL of 6 M guanidine-HCl, 50 mM TrisHCl pH 8, 5 mM Na2EDTA, and incubated with 1.5 µL of 1 M DTT for reduction of disulfide bonds for 1 h at 37°C. Then, 3 µL of 1 M iodoacetamide was added for 1 h in the dark for the alkylation. One µL of 1 M DTT was finally used to quench the alkylation. The samples were diluted in 750 µL of 50 mM Tris HCl pH 8 before the digestion. Four µL of 1 mg/mL Lysyl endopeptidase were added to the samples, which were incubated for 1 h at 37°C. Another 4 µL of Lysyl endopeptidase were added and the samples incubated for an additional 3 h. Five µL of TFA was added at the end to stop the reaction.
Twenty-two µg of digest were injected on column and analyzed by LC-UV/ESI-MS/MS using an Agilent 1200 (Agilent) online connected to an Orbitrap (Thermo Fisher Scientific) mass spectrometer. All fractions were injected in triplicate. The mobile phases were 0.1% TFA in water as A and 0.09% TFA in 90% acetonitrile as B, and the flow rate was set at 0.2 mL/min. The column used for the separation was a Vydac C18 (2.1 × 150 mm, 5 µm, 300 Å) and was operated at 40°C. The UV traces were recorded at 214 and 280 nm. The separation was achieved over 146 min by starting at 2% B and maintaining it for 5 min then ramping to 22% B in 45 min, then to 24% B in 10 min, to 36% B in 28 min, further to 38% B in 10 min and to 90% B in 17 min and maintaining it for 10 min. Finally, one minute later, the composition was set to 2% B and maintained over 20 min to equilibrate the column for the next run. Median standard deviation for the retention time of all peptide peaks was estimated below 0.02 min. The HPLC was directly coupled to the mass spectrometer. The instrument was operating in positive ion mode with a capillary voltage of 3.5 kV. The capillary temperature was set at 250°C. Two replicates were measured with only MS full scan in the Orbitrap. The third one was acquired with MS/MS data: each full scan in the Orbitrap is followed by 3 CID MS/MS events of the 3 most intense ions and by 3 HCD MS/MS of the same ions, all measured in the Orbitrap. Dynamic exclusion was used to prevent fragmentation of the same precursor ion multiple times (repeat counts: 1, repeat duration: 5 s, exclusion duration: 20 s).
PepMap data evaluation using Expressionist
The MS data were directly imported into the Expressionist software without the necessity of prior data conversion. The evaluation was split in 2 separate processes, which were run in parallel for all data sets: 1) Background subtraction followed by retention time alignment, MS peak detection (isotope detection and quantification), charge assignment (MS peak clustering), MS/MS consolidation, and saving of the processed data; and 2) The processed data were checked for the necessity of a normalization step by evaluating the corresponding box plot data (Fig. S4). Based on these results, data were not normalized.
To reduce dependency on single isotope measurements, we summed volumes for all isotope peaks of a specific ion species to calculate the “Cluster volume.” Volumes are defined as the integral of the intensity values inside the peak boundaries, i.e., intensity values × retention time window × m/z width. Scatter plots of all cluster volumes were created and Absent/Present searches performed to identify and compute relative quantification of the differences across CEX fractions. The clusters of interest were searched against the expected sequence with a set of possible modifications (carbamidomethylation, loss of C-Terminal lysine, pyroglutamate formation, glycosylation, deamidation, oxidation). In case the search did not return a hit, a manual interpretation of the MS/MS data was performed.
Disclosure of potential conflicts of interest
FG, BD, ML, HH, and MB were employees of Novartis Pharma AG, and may have had shares in Novartis Pharma AG at the time of publication. PH was an employee of Genedata GmbH at the time of publication.
Supplemental_Figures_1-5.pdf
Download PDF (544.6 KB)Acknowledgments
The authors would like to thank Patrick Wissner, Robin Mende, and Wolfram Kern for technical assistance as well as Dr. Christoph Rösli for the critical review of this manuscript.
Funding
This work was supported by Novartis Pharma AG.
References
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2003; 2:52-62; PMID: 12509759; https://doi.org/10.1038/nrd984
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9:423-30; PMID: 19075682; https://doi.org/10.2174/138920108786786358
- Ludwig DL, Pereira DS, Zhu Z, Hicklin DJ, Bohlen P. Monoclonal antibody therapeutics and apoptosis. Oncogene 2003; 22:9097-106; PMID: 14663488; https://doi.org/10.1038/sj.onc.1207104
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol 2004; 22:1393-8; PMID: 15529164; https://doi.org/10.1038/nbt1026
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426-47; PMID: 17828757; https://doi.org/10.1002/jps.21180
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11-6; PMID: 15903235; https://doi.org/10.1021/bp040016j
- Zhang W, Czupryn MJ. Free sulfhydryl in recombinant monoclonal antibodies. Biotechnol Prog 2002; 18:509-13; PMID: 12052067; https://doi.org/10.1021/bp025511z
- Dick LW, Jr., Kim C, Qiu D, Cheng KC. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol Bioeng 2007; 97:544-53; PMID: 17099914; https://doi.org/10.1002/bit.21260
- Harris RJ. Processing of C-terminal lysine and arginine residues of proteins isolated from mammalian cell culture. J Chromatogr A 1995; 705:129-34; PMID: 7620566; https://doi.org/10.1016/0021-9673(94)01255-D
- Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: Mechanisms and stabilization. J Pharm Sci 2009; 98:4485-500; PMID: 19455640; https://doi.org/10.1002/jps.21746
- Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem 1987; 262:785-94; PMID:3805008
- Horvat S, Jakas A. Peptide and amino acid glycation: New insights into the Maillard reaction. J Pept Sci 2004; 10:119-37; PMID: 15113085; https://doi.org/10.1002/psc.519
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. Mabs 2011; 3:253-63; PMID: 21487244; https://doi.org/10.4161/mabs.3.3.15608
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715-36; PMID: 23134362; https://doi.org/10.1021/ac3032355
- Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by Charge-Sensitive methods. Curr Pharm Biotechnol 2008; 9:468-81; PMID: 19075686; https://doi.org/10.2174/138920108786786402
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl 2001; 752:233-45; PMID: 11270864; https://doi.org/10.1016/S0378-4347(00)00548-X
- Gandhi S, Ren D, Xiao G, Bondarenko P, Sloey C, Ricci MS, Krishnan S. Elucidation of degradants in acidic peak of cation exchange chromatography in an IgG1 monoclonal antibody formed on Long-Term storage in a liquid formulation. Pharm Res 2012; 29:209-24; PMID: 21845507; https://doi.org/10.1007/s11095-011-0536-0
- Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem 2007; 360:75-83; PMID: 17113563; https://doi.org/10.1016/j.ab.2006.10.012
- Tsubaki M, Terashima I, Kamata K, Koga A. C-terminal modification of monoclonal antibody drugs: Amidated species as a general product-related substance. Int J Biol Macromol 2013; 52:139-47; PMID: 23022270; https://doi.org/10.1016/j.ijbiomac.2012.09.016
- Gaza-Bulseco G, Bulseco A, Chumsae C, Liu H. Characterization of the glycosylation state of a recombinant monoclonal antibody using weak cation exchange chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2008; 862:155-60; PMID: 18164669; https://doi.org/10.1016/j.jchromb.2007.12.001
- Cordoba AJ, Shyong BJ, Breen D, Harris RJ. Non-enzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B Analyt Technol Biomed Life Sci 2005; 818:115-21; PMID:15734150; https://doi.org/10.1016/j.jchromb.2004.12.033
- Saleem RA, Affholter BR, Deng SH, Campbell PC, Matthies K, Eakin CM, Wallace A. A chemical and computational approach to comprehensive glycation characterization on antibodies. Mabs 2015; 7:719-31; PMID: 26030340; https://doi.org/10.1080/19420862.2015.1046663
- Quan C, Alcala E, Petkovska I, Matthews D, Canova-Davis E, Taticek R, Ma S. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: Where it is, how it got there, and how it affects charge-based behavior. Anal Biochem 2008; 373:179-91; PMID: 18158144; https://doi.org/10.1016/j.ab.2007.09.027
- Stoll DR, Hannes DC, Danforth J, Wagner E, Guillarme D, Fekete S, Beck A. Direct identification of rituximab main isoforms and subunit analysis by online selective comprehensive Two-Dimensional liquid Chromatography-Mass spectrometry. Anal Chem 2015; 87:8307-15; PMID: 26145446; https://doi.org/10.1021/acs.analchem.5b01578