
ABSTRACT
Amino acid sequence differences in the variable region of immunoglobulin (Ig) cause wide variations in secretion outputs. To address how a primary sequence difference comes to modulate Ig secretion, we investigated the biosynthetic process of 2 human IgG2κ monoclonal antibodies (mAbs) that differ only by one amino acid in the light chain complementarity-determining region 1 while showing ∼20-fold variance in secretion titer. Although poorly secreted, the lower-secreting mAb of the 2 was by no means defective in terms of its folding stability, antigen binding, and in vitro biologic activity. However, upon overexpression in HEK293 cells, the low-secreting mAb revealed a high propensity to aggregate into enlarged globular structures called Russell bodies (RBs) in the endoplasmic reticulum. While Golgi morphology was affected by the formation of RBs, secretory pathway membrane traffic remained operational in those cells. Importantly, cellular protein synthesis was severely suppressed in RB-positive cells through the phosphorylation of eIF2α. PERK-dependent signaling was implicated in this event, given the upregulation and nuclear accumulation of downstream effectors such as ATF4 and CHOP. These findings illustrated that the underlining process of poor Ig secretion in RB-positive cells was due to downregulation of Ig synthesis instead of a disruption or blockade of secretory pathway trafficking. Therefore, RB formation signifies an end of active Ig production at the protein translation level. Consequently, depending on how soon and how severely an antibody-expressing cell develops the RB phenotype, the productive window of Ig secretion can vary widely among the cells expressing different mAbs.
| Abbreviations | ||
| CDR | = | complementarity-determining region |
| CHO | = | Chinese hamster ovary |
| DIC | = | differential interference contrast |
| eIF2α | = | eukaryotic initiation factor 2 α-subunit |
| ER | = | endoplasmic reticulum |
| HEK | = | human embryonic kidney |
| HC | = | heavy chain |
| mAb | = | monoclonal antibody |
| LC | = | light chain |
| PBS | = | phosphate-buffered saline |
| PERK | = | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| RB | = | Russell body |
Introduction
Single amino acid substitution within the complementarity-determining region (CDR) and framework region can have dramatic effects on the overall physicochemical properties and the antigen-binding characteristics of immunoglobulins. Humoral immunity naturally exploits such structure-function relationships to generate higher affinity immunoglobulins by somatic hypermutation on the rearranged variable region genes.Citation1,2 Both in vivo during a secondary immune response and in vitro during an antibody engineering effort, one can envision that some amino acid substitutions may be beneficial in imparting higher affinities toward pathogens or antigens of interest, better physicochemical properties such as higher protein stability, or more efficient biosynthesis resulting in higher secretion outputs. It is also equally likely that amino acid substitutions would produce neutral and deleterious effects on antibody functions or its biosynthetic processes.Citation3-5 Because it is difficult to know a priori what types of amino acid substitutions are favored or disfavored in a given immune response, the immune system relies on a Darwinian selection process.6 Namely, by iterating the expansion of reactive B-cell populations, somatic hypermutations, and the selection of beneficial variants, the antibody repertoire is fine-tuned to suit the need of imminent situations.6 During the very same selection process, however, B cells that come to express harmful and disadvantageous immunoglobulin variants are directed to wastage pathways.Citation5
Examples of harmful single amino acid substitution that affect the specificityCitation7-12 and the affinityCitation13-15 of antigen binding are well documented. Another class of deleterious amino acid substitutions increases aggregation propensity of immunoglobulin proteins by affecting folding stability.Citation16-18 Other disadvantageous amino acid substitution are known to impair the secretion of immunoglobulins.Citation4,19-21 Although defective subunit chain folding and flawed subunit assembly were proposed as the potential reasons for the deficiencies in those previously reported studies, detailed biochemical basis for how a single amino acid substitution affects the secretory outputs has not been investigated to date. The involved signaling pathways, if any, and the underlying cell physiologic processes are also unknown. Given the importance of recombinant monoclonal antibodies (mAbs) as a modality of human therapeutics, it is critical to understand the cell biologic basis for the oft-observed mAb secretion output variance caused by the primary sequence difference.
To investigate the underlying mechanisms for secretion level variance among distinct mAbs, we reasoned that minimizing the sequence difference of the mAbs down to a single amino acid residue would be the most effective approach. During an antibody discovery research program aiming to generate human mAbs that specifically recognize and antagonize human cannabinoid receptor type 1 (CB1), a pair of highly related human IgG2κ mAbs were generated. Although the 2 mAbs differed only by one amino acid residue in the LC-CDR loop-1, their secretion titer difference was ∼20-fold when produced using a HEK293 transient expression system. In this regard, this pair of human IgGs served as an ideal model not only to uncover the effects of one amino acid substitution on physicochemical properties of IgGs, but also to obtain new cell biologic insights into the secretion titer variance. Despite the poor secretion, the low-secreting mAb was not a defective IgG as evidenced from its antigen binding, CB1 antagonistic activity, and protein stability under thermal stress. Structural modeling suggested that the low-secreting mAb had greater solvent-exposed hydrophobic characteristics. During immunoglobulin biosynthesis, the low-secreting mAb revealed a higher propensity to aggregate into enlarged globular structures called Russell bodies (RBs)Citation22-25 in the endoplasmic reticulum (ER). Low secretion output from the RB-positive cells, however, was not caused by the functional disruption of secretory pathway organelles or by the physical blockade of secretory membrane traffic. Instead, cellular protein synthesis was severely attenuated in RB-positive cells through the phosphorylation of translation initiation factor eIF2α. The eIF2α phosphorylation was most likely a result of PERK activation as evidenced by the upregulation and nuclear accumulation of downstream effectors such as ATF4 and CHOP. Collectively, our findings illustrated a detailed picture of how a single amino acid substitution alters mAbs' physicochemical properties and biologic functions, how such changes affect the process of immunoglobulin biosynthesis by inducing RB phenotype, and how RB formation is mechanistically linked to a marked decrease in IgG secretion.
Results
Single amino acid residue difference in immunoglobulin CDR-L1 affects IgG secretion titer ∼20-fold
The 2 model mAbs characterized in this study were human IgG2κ that recognized the extracellular regions of human cannabinoid receptor type 1 (CB1). The only difference between the model mAbs was the substitution of one amino acid in the κLC subunit. Specifically, the amino acid position-35 (AHo numbering systemCitation26) in CDR loop-1 was Asn (N) in one model mAb and Trp (W) in the other mAb. They shared an identical heavy chain (HC) subunit. The closest germline gene segment usage for the VH and VL was VH4|4–31/D3|3–9|RF2/JH6 and VK2|A19/JK1, respectively. In the 2 most closely related germline Vκ-gene segments (i.e., VK2|A19 and VK2|A3), the position-35 was N; while in the next 3 closest germline Vκ-gene segments (i.e., VK2|O1, VK2|O11 and VK2|A1), the position-35 was Asp (D). We extended the survey to 30 additional Vκ-gene segments, but no germline sequence had the W residue at this position. We therefore called N-35 version as the “parental” and W-35 version as “N35W variant” in this study. Amino acid sequences surrounding the N-35 residue were identical to the 2 most closely related germline-encoded gene segments stated above and thus contained no somatic mutations. The primary sequence information can be found in a published patent disclosure.Citation27
The Trp residue of the N35W variant was predicted to be located in the middle of CDR-L1. Structural models of the parental and N35W variant indicated that CDR-L1 might interact directly with CDR-H3 through side chain contacts (). Both of these CDRs were unusually long (CDR-L1: 16 residues; CDR-H3: 17 residues), and were likely to be highly dynamic in the absence of bound antigen based on the structural divergence of low-energy conformations sampled in low-mode molecular dynamics simulations (data not shown). Furthermore, models showed the Trp residue of the N35W variant was solvent-exposed, presenting a hydrophobic patch that could render the antibody prone to non-specific interactions ().
Figure 1. Effects of single amino acid substitution on the secretion of whole IgG and LC subunit. (A) Homology models showing the variable fragment of the parental (left) and the N35W variant (right) mAbs. Surface is colored according to the Eisenberg hydrophobicity scaleCitation47 with red indicating high hydrophobicity and white indicating low hydrophobicity. The N35W variant has more exposed hydrophobic characteristics in the CDRs than does the parental mAb. LC-CDR1 (L1) and HC-CDR3 (H3) are long and are predicted to be highly dynamic in solution. The Trp residue of the N35W variant is likely to be solvent-exposed, presenting a hydrophobic patch that could render the antibody prone to non-specific interactions. (B, C, D) Cell culture media and whole cell lysate samples were prepared on day-7 post transfection and were subjected to SDS-PAGE followed by Western blot using rabbit anti-human IgG (H+L) polyclonal antibody. Samples in lanes 1–2 and lanes 3–5 were analyzed in the non-contiguous lanes of a single gel, but the intervening lanes were digitally removed to create the figures B–D. (B) A sample volume corresponding to the 5 μl of harvested cell culture medium was loaded per lane and analyzed under reducing conditions. Expected band for heavy chain (HC) and light chain (LC) subunits are marked by arrowhead. Determined secretion titers for this mAb production run are shown in lanes 1 and 2. (C) Whole cell lysates equivalent to 12,000–12,500 cells were loaded per lane under reducing conditions. Expected band for HC and LC subunits are marked by arrowhead. A faint band marked by asterisk in lane 5 is apparently a LC-N35W dimer species that was resistant to SDS treatment or disulfide reduction. Anti-GAPDH blot is shown at the bottom as a loading reference. (D) Cell culture media were analyzed under non-reducing conditions. LCs are secreted as a mixture of monomers (mono.) and covalent dimers (di.). Trimer and tetramer species are faintly detectable. (E) On day-2 post transfection, transfected cells were resuspended in fresh growth media with or without 15 μg/ml BFA and maintained in suspension format until day-3 when cell culture media and cell pellets were harvested and analyzed under reducing conditions. The amount of IgGs secreted to the culture medium during the 24 hr period is shown in the left panel. The amount of IgGs detected in the cell lysates is shown in the right panel. Anti-GAPDH blot is shown as a loading reference for cell lysate samples. (F, G) Cell viability and viable cell density were monitored daily by using automated cell counters. Data compiled from 6 independent experiments for the parental IgG and 4 independent runs for the N35W variant were used to calculate the average and standard deviation.
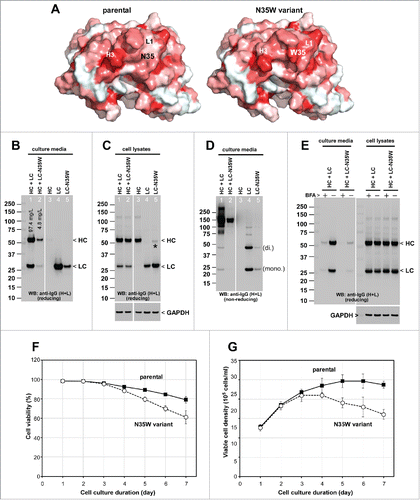
To characterize the biosynthetic processes of the 2 mAb proteins in detail, we recombinantly expressed them using a HEK293 transient expression system. Under identical transfection and cell culture conditions, the secretion titer of the parental mAb [128.4 mg/L ± 61.3 (n = 6)] was consistently 10- to 20-fold higher than that of N35W variant [5.6 mg/L ± 2.7 (n = 4)] (see also ). Similar to many model HCs characterized in the past in our laboratory,Citation24,28,29 the HC subunit (shared by both mAbs) was abundantly synthesized in the cells (), but was not secreted to the culture media in the absence of LC synthesis ( and ). Likewise, similar to many LCs,Citation24,28,29 the parental LC was secreted as a mixture of monomers and covalently-linked dimers even in the absence of HC subunit ( and ). By contrast, although the LC-N35W variant was also detected in the culture media, its secretion level was markedly lower than the parental LC ( and ). Evidently, single amino acid substitution had a profound effect on the secretion of whole IgG and the individually expressed LC subunit. No evidence of unexpected post-translational modification such as N-linked glycosylation or protein cleavage because of the N-35 residue or the N35W substitution was found ().
During our routine 7-day batch cell culture runs, there were noticeable differences in cell viability, cell growth, and peak cell density between the cell population transfected to express the parental IgG and the N35W variant, especially after 72 hr post-transfection (). Because nearly 50% of the cells were non-transfected bystander cells in this expression system,Citation24 we cannot directly interpret the time course data without fully elucidating the growth difference between transfected and non-transfected cells that co-existed in the cell population. However, similar cell growth difference was reported previouslyCitation24 for a different panel of human IgGs where the cells producing high-secreting mAbs maintained higher cell viability than those cells expressing poor-secreting mAbs. Although such cell viability differences between “good expressers” and “bad expressers” has widely been taken for granted, the cell physiologic reasons behind this difference have not been investigated in detail to date (see below for more details).
The poorly secreted N35W variant IgG induces Russell body phenotypes during biosynthesis
Poor secretion titers of various immunoglobulin clones were previously attributed to the formation of RBs both in homologous and heterologous cellular models.Citation24,28,30-32 To address whether the diminished secretion of N35W variant was also associated with the high occurrence of RB phenotype, we examined the subcellular localization of both mAbs by immunofluorescent microscopy.
Under steady-state normal cell growth conditions, the parental IgG localized broadly to the ER and Golgi in transfected cells (). Aggregation of IgGs into globular RB phenotype was observed in 12.9% of the transfected cells (RB-positive/transfected = 16/124). On the other hand, 78.9% (390/494) of the N35W variant-expressing cells developed RB phenotypes (). Similar to our previous studies using different panels of model mAb clones,Citation24,28 the low IgG secretion titers correlated with the high occurrence of RB formation.
Figure 2. Single amino acid substitution leads to a marked increase in Russell body phenotype occurrence during immunoglobulin biosynthesis. Fluorescent micrographs of HEK293 cells expressing the parental IgG or its N35W variant. On day-2 post transfection, HEK293 cells were resuspended in fresh cell culture media with or without 15 μg/ml BFA, then immediately seeded onto poly-lysine coated glass coverslips and statically cultured for 24 hr. On day-3, cells were fixed, permeabilized, and immuno-stained. Co-staining was performed by using FITC-conjugated anti-gamma chain and Texas Red-conjugated anti-kappa chain polyclonal antibodies. Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views. (A) Subcellular localization of gamma-chain and kappa-chain was visualized under steady-state normal cell growth conditions. Two representative image fields for the parental IgG expressing cells are shown in the first 2 rows. Two representative image fields for N35W variant IgG expressing cells are shown in rows 3 and 4. (B) Gamma- and kappa-chains of the parental IgG (first 2 rows) and N35W variant IgG (rows 3 to 5) were visualized after 24 hr BFA treatment. The RB phenotype frequency for each mAb under steady-state or after BFA treatment is stated in the text. Unlabeled scale bar represents 10 μm.
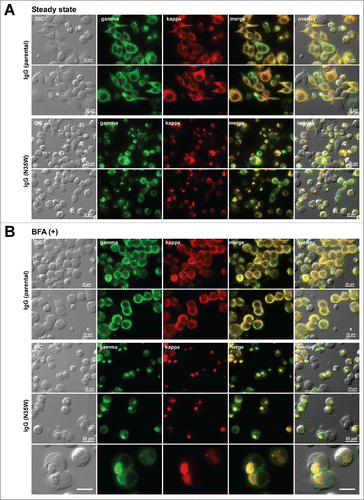
To examine if RB phenotype occurrence would increase under aberrant cell culture conditions, transfected cells were cultured in the presence of brefeldin A (BFA) for 24 hr from day-2 to day-3 post transfection to withhold newly synthesized IgGs in the ER by blocking the ER-to-Golgi cargo transport. During the 24 hr BFA treatment, the secretion of IgGs was almost completely blocked (). Such adverse cell culture conditions were shown previously to increase RB phenotype occurrence by perturbing cellular protein homeostasis.Citation24 For this pair of mAbs, however, the BFA treatment had negligible effect and it only slightly increased the RB phenotype prevalence to 15.2% (75/492) and 82.1% (469/571) for the parental mAb and the variant mAb, respectively ().
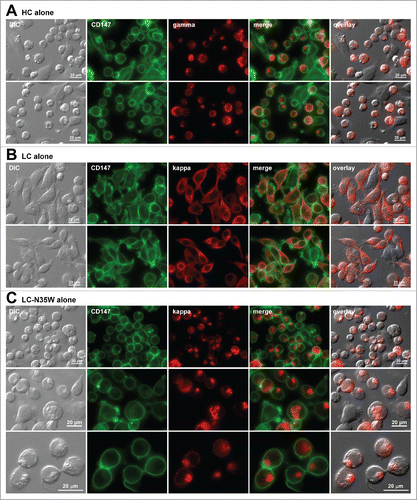
To determine the extent of individual subunit chain contribution toward RB phenotype induction, we individually tested the shared HC, the parental LC, or N35W variant LC subunit for their respective subcellular localization at steady-state. The shared HC subunit induced RB phenotypes very frequently, in 89.0% (269/302) of the expressing cells (). This result was in good agreement with the lack of HC subunit secretion in the absence of LC subunit co-expression (). In stark contrast to HC, the parental LC did not induce detectable RB phenotypes in transfected cells (0%, 0/164), but instead the intracellular LC pool was broadly distributed to the ER and Golgi at steady-state (). This result again agreed with the abundant LC-only secretion in the absence of HC subunit (see above, ). N35W variant LC by itself, on the other hand, was sufficient to induce RB phenotypes frequently in 72.0% (337/468) of the transfected cells (). The propensity of individual subunit chains to induce the RB phenotype was mostly unchanged even after a 24 hr BFA treatment (Supplement 1 and its legend). In summary, this part of the study illustrated the negative effects of N35W substitution on the biosynthetic processes of variant IgG that led to a marked increase in RB phenotype induction. The results also supported the previously suggested inverse correlation of IgG secretion titers and RB phenotype prevalence.
Figure 3. Russell body-inducing propensities of individual subunit chains. Fluorescent micrographs of HEK293 cells transfected with the (A) HC construct alone, (B) LC construct alone, or (C) N35W variant LC construct alone, under steady-state cell growth conditions. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically cultured for 24 hr. On day-3, cells were fixed, permeabilized, and co-stained with anti-CD147 and anti-gamma chain (A). In panels B and C, cells were co-stained with anti-CD147 and anti-kappa chain. Endogenous CD147 was stained to highlight cell shapes. Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘red’ were superimposed to generate ‘overlay’ views.
N35W substitution does not abolish protein stability, antigen binding, or biologic function
One possible reason for the observed poor secretion of N35W variant IgG might be that the amino acid substitution rendered the IgG defective in terms of folding stability or function; and the cell's protein quality control mechanisms discouraged a copious release of such potentially harmful IgG to the extracellular space. To examine the effects of N35W substitution in more detail, we purified a sufficient amount of both mAbs from the harvested cell culture media (see Materials and Methods). On the SDS-PAGE gels, the parental IgG and N35W variant behaved identically under both reducing and non-reducing conditions (). For both mAbs, the measured average masses of reduced LCs and reduced deglycosylated HCs matched those of the theoretical masses within instrumentational deviations (data not shown). Accordingly, there were no detectable gross structural abnormalities in these 2 mAbs. Data obtained from mass spectrometric analysis also excluded the possibility of unexpected acquisition or loss of post-translational modifications as the underlining cause of the secretion titer differences.
Figure 4. Comparison of the parental and N35W variant mAb properties by biochemical and cell-based assays. (A) After purification, the parental IgG and its N35W variant were resolved in SDS-PAGE gels side by side under reducing (lanes 1–2) and non-reducing (lanes 3–4) conditions, then stained with Coomassie blue dye. Protein loading was 10 μg per lane. Protein bands corresponding to the HC subunit, LC subunit, and whole IgG are marked by arrowhead. (B) Purified IgGs were formulated in PBS and analyzed by differential scanning calorimetry. Heat capacity (Cp) is plotted on the y-axis as a function of heat influx on the x-axis. Three discernible peaks are labeled with the domain names corresponding to the respective unfolding events that take place at different temperature range. (C) Size-exclusion chromatograms of purified parental IgG (top) and its N35W variant (bottom) using Superdex G200 column. Protein loading was 10 μg each. The running buffer was PBS at a flow rate of 0.7 ml/min. The elution profile was monitored by UV light at 215 nm. AU, arbitrary unit. Paper-printed chromatograms were scanned and digitally reproduced to create this figure. Retention time is labeled for each elution peak. (D) Flow cytometric binding analysis of the parental and variant mAbs to human CB1 expressed on the surface of stable CHO cell line. After harvesting the adherently cultured CHO cells non-enzymatically, 200,000 cells were stained at testing antibody concentrations of 0.2, 1, and 5 μg/ml. A representative result obtained at 1 μg/ml antibody staining condition is shown. Signal intensity based on the geometric mean values was ∼2-fold higher for N35W variant mAb than for the parental mAb. (E) Results of cell-based assay measuring cellular cAMP levels after CB1 activation in the presence of the parental or N35W variant mAbs. CB1 activation leads to a decrease in intracellular cAMP. If CB1 activation is blocked by antagonistic mAbs, intracellular cAMP level would be restored. In this assay system, the variant mAb showed ∼4-fold higher antagonistic activity than the parental mAb. Four-fold difference in antagonistic activity was reproducible although the IC50 values shifted slightly on different testing occasions. This graph was generated by using a data set obtained in one such representative experiment. Error bar represents ‘standard error of mean’.
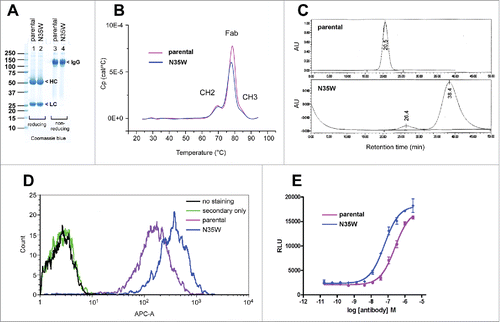
Differential scanning calorimetry for the 2 model mAbs showed that the unfolding profile during thermal stress was similar to each other, with the melting temperature for the Fab domain being ∼77 °C (). The results indicated that an amino acid substitution to a surface-exposed residue (in LC-CDR loop-1) had negligible effects on thermal stability. By contrast, the 2 mAbs behaved rather differently in the elution profiles of size-exclusion chromatography using Superdex G200 column (GE Healthcare). While the parental IgG eluted as a narrow single peak with a retention time of 20.5 min (), N35W variant eluted in 2 peaks consisting of ∼7% pre-peak eluted after 26.4 min and the major broad asymmetric peak eluted after 38.4 min (). Two additional analyses using different size-exclusion chromatography columns, i.e., Superose 12 (GE Healthcare) and TSKgel G3000 (Tosoh Bioscience), gave rise to similar elution profiles (data not shown). The retardation in gel filtration chromatography suggested that N35W variant mAb interacted extensively with matrices due to the surface-exposed aromatic tryptophan residue at position-35.
Specific binding of the model mAbs to stable Chinese hamster ovary (CHO) cells overexpressing human CB1 was evaluated by flow cytometry to determine if there are any effects of N35W substitution on antigen recognition. Live CHO cell staining at 3 different concentrations (0.2, 1, and 5 μg/ml) consistently demonstrated higher staining signal intensity with the N35W variant than with the parental mAb. The flow cytometric binding data for 1 μg/ml staining is shown in . In terms of the geometric mean values, the signal intensity for the N35W variant mAb was ∼2-fold greater than that of the parental mAb at 1 μg/ml staining condition. Similar results were obtained for the staining conditions at 0.2 μg/ml or 5 μg/ml, although the signal intensities varied depending on the testing antibody concentrations, demonstrating a dose-dependent binding of both mAbs (data not shown). Non-specific binding was not observed, as non-transfected CHO cells were not stained by neither mAbs (data not shown).
To test how a stronger binding of N35W variant is translated into measurable activity differences, we compared the antagonistic effects of N35W variant to that of the parental mAb in a cell-based functional assay. Stable CHO cells overexpressing human CB1 were stimulated with forskolin and a synthetic cannabinoid CP 55,940 in the presence of varying concentrations of testing mAbs (see Materials and Methods). Cannabinoid-induced activation of CB1 is known to inhibit adenylyl cyclase activity and was previously shown to decrease intracellular cAMP level.Citation33 Antagonizing the activation of CB1 receptor is therefore expected to restore intracellular cAMP levels in a dose-dependent manner. In this cell-based assay system, the IC50 for the parental mAb and N35W variant was 257.3 nM and 62.1 nM, respectively (). The N35W substitution therefore resulted in ∼4-fold improvement in antagonistic activity.
In summary, although the characterizations conducted here were by no means comprehensive, these studies were nonetheless sufficient to show that N35W variant was not a defective IgG.
Subcellular organelles are affected differently by the formation of Russell bodies
Apparent correlations between RB formation and poor immunoglobulin secretion were documented repeatedly (see above), but precise reasons why the RB formation impairs immunoglobulin secretion remained unknown. Because RBs are large protein aggregates composed of immunoglobulins and ER-resident proteins,Citation23,24,29 we initially suspected that formation of RBs would disrupt the organization of secretory organelles and block secretory pathway membrane traffic, thereby resulting in diminished IgG secretion. To test this view, we examined whether there are detectable abnormalities in the subcellular organizations in RB-positive cells.
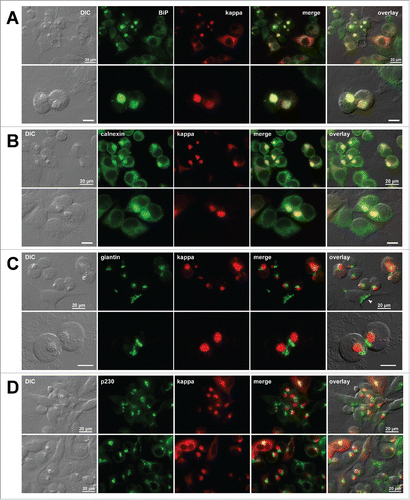
As repeatedly shown in the past,Citation23,24,29 immunoglobulins and ER-resident proteins such as BiP and calnexin co-localized in the spherical structures (,). In fact, this is one of the hallmark characteristics that defines RBs—except for a mouse μ-HC mutant lacking the CH1 domainCitation25 in which RBs seemed to be segregated from the ER resident proteins. Although ER resident proteins co-localized with the RBs, the tubular and reticular portions of the ER membranes appeared intact in the cell periphery (). This suggested that not all the ER membranes and ER-resident proteins were affected or became absorbed into the enlarged globular structures. When visualized by giantin, the cis-/medial-Golgi membranes were condensed and placed in close juxtaposition to RBs () instead of showing a characteristic ribbon-like appearance (). Similarly, a trans-Golgi marker p230 distributed to a clustered localization next to RBs (). These morphological data demonstrated that the formation of RBs in the ER affected the distribution of Golgi membranes.
Figure 5. Organization of secretory pathway organelles in Russell body-positive cells. Fluorescent micrographs of HEK293 cells transfected with N35W variant LC construct. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically incubated for 24 hr. On day-3, cells were fixed, permeabilized, and co-stained with Texas Red-conjugated anti-kappa chain polyclonal antibody and specific antibodies against various organelle markers. (A, B) ER markers BiP and calnexin. (C) A cis-/medial-Golgi marker giantin. (D) A trans-Golgi marker p230. Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views. In panel C top row, arrowhead points to a normal ribbon-like Golgi morphology in a non-transfected cell. Unlabeled scale bar represents 10 μm.
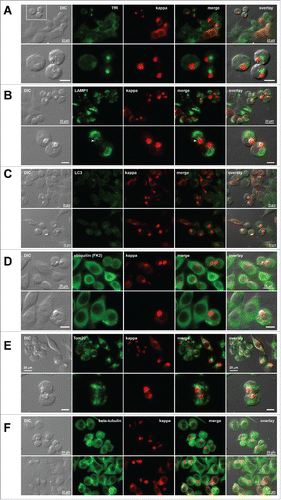
We also examined the organelles of the endocytic pathway and other important subcellular structures to determine if there are any other notable changes in subcellular organization. Transferrin receptor (TfR), a recycling endosome marker, redistributed from the normal cytoplasmic puncta and cell surface localization to a diffused punctated structure locating next to RBs (). Although some lysosomal membranes (visualized by LAMP1) were in contact with RBs (, bottom row, arrowhead), the majority of lysosome membranes occupied the cytoplasmic space that did not overlap with RBs (). Autophagosome formation (visualized by LC3) was not enhanced in RB-positive cells (), although we initially considered an intriguing possibility that RBs are targeted to the autophagic route similar to those reported in the clearance of aggresomes.Citation34 Ubiquitin staining (detected by a mAb clone FK2Citation35) showed that poly-ubiquitinated proteins distributed throughout the cytosol regardless of the RB phenotype (). This observation clearly differentiated the ER-derived RBs from the cytosolic inclusion bodies such as aggresomesCitation36 and Lewy bodies,Citation37 both of which are known to co-accumulate poly-ubiquitinated proteins. Mitochondria (stained by Tom20) also occupied the cytoplasmic space different from the RB structures; but similar to those of lysosomes, a subset of mitochondrial membranes appeared contacting RBs (). Lastly, microtubules surrounded RBs like a cage, but their arrangement and overall intracellular distribution (visualized by a mAb against β-tubulin) did not show readily detectable abnormalities (). Except for the TfR that redistributed to a large puncta in the Golgi region, we did not detect any other notable differences from these evaluations.
Figure 6. Distribution of additional subcellular markers in Russel body-positive cells. Fluorescent micrographs of HEK293 cells transfected with N35W variant LC construct. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically incubated for 24 hr. On day-3, cells were fixed, permeabilized, and co-stained with Texas Red-conjugated anti-kappa chain polyclonal antibody and specific antibodies against various subcellular markers. (A) A recycling endosome marker, transferrin receptor (TfR). The boxed area in the first row is digitally magnified and shown in the second row. (B) A lysosome marker, LAMP1. Arrowhead shown in the second row points to lysosome–RB contact area. (C) An autophagosome marker, LC3. Regardless of the RB phenotype, LC3 is not upregulated under normal cell culture conditions used. (D) Poly-ubiquitinated proteins were stained with anti-ubiquitin monoclonal antibody (clone FK2). (E) A mitochondrial membrane marker, Tom20. (F) A microtubule component β-tubulin. Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views. Unlabeled scale bar represents 10 μm.
Secretory membrane traffic is not obliterated in Russell body-positive cells
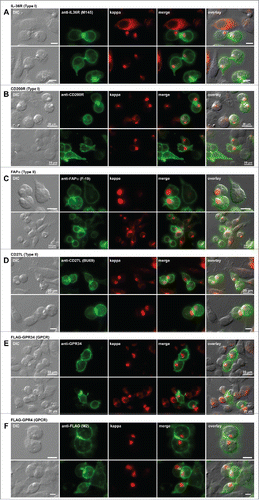
Because mere examinations of organelle integrity did not give us direct clues to understand the diminished IgG secretion when RBs are formed, we next tested whether the flow of cargo trafficking in secretory pathway was impaired in RB-positive cells. In the literature, the arrival of endogenously expressed plasma membrane proteins to the cell surface was taken as the evidence of normal secretory pathway trafficking in RB-positive cells.Citation30,31 In fact, endogenously expressed CD147 was also detectable at the surface of RB-positive cells in this study (see ). However, the plasma membrane cargo could have easily reached the cell surface well before the onset of RB formation. To avoid this pitfall, we co-expressed the RB-inducing N35W variant LC and one of the 6 different membrane-anchored proteins destined to travel to the cell surface. To use a wide variety of cargo classes, we chose IL-36R and CD200R from the type I membrane proteins; FAPα and CD27L to represent the proteins of type II membrane topology; and GPR34 and GPR4 as examples of polytopic membrane proteins. After co-transfection, cells were examined to determine whether the membrane-anchored cargo reached the cell surface in RB-housing cells. In this way, we can temporally synchronize the synthesis of comparing RB-inducing LC subunit and the recombinant cell surface proteins. In all 6 cases, the membrane-anchored proteins reached the cell surface regardless of the RB phenotypes (). Although these results did not address the membrane trafficking kinetic differences, if any, between the RB-positive and RB-negative cells, the results nonetheless demonstrated the flow of secretory membrane trafficking was not hindered in RB-positive cells.
Figure 7. Cell surface trafficking of membrane-anchored proteins in Russell body-positive cells. Fluorescent micrographs of HEK293 cells co-transfected with N35W variant LC construct and one of the expression constructs encoding the following plasma membrane protein. Type I membrane proteins: (A) human IL-36R and (B) human CD200R. Type II membrane proteins: (C) human FAPα and (D) human CD27L. Polytopic membrane proteins: (E) N-terminally FLAG-tagged human GPR34 and (F) N-terminally FLAG-tagged human GPR4. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically incubated for 24 hr. On day-3, cells were fixed, permeabilized, and co-stained with Texas Red-conjugated anti-kappa chain polyclonal antibody and the following antibodies. (A) Mouse anti-human IL-36R monoclonal antibody, clone M145. (B) Mouse anti-human CD200R monoclonal antibody, clone 380525. (C) Mouse anti-human FAPa monoclonal antibody clone F19. (D) Mouse anti-human CD27L monoclonal antibody, clone BU69. (E) Mouse anti-human GPR34 monoclonal antibody, clone 419859. (F) Mouse anti-FLAG monoclonal antibody, clone M2. Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views. Unlabeled scale bar represents 10 μm.
Protein synthesis is repressed in Russell body-positive cells
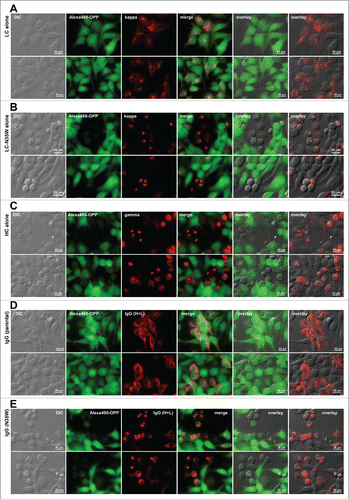
Secretory pathway membrane trafficking appeared operational in RB-positive cells, yet the IgG secretion level dropped significantly when RB phenotypes were induced. Another possible way to explain the correlation between low immunoglobulin secretion and high-level induction of RBs is that immunoglobulins are no longer actively synthesized in RB-positive cells. We reasoned that downregulation of IgG synthesis at the translation level could also cause poor secretory output even when the secretory pathway trafficking was operational. To directly measure the extent of active protein synthesis in situ by fluorescent imaging, we used a protein synthesis assay kit that was based on O-propargyl-puromycin (OPP) and click chemistry reactions (see Materials and Methods).
In the cells expressing the parental LC alone, the LC was distributed to the ER and Golgi at steady-state and the RB phenotype was rarely induced as before (). When the degree of active protein synthesis was measured in the same cells, the fluorescent signals, which reflect the active incorporation of OPP to polypeptide chains during the 30 min labeling period, were indistinguishable between the LC-expressing cells and non-transfected cells in the same image fields (). By contrast, the cells expressing the LC-N35W variant or the HC subunit alone induced RB phenotypes at high frequency as before (); in those RB-positive cells, fluorescent signals corresponding to the degree of active protein synthesis were hardly detectable (). Non-transfected cells in the same image fields were actively synthesising the proteins under the same labeling condition (). Similarly, while the cells expressing the parental mAb showed active protein synthesis (), the cells expressing the variant mAb induced RB phenotypes and the protein synthesis was suppressed when RBs are formed (). Regardless of whether the cells expressed whole IgGs or individual subunits, when cells developed RB phenotypes, the cellular protein synthesis was severely suppressed.
Figure 8. Protein translation is suppressed in Russell body-positive cells. Fluorescent micrographs of HEK293 cells transfected with the following construct(s): (A) LC construct alone; (B) LC-N35W construct alone; (C) HC construct alone; (D) parental IgG; (E) N35W variant IgG. Transfected cells were cultured under steady-state growth conditions and labeled for 30 min using Alexa Fluor 488 Click-iT® Plus OPP reagent to measure actively ongoing protein translation in situ (see Materials and Methods). The click-labeled cells were then immuno-stained with (A, B) Texas Red-conjugated anti-kappa chain, (C) Texas Red-conjugated anti-gamma chain, or (D, E) Alexa Fluor 594-conjugated anti-human IgG (H+L). Green and red image fields were superimposed to create ‘merge’ views. DIC and green or DIC and red images were superimposed to generate ‘overlay’ views.
To exclude a possibility that RB-positive cells were already dead (and thus they stopped synthesising proteins), we performed an imaging-based cell viability assay using a fixable viability dye (see Materials and Methods). At steady-state normal cell growth conditions, RB-positive cells were viable at 72 hr post transfection at which time point the cells were fixed and processed for imaging (Supplement 2).
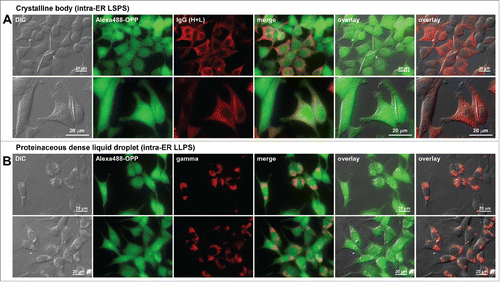
At least 2 additional types of intracellular inclusion bodies are known to develop in the ER of immunoglobulin overexpressing cells. These are crystalline body29,38 and dense liquid protein droplet.Citation39 We showed previously that these inclusion body phenotypes were not necessarily associated with low immunoglobulin secretion titers.Citation29,38,39 If the correlation between an attenuated protein synthesis and a diminished immunoglobulin secretion holds true, the protein translation would remain active in those cells despite the formation of prominent inclusion bodies. As predicted from this view, the level of active protein translation was not affected in the cells that house those inclusion bodies ().
Figure 9. Protein translation is not attenuated in cells showing different classes of immunoglobulin inclusion body phenotype. Fluorescent micrographs of HEK293 cells expressing (A) a human IgG4 clone that induces crystalline body via intra-ER liquid-solid phase separation (LSPS)Citation29 and (B) a scFv-Fc construct that induces morula cell phenotype via intra-ER liquid-liquid phase separation (LLPS).Citation39 Transfected cells were first labeled for 30 min using Alexa Fluor 488 Click-iT® Plus OPP reagent to detect actively ongoing protein translation in situ (see Materials and Methods). The click-labeled cells were then immuno-stained with (A) Alexa Fluor 594-conjugated anti-human IgG (H+L) or (B) Texas Red-conjugated anti-gamma chain polyclonal antibodies. Green and red image fields were superimposed to create ‘merge’ views. DIC and green or DIC and red images were superimposed to generate ‘overlay’ views.
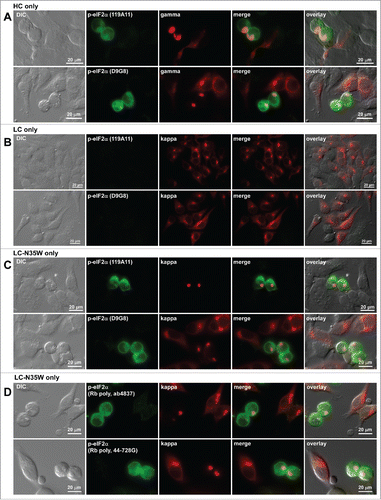
Translation initiation factor eIF2α is phosphorylated in RB-positive cells
One arm of unfolded protein response signaling pathway mediated by PERK is known to downregulate protein synthesis by phosphorylating the Ser-51 residue of eIF2α.Citation40 We were thus prompted to examine the phosphorylation status of eIF2α in RB-positive cells. Testing with 2 independent mAbs (clones 119A11 and D9G8) that specifically recognize phosphorylated eIF2α revealed that eIF2α was indeed phosphorylated in RB-positive cells regardless of whether the phenotype was induced by HC subunit alone (), by LC-N35W alone (), or by N35W variant IgG (data not shown). Comparison with non-transfected cells and RB-negative transfected cells in the same image fields illustrated that eIF2α phosphorylation occurred selectively in RB-positive cells (). By contrast, the cells expressing the parental LC subunit alone did not show detectable eIF2α phosphorylation at steady-state (). To lend stronger support for this finding, we additionally probed with 2 independent polysera raised against phosphorylated eIF2α and obtained identical results (). The evidence demonstrated that downregulation of protein translation in RB-positive cells was attributable to the phosphorylation of eIF2α.
Figure 10. Translation initiation factor eIF2α is phosphorylated in Russell body-positive cells. Fluorescent micrographs of HEK293 cells transfected with the following constructs: (A) HC construct alone; (B) LC construct alone; (C, D) N35W variant LC construct alone. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically incubated for 24 hr. On day-3, cells were fixed, permeabilized, and stained with Texas Red-conjugated anti-kappa chain or Texas Red-conjugated anti-gamma chain polyclonal antibodies. In panels A through C, cells were co-stained with 2 different rabbit anti-phosphorylated eIF2α monoclonal antibodies, clones 119A11 (first row of each panel) or D9G8 (second row of each panel). In panel D, cells were co-stained with 2 rabbit anti-phospho-eIF2α polysera obtained from 2 different sources (see Materials and Methods). Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views.
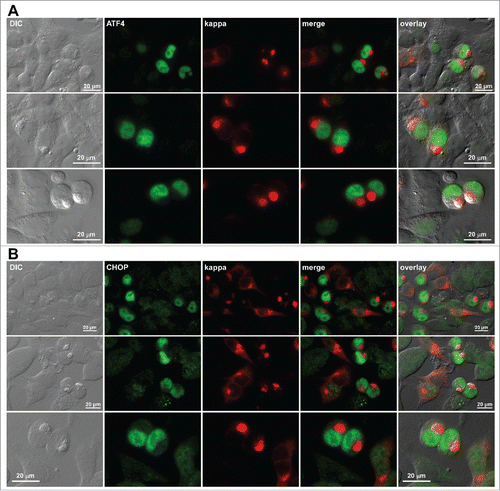
We attempted to visualize the phosphorylation status of PERK itself in individual cells but were limited by the lack of reliable antibody reagents that can report the phosphorylation status of PERK by immunofluorescent imaging. This led us instead to probe 2 key downstream PERK effectors,Citation41 ATF4 and CHOP. Upregulation and nuclear accumulation of both ATF4 and CHOP () implicated that PERK was involved in the signaling pathway leading to eIF2α phosphorylation in RB-positive cells. Direct evidence of PERK activation was not obtained in this study and will require further investigation.
Figure 11. Upregulation and nuclear accumulation of PERK downstream effectors ATF4 and CHOP in Russell body-positive cells. Fluorescent micrographs of HEK293 cells transfected with N35W variant LC construct. On day-2 post transfection, suspension cultured cells were seeded onto poly-lysine coated glass coverslips and statically incubated for 24 hr. On day-3, cells were fixed, permeabilized, and co-stained with Texas Red-conjugated anti-kappa polyclonal antibody and specific antibodies against ATF4 (A) and CHOP (B). Green and red image fields were superimposed to create ‘merge’ views. DIC and ‘merge’ were superimposed to generate ‘overlay’ views.
Discussion
The pair of human IgG clones examined in this study showed that single amino acid substitution in the variable region was sufficient to alter the efficiency of immunoglobulin biosynthesis. The 2 mAbs differed only by one amino acid in the LC's CDR1. Despite the near-identity of their primary sequences, the parental mAb secreted copious amounts of IgG to the culture media, while the variant mAb induced RB phenotypes extensively and secreted 20-fold less IgG. Importantly, the 2 model IgGs were by no means abnormal or defective as mAbs. It is quite intriguing to see a profound impact of a single amino acid substitution on immunoglobulin biosynthesis.
The effect of single amino acid substitution was not limited to the biosynthetic processes. For the current 2 model mAbs, in exchange for a compromised secretion titer, the variant mAb acquired stronger binding to its antigen human CB1 (as determined by FACS geometric mean because the binding affinity was difficult to measure on full-length, cell-surface expressed receptors), which corresponded with 4-fold higher potency in antagonizing CB1 activation. At the same time, the variant mAb showed extensive interactions with the matrices used in size-exclusion chromatography. Partly because the substitution was at a surface-exposed amino acid residue, the substitution itself did not compromise protein stability measured by thermal unfolding. If we could borrow a word from genetics, it was evident that the single amino acid substitution had pleiotropic effects on various aspects of immunoglobulin properties. In this respect, changing a desirable property to an undesirable one, and vice versa, can be as simple as a single amino acid substitution. To obtain desired outcomes, the main difficulty is our inability to know a priori which position is the right place to make a substitution and which amino acid should be the substitute residue.
This study unraveled a sequence of events that apparently linked RB formation and the suppression of immunoglobulin secretion. We initially assumed that enlarged aggregate formation in the ER disrupts the organization of secretory organelles or obstructs the secretory pathway membrane trafficking. Although the Golgi membrane distribution was somewhat altered in RB-positive cells, the membrane trafficking from the ER to the plasma membrane was operational even when the cells developed RB phenotypes. Instead, we found that the amount of cellular protein synthesis was severely downregulated in RB-positive cells through the phosphorylation of eIF2α, potentially by way of PERK activation. These findings led us to propose that the amount of immunoglobulin synthesis declines as a function of RB formation in the ER. Simply because fewer immunoglobulin molecules are synthesized, the cellular secretory output drops accordingly even when secretory pathway trafficking is operational. Therefore, depending on how soon and how severely the RB phenotypes are induced after the onset of massive immunoglobulin synthesis, the productive window of immunoglobulin secretion varies significantly. For instance, our previous studies showed that recombinant human IgG secretion can be as low as < 1 mg/L or as high as > 300 mg/L depending on the individual mAb clones, despite using the identical HEK293 cell-based batch production method.Citation24,28,29,38,39 As soon as cells develop RBs, they stop contributing to amass the secreted IgGs in cell culture medium during a cell culture process. Consequently, depending on how many cells are to develop RB phenotype in a given cell population and how early it happens, the volumetric titers can vary widely. In essence, the induction of RB phenotypes effectively signals an end of immunoglobulin production in afflicted cells.
Studies in the 1970s and 1980s led to a view that the RB phenotype might be a sign of plasma cell senescence and the terminal differentiation state of plasma cell life cycle when the cell's secretory functions come to an end.Citation42,43 Alanen and coworkers' findings that RB phenotypes were associated with low or no immunoglobulin secretion in Mott cell hybridomasCitation30,31 were viewed as the supportive evidence for this hypothesis. The current study not only supported these views, but also provided a potential underlining cell physiologic process that linked the induction of RB phenotype and diminished immunoglobulin secretion. Given that IgGs constitute a large proportion of total cellular protein synthesis during overexpression, we argue that the global attenuation of protein synthesis would most profoundly affect the amount of IgG protein synthesis both in plasma cells and recombinant cells alike. Although we suggest that attenuation of protein synthesis played a major role in the downregulation of IgG secretion output, we cannot exclude the possibility that the kinetics of secretory membrane traffic was also affected. This could lead to an even more precipitous way to shutdown immunoglobulin secretion upon RB formation.
Although intracellular inclusion bodies now referred to as RBs were first described in 1890Citation22 and several studies were conducted in animal models and cell culture systems,Citation23 some fundamental aspects of the RB phenotype are still enigmatic. For example, whether RB formation is indeed the terminal cellular state and whether the phenotype can be reversed to restore protein synthesis and secretion are physiologically important questions that remain unanswered. Pathophysiological reasons why RBs and Mott cell phenotypes are frequently reported in patients of plasma cell dyscrasias are still largely unknown. Cellular health was affected when RBs were induced in a transient burst expression system using heterologous cell hosts, but there are examples in hybridoma cell linesCitation30,31 and multiple myeloma in plasma cell dyscrasiasCitation23 where the RB-harboring cells continued to propagate without losing Mott cell phenotypes. What allowed these RB-housing cells to remain viable (in the wake of chronic intracellular immunoglobulin deposition) is currently unknown. We speculate that some unidentified signaling pathway rewiring might have taken place in these cells that allowed them to circumvent cell death. If we are able to elucidate the genetic or epigenetic basis of such cell capacity enhancement or cell physiology changes, the findings can be applied to recombinant mAb production biotechnology through cell engineering.
Materials and methods
Detection antibodies
Mouse monoclonal anti-FLAG (M2) (cat. F1804) and rabbit polyclonal anti-calnexin (cat. C4731, lot 095M4787V) were purchased from Sigma-Aldrich. Mouse monoclonal anti-GAPDH (cat. MAB374) and rabbit polyclonal anti-transferrin receptor (cat. CBL47, lot 2207119) were obtained from Chemicon. Mouse monoclonal anti-CD147 (cat. 555961) and mouse monoclonal anti-p230 (cat. 611280) were from BD Biosciences. Rabbit polyclonal anti-giantin (cat. PRB-114P, lot E10388KF) was from Covance. Mouse monoclonal anti-LAMP1 (H4A3) (cat. sc-20011) and rabbit polyclonal anti-Tom20 (FL-145) (cat. sc-11415, lot D1409) were from Santa Cruz Biotechnology. Mouse monoclonal anti-LC3 (4E12) (cat. M152–3) was from MBL International Corporation. Monoclonal anti-ubiquitin (FK2) (cat. 04–263) was from Millipore. Mouse monoclonal anti-β-tubulin (E7) (cat. E7) was obtained from Developmental Studies Hybridoma Bank. Mouse monoclonal anti-IL36R (M145) was produced using hybridoma generated at Amgen Inc.Citation44 Mouse monoclonal anti-CD200R (380525) (cat. MAB3414) and mouse monoclonal anti-GPR34 (419859) (cat. MAB4617) were from R&D Systems. Mouse monoclonal anti-human FAPα (F19) was produced and purified using a hybridoma (cat. CRL-2733) obtained from ATCC. Two independent rabbit monoclonal antibodies (clones D9G8 and 119A11) (cat. 3398 and cat. 3597, respectively) against phospho-eIF2α (Ser51), rabbit monoclonal anti-ATF4 (D4B8) (cat. 11815), and mouse monoclonal anti-CHOP (L63F7) (cat. 2895) were obtained from Cell Signaling Technology. Rabbit polyclonal anti-BiP (cat. ab21685, lot GR127527–1), mouse monoclonal anti-CD27L (BU69) (cat. ab77868), and rabbit polyclonal anti-phospho-eIF2α (Ser51) (cat. ab4837, lot GR270408–1) were from abcam. Another rabbit polyclonal anti-phospho-eIF2α (Ser51) (cat. 44–728G, lot RD226932) was obtained from Thermo Fisher Scientific. Affinity purified rabbit polyclonal anti-human IgG (H+L) (cat. 309–005–082) used for Western blotting was from Jackson ImmunoResearch Laboratories. FITC- or Texas Red-conjugated goat polyclonal anti-human gamma-chain (cat. 2040–02, cat. 2040–07) and anti-human kappa-chain (cat. 2060–02, cat. 2060–07) were from Southern Biotech. Alexa Fluor®-488, -594, -647, and -680 conjugated goat polyclonal anti-mouse IgG (H+L), rabbit IgG (H+L), and human IgG (H+L) antibodies were purchased from Thermo Fisher Scientific.
Expression constructs
Coding sequences for both LC and HC of the human anti-human CB1 monoclonal IgGs were obtained from hybridoma cell lines derived from immunized Xenomouse™ by using the molecular cloning method described previously.Citation24 A pair of anti-human CB1 mAbsCitation27 characterized in this study recognized the extracellular regions of human CB1 and blocked cannabinoid-induced activation of CB1 signaling (see Results section). LC and HC coding sequences were individually subcloned into pTT5 expression vectors (licensed from National Research Council of Canada) by a commonly used molecular cloning technique using Sal I and Not I restriction sites. The closest germline gene segment usage for the VH and VL regions was determined by using VBASE.Citation45
Mammalian expression vectors encoding human FAPα (NM_004460), human CD27L (NM_001252), human CD200R (NM_138806), and human IL-36R (NM_003854) were purchased from OriGene Technologies. All purchased expression constructs were maxiprepped and sequence-verified at GENEWIZ (South Plainfield, NJ, USA). Expression vector encoding N-terminally FLAG-tagged human GPR34 was described previously.Citation46 To create an N-terminally FLAG-tagged human GPR4 expression vector, FLAG tag-coding sequence was genetically inserted immediately after the initiating methionine of the GPR4 open reading frame (NM_005282) by a PCR-based method, and subcloned into pTT5 vector using Sal I and Not I restriction sites.
Cell culture, transient transfection, and protein production method
HEK293-EBNA1(6E) cells (hereafter HEK293 cells) were licensed from National Research Council of Canada for research use. HEK293 cells were cultured in a humidified Reach-In CO2 incubator (37 °C, 5% CO2) using FreeStyle™ 293 Expression Medium (Thermo Fisher Scientific). HEK293 cells were maintained in suspension culture format in Corning® Erlenmeyer cell culture flasks placed on an Innova 2100 shaker platform (New Brunswick Scientific) rotating at 110 rpm. Expression constructs were transfected into HEK293 cells using the protocol described in detail previously.Citation24 To express and produce whole IgGs, a cognate HC and LC construct pair was co-transfected at the plasmid DNA ratio of one-to-one (normalized by mass). When individual subunit chains were evaluated, 50% of the total DNA was substituted with an empty pTT5 vector or the expression vector encoding one of the 6 membrane-anchored cell surface proteins (see above). At 24 hr post transfection, cells were fed with Difco yeastolate cell culture supplement (BD Biosciences). Cell viability and viable cell density were monitored daily using Vi-CELL XR cell counters (Beckman Coulter) during a 7-day batch cell culture runs. The accuracy of cell counting decreased toward the end of cell culture duration because cells tended to clump and stick to the wall of shaker flasks, sometimes forming a white ring. Cell culture media were harvested and the whole cell lysates were prepared on day-7 post transfection for various analytical purposes including IgG titer determination, Western blotting, and IgG purification. Concentration of human IgG in the harvested cell culture media was determined by bio-layer interferometry with an Octet RED96 (ForteBio) using Protein A biosensor probes according to manufacturer's recommendations.
Immunofluorescent microscopy
To carry out cell imaging assays at steady-state or under ER-to-Golgi transport block conditions, growth media of HEK293 suspension cell culture were replaced with fresh media without or with 15 μg/ml brefeldin A (BFA) (Sigma-Aldrich) at 48 hr post transfection. Immediately after the media replacement, one million cells were withdrawn from each shake flask and seeded onto poly-L-lysine coated glass coverslips placed in 6-well plates. Each well was pre-filled with 3 ml/well of corresponding fresh cell culture media without or with BFA. After 24 hr of static cell culture, cells were fixed in 0.1 M sodium phosphate buffer (pH 7.2) containing 4% paraformaldehyde for 30 min at room temperature. After washing steps in PBS containing 0.1 M glycine, fixed cells were immersed in permeabilization buffer (PBS containing 0.4% saponin, 1% BSA, 5% fish gelatin) for 15 min, followed by incubation with primary antibody in permeabilization buffer for 60 min. After 3 washes in permeabilization buffer, the cells were stained with secondary antibodies for 60 min again in permeabilization buffer. Coverslips were mounted to microscope slide-glass using Vectashield mounting media (Vector Laboratories) and cured overnight at 4 °C. The slides were analyzed on Nikon Eclipse 80i microscope or Eclipse Ti-E microscope with a 100× or 60× CFI Plan Apocromat oil objective lens and Chroma FITC-HYQ or Texas Red-HYQ filter. Images were acquired using a Cool SNAP HQ2 CCD camera (Photometrics) and Nikon Elements imaging software. To determine RB phenotype prevalence in a population of transfected cells, microscopic images of 10–50 fields were captured for each construct set using 60× objective lens. Based on the anti-gamma and/or anti-kappa staining, the total number of transfected cells was first determined for each condition. The number of RB phenotype-positive cells was then determined by visually inspecting individual images.
Cell imaging-based assays to measure cell viability and active protein synthesis
A fixable VivaFix™ 498/521 cell viability dye (BIO RAD) was used to determine cell viability in a population of transfected HEK293 cells by fluorescent microscopy in conjunction with RB phenotype detection. At 72 hr post transfection, staining solution was prepared by diluting the dye stock in growth medium according to manufacturer's instruction. Transfected cells were cultured in the staining solution for 30 min under normal cell culture condition. After aspirating the dye, the labeled cells were washed twice in pre-warmed cell culture medium before the cells were fixed and processed for immunofluorescent staining to visualize RBs.
Alexa Fluor 488 Click-iT® Plus OPP protein synthesis assay kit (Thermo Fisher Scientific) was used to determine the cell-to-cell differences in actively ongoing protein synthesis in a population of transfected HEK293 cells by fluorescent imaging. In brief, the cells were labeled in 20 μM OPP working solution for 30 min under normal growth condition according to manufacturer's protocol. After aspirating the labeling solution, the cells were washed 3 times in pre-warmed cell culture medium before fixation and permeabilization using recommended conditions. A freshly prepared OPP reaction cocktail was then overlaid on the cells and the click reaction was allowed to take place for 30 min at room temperature in the dark. The reaction was quenched by aspirating the reaction cocktail followed by adding a designated rinse buffer. After the quenching, the cells were processed for immunofluorescent staining to visualize RBs.
SDS-PAGE and western blotting
On day-7 post transfection, an aliquot of suspension cell culture was withdrawn from shake flasks, and the culture media were separated from cell pellets by a centrifugation step at 1000 g for 5 min. Harvested cell culture media were mixed with an equal volume of 2× lithium dodecyl sulfate (LDS) sample buffer (Thermo Fisher Scientific). Likewise, cell pellets were directly lysed in 1× LDS sample buffer. Both samples were heat treated at 75 °C for 5 min. When preparing protein samples under reducing conditions, β-mercaptoethanol was included in the sample buffer to a final concentration of 5% (v/v). For non-reducing sample preparation, reducing agents were omitted, but 2 mM N-ethylmaleimide was added as an alkylating agent. To normalize the sample loading, whole cell lysates corresponding to 12,000–12,500 cells were analyzed per lane. To compare the differences in volumetric secretion titers between different mAbs, a sample volume equivalent to the 5 μl of harvested culture media was analyzed per lane. SDS-PAGE was performed using NuPAGE 4–12% Bis-Tris gradient gel and a compatible MES SDS buffer system (both from Thermo Fisher Scientific). Resolved proteins were electrotransferred to a nitrocellulose membrane, blocked with Odyssey® blocking buffer (LI-COR Biosciences), and probed with primary antibodies of choice (see above). After 3 washes in PBS containing 0.05% (v/v) Tween-20, the nitrocellulose membranes were probed with Alexa Fluor 680-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 hr. After 3 round of washing steps, probed membranes were scanned to acquire imaging data using Odyssey® infrared imaging system (LI-COR Biosciences).
IgG purification
Human IgG mAbs (both the parental and N35W variant) secreted to the culture media were first purified by protein A affinity chromatography using MabSelect resin (GE Healthcare) by following manufacturer's instruction. Eluted IgG was subjected to cation exchange chromatography using SP Sepharose High Performance resin (GE Healthcare). The eluted fractions containing IgG were pooled and concentrated by using Jumbosep™ centrifugal device (Pall Corporation). The purified human IgG was then buffer-exchanged to PBS by dialysis. The final protein concentration was 29.2 mg/ml and 16.9 mg/ml for the parental mAb and N35W variant, respectively.
Size-exclusion chromatography
Size-exclusion HPLC was performed on Agilent 1100 system using Superdex G200 column (GE Healthcare), Superose 12 (GE Healthcare), and TSKgel G3000 (Tosoh Bioscience) at a flow rate of 0.7 ml/min for 40 to 50 min, using PBS as running buffer. Purified human IgG mAbs (10 μg each) were injected to the column at 1 mg/ml concentration. The elution profile was monitored by UV light at 215 nm.
Differential scanning calorimetry
MicroCal VP-Capillary DSC system (GE Healthcare) was used to assess unfolding profiles of testing mAbs under thermal stress by modifying a described previously method.Citation38 Briefly, concentration of the testing mAb samples were adjusted to 0.5 mg/ml in PBS and 400 μl each of mAb-containing solution was analyzed from 20 to 100 °C at a heating rate of 1 °C per minute. Blank PBS solution was measured for baseline subtraction.
Flow cytometry
Adherent stable CHO-K1 cell line expressing recombinant human CB1 (CHO-hCB1) was obtained from Euroscreen S.A. (Gosselies, Belgium) and was grown quiescently in a humidified 5% CO2 incubator using a complete DMEM medium (containing 2 mM L-glutamine, 100 units/ml penicillin G, 100 µg/ml streptomycin, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate) supplemented with 10% fetal bovine serum and 400 μg/ml G418. To carry out cell staining, the cells were harvested non-enzymatically by using versene cell dissociation solution (Thermo Fisher Scientific) and were suspended in an ice-chilled assay buffer (PBS with 1% goat serum) before seeding to a U-bottom 96-well plate at 200,000 cells per well. The cells were stained with 0.2, 1, and 5 μg/ml anti-human CB1 mAbs (the parental and N35W variant) for 60 min at 4 °C in 200 μl volume. After 2 washes in the assay buffer, the cells were stained with Alexa Fluor 647-labeled goat anti-human IgG (H+L) polyclonal antibody (Thermo Fisher Scientific) at 2 μg/ml for 60 min at 4 °C. Cells were again washed twice and then resuspended in the final staining assay buffer including YO-PRO®-1 iodide dye (Thermo Fisher Scientific) to label the dead cells and exclude them from analysis. Data were acquired on a BD LSR II flow cytometer (BD Biosciences) and analyzed by using FCS Express software (De Novo Software).
Cell-based cAMP assay
Stable CHO-hCB1 cells (see above) were seeded onto 96-well plates at 10,000 cells per well using the complete DMEM cell growth media (see above). The seeded plates were placed quiescently in a humidified 5% CO2 incubator at 37 °C. After 24 hr of incubation, the volume of cell culture medium was adjusted to 40 μl/well using fresh medium. Then, 10 μl/well of the following assay cocktail was added after premixed in assay buffer (10 mM acetic acid, pH 5.0, 150 mM NaCl). The assay cocktail was made of 3 components: (i) varying concentrations of testing antagonistic anti-CB1 mAbs (the parental or N35W variant); (ii) 1.25 nM of synthetic cannabinoid CP 55,940; (iii) 75 μM of forskolin. Cells were returned to normal growth conditions for 30 min before they were processed to measure intracellular cAMP production levels using HitHunter™ cAMP XS+ Assay Kit (DiscoveRX). Luminescent signals were acquired using ViewLux Microplate Imager from PerkinElmer. Each condition was tested in duplicate and ‘standard error of mean’ was shown as error bar at each data point.
Structural modeling of IgG variable fragment (Fv)
Homology models of the parental and N35W variant mAbs were generated using the Molecular Operating Environment (MOE; Chemical Computing Group Inc., Montreal, Canada) using the Amber10:EHT forcefield. Template selection was performed using the Antibody Modeling module in MOE, which considers resolution and B-factors of the template structure in addition to identity to query sequence. The framework regions were modeled using template PDB ID, 5BMF. HC-CDRs were modeled from the following template CDRs: 2VXQ.H for H1; 5E94.D for H2; and 3X3F.H for H3. Likewise, LC-CDRs are modeled using 3BKJ.L or 1Q72.L for L1; 4LRI.A for L2; and 4LRI.A for L3. For each Fv, 10 models were generated and the lowest scoring model was selected for further refinement and visual inspection. Because CDRs L1 and H3 were long, the conformations of these CDRs were further investigated using the low-mode MD Conformational Search module in MOE. Figures were rendered in the PyMOL Molecular Graphics System, version 1.8 (Schrödinger, LLC). Surface hydrophobicity was colored according to the Eisenberg hydrophobicity scale.Citation47
Disclosure of potential conflicts of interest
No potential conflict of interest were disclosed.
Author contributions
HH designed the work. HH, AS, CET, KS, AAN, MMT performed the experiments and analyzed the data. HH wrote the manuscript. All the authors critically reviewed the manuscript.
Supplemental_Materials.zip
Download Zip (8 MB)Acknowledgments
The authors thank colleagues in Amgen British Columbia for generating anti-CB1 mAbs from hybridoma campaigns, Michelle Hortter for generating anti-CB1 IgG expression constructs, Anne Abrajano for making FLAG-GPR4 construct, Dao Kemp for mAb production cell culture assistance, and Ling Cai for mAb purification support.
References
- Tonegawa S. Somatic generation of antibody diversity. Nature 1983; 302:575-81; PMID:6300689; https://doi.org/10.1038/302575a0
- French DL, Laskov R, Scharff MD. The role of somatic hypermutation in the generation of antibody diversity. Science 1989; 244:1152-7; PMID:2658060; https://doi.org/10.1126/science.2658060
- Chen C, Roberts VA, Stevens S, Brown M, Stenzel-Poore MP, Rittenberg MB. Enhancement and destruction of antibody function by somatic mutation: unequal occurrence is controlled by V gene combinatorial associations. EMBO J 1995; 14:2784-94; PMID:7796805
- Wiens GD, Heldwein KA, Stenzel-Poore MP, Rittenberg MB. Somatic mutation in VH complementarity-determining region 2 and framework region 2: differential effects on antigen binding and Ig secretion. J Immunol 1997; 159:1293-302; PMID:9233625
- Wiens GD, Roberts VA, Whitcomb EA, O'Hare T, Stenzel-Poore MP, Rittenberg MB. Harmful somatic mutations: lessons from the dark side. Immunol Rev 1998; 162:197-209; PMID:9602365; https://doi.org/10.1111/j.1600-065X.1998.tb01442.x
- DeFranco AL. The germinal center antibody response in health and disease. F1000Res 2016; 5; PMID:27303636; https://doi.org/10.12688/f1000research.7717.1
- Rudikoff S, Giusti AM, Cook WD, Scharff MD. Single amino acid substitution altering antigen-binding specificity. Proc Natl Acad Sci U S A 1982; 79:1979-83; PMID:6804947; https://doi.org/10.1073/pnas.79.6.1979
- Bruggemann M, Muller HJ, Burger C, Rajewsky K. Idiotypic selection of an antibody mutant with changed hapten binding specificity, resulting from a point mutation in position 50 of the heavy chain. EMBO J 1986; 5:1561-6; PMID:3017703
- McKean PG, O'Dea K, Brown KN. A single amino acid determines the specificity of a monoclonal antibody which inhibits Plasmodium chabaudi AS in vivo. Mol Biochem Parasitol 1993; 62:211-21; PMID:7511215; https://doi.org/10.1016/0166-6851(93)90110-J
- Casson LP, Manser T. Evaluation of loss and change of specificity resulting from random mutagenesis of an antibody VH region. J Immunol 1995; 155:5647-54; PMID:7499849
- Iba Y, Hayashi N, Sawada J, Titani K, Kurosawa Y. Changes in the specificity of antibodies against steroid antigens by introduction of mutations into complementarity-determining regions of the V(H) domain. Protein Eng 1998; 11:361-70; PMID:9681868; https://doi.org/10.1093/protein/11.5.361
- Winkler K, Kramer A, Kuttner G, Seifert M, Scholz C, Wessner H, Schneider-Mergener J, Höhne W. Changing the antigen binding specificity by single point mutations of an anti-p24 (HIV-1) antibody. J Immunol 2000; 165:4505-14; https://doi.org/10.4049/jimmunol.165.8.4505
- Hall BL, Zaghouani H, Daian C, Bona CA. A single amino acid mutation in CDR3 of the 3-14-9 L chain abolished expression of the IDA 10-defined idiotope and antigen binding. J Immunol 1992; 149:1605-12; PMID:1380535
- Xiang J, Chen Z, Delbaere LT, Liu E. Differences in antigen-binding affinity caused by a single amino acid substitution in the variable region of the heavy chain. Immunol Cell Biol 1993; 71(Pt 4):239-47; PMID:8225393; https://doi.org/10.1038/icb.1993.28
- Schildbach JF, Near RI, Bruccoleri RE, Haber E, Jeffrey PD, Ng SC, Novotny J, Sheriff S, Margolies MN. Heavy chain position 50 is a determinant of affinity and specificity for the anti-digoxin antibody 26–10. J Biol Chem 1993; 268:21739-47; PMID:7691815
- Stevens FJ, Myatt EA, Chang CH, Westholm FA, Eulitz M, Weiss DT, Murphy C, Solomon A, Schiffer M. A molecular model for self-assembly of amyloid fibrils: immunoglobulin light chains. Biochemistry 1995; 34:10697-702; PMID:7662653; https://doi.org/10.1021/bi00034a001
- Villalba MI, Canul-Tec JC, Luna-Martinez OD, Sanchez-Alcala R, Olamendi-Portugal T, Rudino-Pinera E, Rojas S, Sánchez-López R, Fernández-Velasco DA, Becerril B. Site-directed mutagenesis reveals regions implicated in the stability and fiber formation of human lambda3r light chains. J Biol Chem 2015; 290:2577-92; PMID:25505244; https://doi.org/10.1074/jbc.M114.629550
- Helms LR, Wetzel R. Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J Mol Biol 1996; 257:77-86; PMID:8632461; https://doi.org/10.1006/jmbi.1996.0148
- Dul JL, Argon Y. A single amino acid substitution in the variable region of the light chain specifically blocks immunoglobulin secretion. Proc Natl Acad Sci U S A 1990; 87:8135-9; PMID:2122454; https://doi.org/10.1073/pnas.87.20.8135
- Wiens GD, Lekkerkerker A, Veltman I, Rittenberg MB. Mutation of a single conserved residue in VH complementarity-determining region 2 results in a severe Ig secretion defect. J Immunol 2001; 167:2179-86; PMID:11490003; https://doi.org/10.4049/jimmunol.167.4.2179
- Wu GE, Hozumi N, Murialdo H. Secretion of a lambda 2 immunoglobulin chain is prevented by a single amino acid substitution in its variable region. Cell 1983; 33:77-83; PMID:6432336; https://doi.org/10.1016/0092-8674(83)90336-7
- Russell W. An Address on a Characteristic Organism of Cancer. Brit Med J 1890; 2:1356-60; PMID:20753194; https://doi.org/10.1136/bmj.2.1563.1356
- Hasegawa H. Aggregates, crystals, gels, and amyloids: intracellular and extracellular phenotypes at the crossroads of immunoglobulin physicochemical property and cell physiology. Int J Cell Biol 2013; 2013:604867; PMID:23533417; https://doi.org/10.1155/2013/604867
- Stoops J, Byrd S, Hasegawa H. Russell body inducing threshold depends on the variable domain sequences of individual human IgG clones and the cellular protein homeostasis. Biochim Biophys Acta 2012; 1823:1643-57; PMID:22728328; https://doi.org/10.1016/j.bbamcr.2012.06.015
- Valetti C, Grossi CE, Milstein C, Sitia R. Russell bodies: a general response of secretory cells to synthesis of a mutant immunoglobulin which can neither exit from, nor be degraded in, the endoplasmic reticulum. J Cell Biol 1991; 115:983-94; PMID:1955467; https://doi.org/10.1083/jcb.115.4.983
- Honegger A, Pluckthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol 2001; 309:657-70; PMID:11397087; https://doi.org/10.1006/jmbi.2001.4662
- Coward P, Moore SA, Meng SY, Tsai MM, King CT, Nazarian AA. Cb-1 receptor antigen-binding proteins and uses thereof. source>. US 20160145333 A1, 2016
- Hasegawa H, Woods CE, Kinderman F, He F, Lim AC. Russell body phenotype is preferentially induced by IgG mAb clones with high intrinsic condensation propensity: relations between the biosynthetic events in the ER and solution behaviors in vitro. mAbs 2014; 6:1518-32; PMID:25484054; https://doi.org/10.4161/mabs.36242
- Hasegawa H, Forte C, Barber I, Turnbaugh S, Stoops J, Shen M, Lim AC. Modulation of in vivo IgG crystallization in the secretory pathway by heavy chain isotype class switching and N-linked glycosylation. Biochim Biophys Acta 2014; 1843:1325-38; PMID:24703881; https://doi.org/10.1016/j.bbamcr.2014.03.024
- Alanen A, Pira U, Colman A, Franklin RM. Mott cells: a model to study immunoglobulin secretion. Eur J Immunol 1987; 17:1573-7; PMID:2890529; https://doi.org/10.1002/eji.1830171108
- Alanen A, Pira U, Lassila O, Roth J, Franklin RM. Mott cells are plasma cells defective in immunoglobulin secretion. Eur J Immunol 1985; 15:235-42; PMID:3979421; https://doi.org/10.1002/eji.1830150306
- Weiss S, Burrows PD, Meyer J, Wabl MR. A Mott cell hybridoma. Eur J Immunol 1984; 14:744-8; PMID:6432555; https://doi.org/10.1002/eji.1830140814
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 1995; 48:443-50; PMID:7565624
- Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL, Dawson TM, Cuervo AM, Lim KL. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet 2008; 17:2570-82; PMID:18502787; https://doi.org/10.1093/hmg/ddn157
- Fujimuro M, Sawada H, Yokosawa H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett 1994; 349:173-80; PMID:7519568; https://doi.org/10.1016/0014-5793(94)00647-4
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; https://doi.org/10.1083/jcb.143.7.1883
- Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 2003; 278:44405-11; PMID:12923179; https://doi.org/10.1074/jbc.M308041200
- Hasegawa H, Wendling J, He F, Trilisky E, Stevenson R, Franey H, Kinderman F, Li G, Piedmonte DM, Osslund T, et al. In vivo crystallization of human IgG in the endoplasmic reticulum of engineered Chinese hamster ovary (CHO) cells. J Biol Chem 2011; 286:19917-31; PMID:21464137; https://doi.org/10.1074/jbc.M110.204362
- Hasegawa H, Patel N, Lim AC. Overexpression of cryoglobulin-like single-chain antibody induces morular cell phenotype via liquid-liquid phase separation in the secretory pathway organelles. FEBS J 2015; 282:2777-95; PMID:26036200; https://doi.org/10.1111/febs.13332
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397:271-4; PMID:9930704; https://doi.org/10.1038/16729
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6:1099-108; PMID:11106749; https://doi.org/10.1016/S1097-2765(00)00108-8
- Wight PA, Burns RB, Rothwell B, Mackenzie GM. The Harderian gland of the domestic fowl. I. Histology, with reference to the genesis of plasma cells and Russell bodies. J Anat 1971; 110:307-15; PMID:4111364
- Posnett DN, Mouradian J, Mangraviti DJ, Wolf DJ. Mott cells in a patient with a lymphoproliferative disorder. Differentiation of a clone of B lymphocytes into Mott cells. Am J Med 1984; 77:125-30; PMID:6377889; https://doi.org/10.1016/0002-9343(84)90446-7
- Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, Sims JE. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36 gamma) or antagonist (IL-36Ra) activity. J Biol Chem 2011; 286:42594-602; PMID:21965679; https://doi.org/10.1074/jbc.M111.267922
- Retter I, Althaus HH, Munch R, Muller W. VBASE2, an integrative V gene database. Nucleic Acids Res 2005; 33:D671-4; PMID:15608286; https://doi.org/10.1093/nar/gki088
- Hasegawa H, Patel N, Ettehadieh E, Li P, Lim AC. Topogenesis and cell surface trafficking of GPR34 are facilitated by positive-inside rule that effects through a tri-basic motif in the first intracellular loop. Biochim Biophys Acta 2016; 1863:1534-51; PMID:27086875; https://doi.org/10.1016/j.bbamcr.2016.04.010
- Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 1984; 179:125-42; PMID:6502707; https://doi.org/10.1016/0022-2836(84)90309-7