
ABSTRACT
Aggregation and self-association in protein-based biotherapeutics are critical quality attributes that are tightly controlled by the manufacturing process. Aggregates have the potential to elicit immune reactions, including neutralizing anti-drug antibodies, which can diminish the drug's efficacy upon subsequent dosing. The structural basis of reversible self-association, a form of non-covalent aggregation in the native state, is only beginning to emerge for many biologics and is often unique to a given molecule. In the present study, crystal structures of the infliximab (Remicade) Fc and Fab domains were determined. The Fab domain structures are the first to be reported in the absence of the antigen (i.e., tumor necrosis factor), and are consistent with a mostly rigid complementarity-determining region loop structure and rotational flexibility between variable and constant regions. A potential self-association interface is conserved in two distinct crystal forms of the Fab domain, and solution studies further demonstrate that reversible self-association of infliximab is mediated by the Fab domain. The crystal structures and corresponding solution studies help rationalize the propensity for infliximab to self-associate and provide insights for the design of improved control strategies in biotherapeutics development.
Abbreviations
| AUC | = | analytical ultracentrifugation |
| CDR | = | complementarity determining region |
| Fab | = | Fragment antigen binding |
| Fc | = | fragment crystallizable |
| rpm | = | rotations per minute |
| SEC | = | size exclusion chromatography |
| TNF | = | tissue necrosis factor |
In the three decades since the first therapeutic monoclonal antibody (mAb) was approved by the US Food and Drug Administration, mAb therapies have proven to be essential treatments for patients with cancer, autoimmune diseases, and a host of other conditions. Therapeutic antibody revenues continue to dominate among biologics and other pharmaceuticals, with Humira® (adalimumab), Remicade® (infliximab) and Rituxan/MabThera® (rituximab) among the all-time top selling pharmaceuticals. MAbs offer several advantages over other biologics, including prolonged stability, desirable bio-availability, and specific, high-affinity antigen targeting, all of which are directly related to their molecular structure. It is therefore critical from a development standpoint to obtain and leverage a structural understanding of biotherapeutics in an effort to retain desired qualities and minimize undesirable attributes in developing improved biotherapeutics.
Therapeutic protein aggregation is a closely monitored attribute during drug discovery and throughout development. Aggregation poses an increased risk for an immunogenic response and generation of anti-drug antibodies, potentially diminishing the therapeutic potency upon repeat administrations.Citation1 Indeed, a substantial amount of research goes into predictive algorithms for aggregation propensity,Citation2-4 developing appropriate analytical tools to monitor aggregationCitation5-7 and preventing self-association through rational design and formulation of biotherapeutics.Citation8,Citation9 Despite these efforts, it is often the case that aggregation mechanisms are unique for a given antibody or remain poorly characterized. Recent studies demonstrate diversity in the mechanism for mAb aggregation,Citation10-13 and additional insights are being gleaned with the emergence of improved characterization methods.Citation14,Citation15
Infliximab, a mouse/human chimeric IgG1, is prescribed for the treatment of Crohn's disease, psoriasis, rheumatoid arthritis and other autoimmune diseases.Citation16,Citation17 The global revenue for infliximab exceeded US $9B in 2014, making it a prime candidate for biosimilar development. One biosimilar of infliximab (Inflectra™) has been approved in the US, two (Inflectra™/Remsima®; Flixabi®) have been approved in the European Union, and additional biosimilar versions are in clinical development. The intense interest in infliximab biosimilars underscores the need for a heightened understanding of its mechanism of action and the quality attributes of the originator, Remicade®.
Infliximab functions by binding to the inflammation-regulating cytokine tumor necrosis factor (TNF), and inhibiting receptor interactions, thereby decreasing inflammation and autoimmune response in patients.Citation18 The structural basis for TNF neutralization by infliximab was revealed previously in the crystal structure of the TNF trimer in complex with the infliximab antigen-binding fragment (Fab) domain.Citation19 In the complex, three Fab domains are positioned 120° apart, around the three-fold symmetry axis of the TNF trimer, with one Fab bound per TNF monomer. In addition, a comparison of Fab-bound crystal structures demonstrated that the infliximab and adalimumab epitopes on TNF are distinct;Citation20 however, high resolution structures of infliximab in the absence of its antigen have not been reported.
Here, we report the first crystal structures of the infliximab Fab domain in the absence of the TNF antigen, as well as the structure of the crystallizable fragment (Fc) domain in a new crystal form. The structures reveal a potential Fab domain-mediated self-association mechanism, which was not readily apparent in the Fab:TNF complex structure. We then confirm that self-association of the full-length infliximab is mediated through the Fab domain in solution using biochemical and biophysical methods. The identification of the self-association interface allows one to rationalize the propensity for infliximab aggregation, and provides insights into future design of improved biotherapeutics.
Results
Infliximab crystal structures
To better understand the physicochemical properties of infliximab, we determined crystal structures of the Fab and Fc domains. The intact infliximab antibody was digested with papain and the purified Fab and Fc domains were crystallized. Crystal structures were solved by molecular replacement methods and the final refinement statistics are shown in . All structures are of high quality for the resolution of their data, as determined by MolProbity scores ().
Table 1. Crystallographic data collection and refinement statistics.
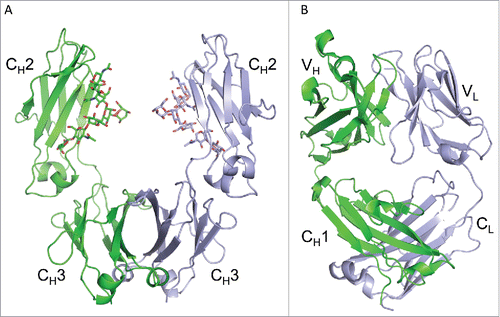
The Fc structure was determined in a C-centered orthorhombic crystal form, which does not appear to have been reported previously in the PDB for other human IgG1-Fc crystal structures (which have 100% sequence identity). The asymmetric unit contained one Fc chain, and the second Fc chain of the biological dimer was related via crystallographic symmetry (). Based on related published crystal structures such as PDB ID 4CDH, we built the N-linked glycan into the well resolved omit map (|Fo|-|Fc|) positive density. The glycan anchored to Asn300 corresponds to the G1F minus GlcNAc, where the terminal galactose is the least well-ordered, having the highest B-factors. Additional sugars could be present in the glycan, but are not sufficiently well-ordered in the electron density and could not be modeled.
Figure 1. Crystal structures of infliximab Fc domain (A) and Fab domain (B). The Fc dimer is shown in (A), with one heavy chain shown in green and the symmetry-related chain shown in silver. N-linked glycans are shown in stick representation and the CH2 and CH3 sub-domains are labeled. The I212121 form of the Fab domain is shown in (B), with the heavy chain in green and the light chain in silver. Variable (VH and VL) and constant (CH1 and CL) regions are labeled.
Crystal structures of the Fab domain of infliximab were determined to 2 Å resolution in two distinct crystal forms: I212121 and C2221 ( and ). Both crystal forms contain two Fab domains in the asymmetric unit, and the Fab variable and constant regions are highly similar in both crystal forms. The all atom root-mean-square deviations of the LSQ superimposed crystal forms are 0.47–0.53 Å and 0.74–0.85 Å for the variable and constant regions, respectively.
Figure 2. Flexibility between the variable and constant regions of the infliximab Fab domain. Fab structures determined in C2221 (blue) and I212121 (green) crystal forms, aligned by their variable regions, show a ∼40° relative rotation of their constant regions, indicating intramolecular flexibility within the Fab domain.
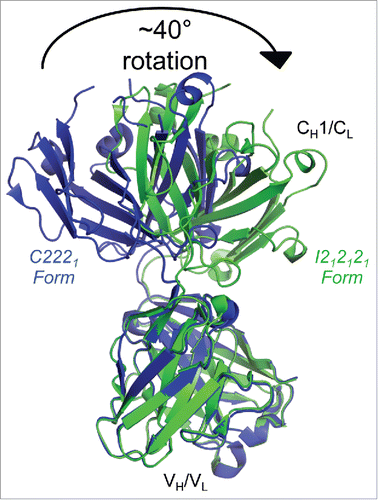
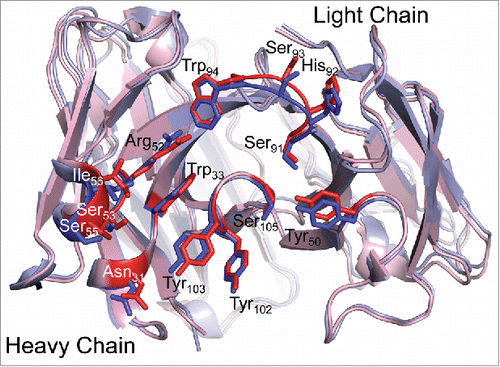
A comparison of the I212121 and C2221 Fab structures reveals apparent intramolecular flexibility between the variable and constant regions (). An elbow rotation of ∼40° was observed between the Fab variable and constant regions in the two crystal forms. This is not surprising, as antibodies typically display conformational flexibility between the two domains.Citation21 However, the variable domains and particularly the complementary determining regions (CDRs) appear in a nearly identical conformation in both unbound Fab structures and in the TNF bound crystal structure (). The largest differences among antigen binding residues in the bound and free Fab structures are less than 1 Å. These differences are found in Ser53, Ser55 and Ile56, which have ∼2-fold higher relative B-factors in the bound form compared to the unbound form. B-factors among the remaining antigen-binding residues are similar in the bound and unbound forms, and are slightly below the average B-factors for the molecule. Together, the structural similarity and relatively low B-factors indicate that the infliximab antigen-binding site adopts a mostly rigid conformation that does not change significantly upon TNF binding.
Figure 3. TNF binding does not affect the infliximab CDR loop structure. Fab structures in the unbound (blue; I212121 form) and TNFα-bound (red) states are overlaid. CDR loop residues involved in TNF binding are shown as sticks in darker coloring and are labeled. The antigen binding interface structure is unchanged in the bound and free states, indicating that antigen binding does not affect the structure of infliximab.
Identification of a potential self-association interface on infliximab
Both the I212121 and C2221 crystal forms contain two copies of the Fab heterodimer in the asymmetric unit. Despite the differences in their space group geometries, we observed several similarities in the asymmetric units of the two crystal forms. For both forms, the packing interactions between the two domains in the asymmetric unit occur exclusively via the light chains, and the two heterodimers adopt a “head-to-tail” orientation (, and ). While the exact light chain:light chain (LC:LC) interactions in the I212121 are different than those observed in the C2221 crystal form, a majority of the interface residues are common in both crystal forms (see below). Finally, the LC:LC contact surface area (1083 Å2 and 1066 Å2 in the I212121 and C2221 forms, respectively) is considerably greater than the surface areas of other crystal contacts (< 500 Å2 per chain). Indeed, the LC:LC contact surface area is more similar to that of solution oligomer interfaces than to crystal contact interfaces.Citation22 For comparison, the biological interfaces in the Fab domain structures (heavy chain:light chain interfaces) range from 1593–1672 Å2, and the HC:HC interface in the Fc domain is 1010 Å2. Together, these observations led us to hypothesize that the light chain interface observed in both I212121 and C2221 crystal forms represents a self-association interface in solution.
Figure 4. The Fab asymmetric unit contains a head-to-tail dimer, associated through light chain:light chain interactions in both crystal forms. Two Fab molecules (#1 and #2) and their variable and constant regions are labeled in the I212121 form (left) and the C2221 form (right). Heavy chains are shown in shades of green and light chains in shades of blue. In both crystal forms, Fab #1 is oriented with variable regions at the top, while Fab #2 variable regions are positioned at the bottom. The large contact area (> 1000 Å2) between light chains is larger than would be expected for typical crystal contacts, indicating a potential self-association interface in solution.
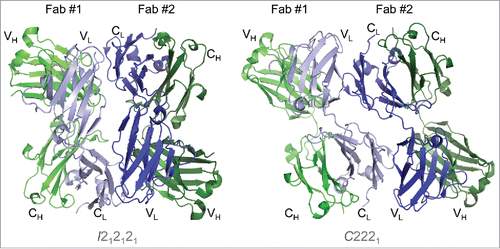
Figure 5. Fab:Fab contact interface in the I212121 crystal form. (A) A surface representation of the Fab asymmetric unit (dimer) is shown with heavy chains colored in shades of green and light chains in shades of blue. Interface residues are colored by residue type (green = polar, blue = basic, red = acidic, magenta = hydrophobic). (B) Each Fab domain is rotated outward 90° to expose the light chain:light chain interface residues, which are highlighted below in the light chain sequence. Residues common to both crystal form interfaces are underlined in the sequence. (C) Close-up view of Leu3 from one light chain positioned into a pocket created by Thr197, Lys145 and Glu143 in the adjacent light chain in the I212121 crystal form.
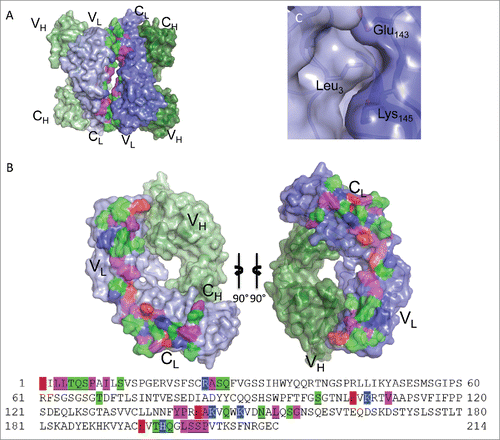
Figure 6. Fab:Fab contact interface in the C2221 crystal form. (A) A surface representation of the Fab asymmetric unit (dimer) is shown with heavy chains colored in shades of green and light chains in shades of blue. Interface residues are colored by residue type (green = polar, blue = basic, red = acidic, magenta = hydrophobic). (B) Each Fab domain rotated outward 90° to expose the light chain:light chain interface residues, which are highlighted below in the light chain sequence. Residues common to both crystal form interfaces are underlined. (C) A close-up view of Leu3 in the C2221 interface, positioned within a pocket created by Val191, Asp151, Asn152 and Lys190 of the adjacent Fab. The surrounding hydrogen bond interactions are shown as dashed lines.
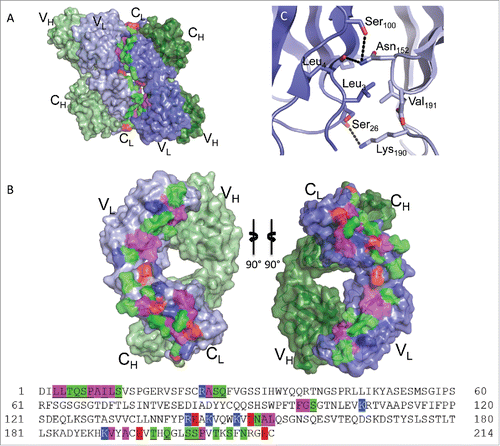
The self-association interface involves 37 residues in the I212121 form () and 39 in the C2221 form (). While the exact interactions are not conserved between the crystal forms, 25 common residues participate in light chain:light chain contacts in both crystal forms (underlined in and ). Of the 25 common residues, seven are non-aromatic hydrophobic residues, twelve are polar and six are charged. The charged residues are not involved in salt bridges in either crystal form. The structures indicate that dimerization is facilitated mainly through hydrogen-bonding and hydrophobic interactions among aliphatic side-chains and backbone carbon atoms. The full self-association interface is composed of two smaller patches related by the head-to-tail symmetry of the Fab dimer, forming identical VL:CL and CL:VL region interfaces. These smaller patches involve residues Leu3, Leu4, Thr5, Gln6, Ser7, Pro8, Ile10, Ser12, Arg24, Ala25, Ser26, Gln27, Ser100 and Lys107 on one Fab domain (VL region) and residues Glu143, Lys145, Lys149, Asn152, Leu154, Glu195, Thr197, Gln199, Ser202, Ser203, and Pro204 on the second Fab domain (CL region).
The positioning of Leu3 (in the VL region) exemplifies an interface residue that has distinct interactions in each crystal form with the adjacent Fab CL region residues. In the I212121 form, Leu3 is stacked in a pocket created by the aliphatic regions on Thr197, Lys145 and Glu143 of the opposing light chain (). Hydrogen bonding interactions between Arg24 O: Gln199 Oϵ1, Ala25 O:Gln199 Nϵ2, Ser26 O:Leu201 O (water-mediated) and Ser100 Oγ:Glu143 Oϵ2 surround the hydrophobic pocket involving Leu3. In the C2221 form, Leu3 is again positioned within a pocket of the adjacent Fab domain, but here the pocket is formed by Val191 and the aliphatic regions of Asp151, Asn152, and Lys190 (). The Leu3 contact region in the C2221 form is flanked by hydrogen bonding interactions between Ser26 O:Lys190 Nζ, Leu4 O:Asn152 Nδ2, Ser100 Oγ:Asn152 Nδ2, and Ala9 N:Glu195 Oϵ1. Together, the structures indicate that multiple configurations are possible, utilizing a conserved set of light chain residues, to drive infliximab reversible self-association in solution.
Infliximab shows reversible self-association in solution
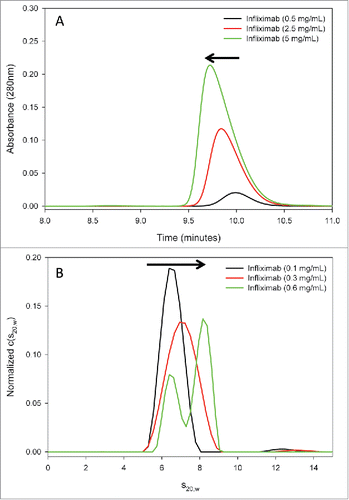
Reversible self-association of protein biotherapeutics is typified by concentration-dependent, non-covalent interactions in the native conformation. To test infliximab for reversible self-association in solution, increasing concentrations of the antibody were evaluated using size-exclusion chromatography (SEC) and sedimentation velocity analytical ultracentrifugation (AUC-SV). The results from both methods indicate that infliximab self-associates in a concentration-dependent manner (). In the SEC studies, the infliximab elution peak shifted to earlier retention times at increasing concentrations. In addition, the overall peak shape became asymmetric, with the mass distribution shifting to earlier retention times with increasing concentration. Both of these trends are hallmarks of reversible self-association. Because sample dilution on the SEC column can shift the monomer:dimer equilibrium to the monomer species in SEC analysis, AUC-SV was used as an orthogonal technique to evaluate self-association. AUC-SV provides size and shape information without sample dilution or nonspecific stationary phase interactions. Similar to SEC, AUC-SV analysis of infliximab reveals evidence of reversible self-association. The sedimentation coefficient of infliximab increased from a value typical of an antibody monomer (∼6.6 S at 0.1 mg/mL) to that between the monomer and the dimer (∼7.0 S at 0.3 mg/mL), representing an averaged sedimentation rate between that of the monomer and of the dimer. This peak shifting is indicative of relatively rapid self-association kinetics.Citation23,Citation24 At a higher concentration (0.6 mg/mL), the monomer-dimer equilibrium shifted sufficiently toward the dimer sedimentation coefficient to allow the monomer peak to be resolved in the c(s) distribution. Based on the signal weighted average sedimentation coefficients (sw(20,w)), the peaks likely represent the monomer species (6.6 S) becoming resolved from the monomer/dimer interaction boundary (at 8.2 S) at the highest measured concentration. A fully dimerized species would be expected to sediment at a higher sedimentation coefficient than was observed here (i.e., > 8.2 S). The SEC and AUC-SV results indicate that the self-association of infliximab is reversible with fast kinetics and arises from low affinity interactions mainly between monomer and dimer species in the concentration range that can be tested by these methods.
Figure 7. Reversible self-association of full-length infliximab in solution. (A) SEC analysis of infliximab at increasing concentrations (0.5, 2.5, and 5 mg/mL) reveals a corresponding shift in the main peak to earlier retention times and a progressive asymmetry, indicative of self-association. (B) AUC-SV analysis of infliximab demonstrates that the sedimentation coefficient increases with concentration, and that two peaks become resolved at the highest concentration measured. Results from both methods are consistent with concentration-dependent, reversible self-association of infliximab.
Sedimentation equilibrium analytical ultracentrifugation (AUC-SE) was performed to determine the equilibrium dissociation constant(s) among the predominant species in the concentration range of 0.07 to 0.6 mg/mL. The rotor speed was also varied (7500, 10000, 12500, and 15000 rpm) to broaden the data range for more accurate analysis. Global fitting was performed with various models using the SedAnal program, first to establish the most appropriate equilibrium model, as well as to fit for the dissociation constant (KD). A one-component model resulted in a poor fit to the data. A two-component model was subsequently tested and resulted in an improved fit, as assessed by a significantly reduced standard deviation. Moreover, the measured molar masses were in good agreement with the theoretical mass of the monomer (∼145,880 Da) and the dimer (∼301,320 Da). The fit to a three-component model with the monomer molecular mass (sequence-based) fixed returned two molar mass values, one close to a dimer and the other close to a tetramer. To explore whether there is a trimer in the solution, a model of four-component was attempted, but failed to converge. The data therefore indicate that, in the tested concentration range, infliximab consists of mainly monomer, dimer, and a trace amount of the tetramer in solution ().
Table 2. Infliximab species measured by AUC-SE.
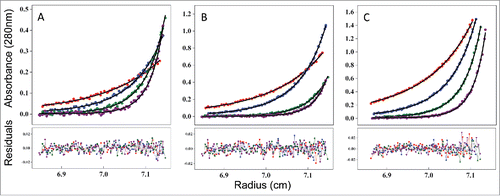
The data were then fit to the monomer-dimer equilibrium model to obtain the monomer-dimer dissociation constant (KD). To reduce the number of fitting parameters, the sequence-based molecular weight for both the monomer and the dimer were fixed. The experimental and fitted AUC-SE profiles are shown in . The fitted curves overlay well with the experimental profiles, with acceptable residual distribution, indicating that the equilibrium equations used by the software were adequate and the results obtained by these analyses were reliable. The measured KD value for the monomer-dimer equilibrium was 21 μM. A KD in μM range indicates weak association interactions, consistent with the monomer/dimer interaction boundary observed by SEC and AUC-SV above, as opposed to discrete monomer and dimer species that would be expected for a higher affinity complex.
Figure 8. Measuring the affinity of infliximab self-association using AUC-SE. Experimental data (scatter plots) were fit to a monomer-dimer equilibrium model (C(cal), solid lines) to determine the equilibrium dissociation constant (KD) of 21 μM. Infliximab was evaluated at 0.07 mg/mL (A), 0.2 mg/mL (B), and 0.6 mg/mL (C). Samples were run at rotor speeds of 7500 (red), 10000 (blue), 12500 (green) and 15000 rpm (purple).
Infliximab self-association in solution is localized to the Fab domain
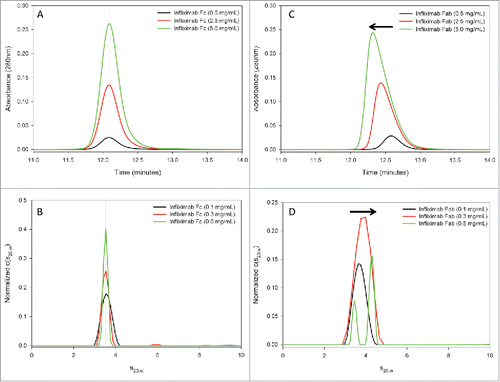
To test our structural hypothesis that the Fab domain drives infliximab self-association, SEC and AUC-SV were repeated using the isolated Fab and Fc domains (). Similar to the intact infliximab analysis, samples were analyzed at 0.5, 2.5, and 5 mg/mL by SEC and at 0.1, 0.3 and 0.6 mg/mL by AUC-SV. Neither SEC, nor AUC-SV revealed evidence of concentration-dependent self-association in the Fc domain ( and , respectively). The SEC elution peak retention times and AUC-SV sedimentation coefficients were consistent at all concentrations for the Fc. In contrast, both techniques demonstrated that the Fab domain of infliximab self-associates in solution. Similar to the SEC results for intact infliximab, the SEC peak for the infliximab Fab domain shifted to earlier retention times with increasing concentration and showed a corresponding loss of peak symmetry, indicative of self-association (). Further, concentration-dependent changes in the Fab domain c(s) distributions mirror those observed with the intact antibody. The sedimentation coefficient value for the Fab domain increased with concentration, with two peaks being resolved at the highest concentration tested (0.6 mg/mL). Similar to the results for intact infliximab, the data indicate that the monomer-dimer equilibrium of the Fab domain at 0.6 mg/mL shifted sufficiently toward the dimer sedimentation coefficient to allow the monomer peak to be resolved in the c(s) distribution (). No differences were observed in the AUC-SV analysis of the Fc domain, when NaCl was substituted in the sample buffer with sucrose. In contrast, the concentration-dependent shift in the Fab domain sedimentation coefficient was less drastic, when NaCl was replaced with sucrose (data not shown). The effect of salt on self-association in the Fab domain is consistent with the observed structural interface composed of weak hydrophobic interactions. Combined, the SEC and AUC-SV results demonstrate that reversible self-association of infliximab is mediated through the Fab domain, consistent with the crystallographic observations.
Figure 9. Infliximab self-association is localized to the Fab domain in solution. The infliximab Fc fragment does not show evidence of self-association by SEC (A) or AUC-SV (B). The SEC elution peak is symmetric and the retention time is consistent at all measured concentrations (A). Similarly, the sedimentation coefficient of the Fc fragment is consistent at all measured concentrations (B). In contrast, the Fab fragment shows evidence of reversible self-association by both SEC (C) and by AUC-SV (D), similar to the intact infliximab antibody ().
Discussion
The results of our study reveal new insights into the structural and solution properties of infliximab. A putative self-association interface observed in the crystal structures indicates that the Fab domain drives self-association, and we confirmed this observation by comparing self-association of the intact infliximab to the Fab and Fc domains. The self-association interface was not observed in the prior crystal structure of the Fab domain in complex with TNF,Citation19 although the antigen binding site and self-association interface are distinct from each other. In the complex structure, three infliximab Fabs are bound to a single TNF trimer. The planar geometry of the antigen trimer positions the Fabs 120° apart from one another, precluding the head-to-tail orientation observed in the unbound structures.
The residues that comprise the infliximab self-association interface reside outside of the CDR loops, suggesting that related antibodies might also self-associate through a similar interface. Cetuximab, a mouse/human chimeric IgG1 antibody, shares 200 of the 214 light chain residues in common with infliximab, including 24 of the 25 residues that comprise the core infliximab self-association interface described above. Interestingly, in the cetuximab Fab domain crystal structure,Citation25 the crystal contains two Fab domains positioned with a LC:LC interface, although the cetuximab Fab domains in the crystal are “face-to-face” and not in the “head-to-tail orientation” observed for infliximab. Despite these similarities and high degree of light chain sequence identity, we did not observe evidence of self-association in cetuximab by AUC-SV (data not shown), indicating that the self-association interface characterized here is unique to infliximab (or of much lower affinity in related antibodies).
Because infliximab self-association is reversible, of low affinity, and not observed in the TNF complex, it is unlikely that Fab-driven self-association would affect the pharmacokinetic properties in patients, although the consequences of reversible self-association on immunogenicity are unknown. An improved knowledge of the structural and biophysical properties of infliximab and other therapeutic antibodies will benefit our understanding of existing medicines and support future development of the next generation of biotherapeutics.
Materials & methods
Materials
Remicade Lots CJM76016P1 and BGS47015P1 were purchased from Janssen Biotech, Inc. and reconstituted according to manufacturer's instructions.
Papain digestion and fragment purification
30 mg of infliximab (Lot CJM76016P1) was digested with immobilized papain slurry (Pierce #20341) for 5 hours at 37°C, using a 1:60 ratio of enzyme to antibody. Enzyme activation was performed following manufacturer's instructions. The Fab was separated from the Fc using MabSelect Protein A affinity chromatography (GE Life Sciences), and both fragments were purified further by size-exclusion HPLC using a YMC-Pack Diol-200 column (Waters). Samples were concentrated to 10 mg/mL in 20 mM succinate, 150 mM sodium chloride, pH 6 and frozen prior to crystallization and analysis by SEC and AUC-SV.
Crystallization, data collection and structure determination
Crystals of the infliximab Fab were obtained in I-centered orthorhombic (I212121) and C-centered orthorhombic (C2221) crystal forms. Crystals in the I212121 form were grown in 2.16 M sodium malonate, 10 mM NAD and were cryo-protected with 20% v/v glycerol prior to data collection. Crystals in the C2221 form were grown in 20% w/v PEG 3350, 0.2 M potassium citrate tribasic and were cryo-protected with 20% v/v ethylene glycol prior to data collection.
Crystals of the infliximab Fc were obtained in a C-centered orthorhombic (C2221) crystal form. Crystals were grown in 10% v/v isopropanol, 0.2 M zinc acetate, 0.1 M sodium cacodylate pH 6.5, and were cryo-protected with 20% v/v ethylene glycol prior to data collection.
Crystals were harvested and complete X-ray data sets were obtained at the Advanced Photon Source (APS), Life Sciences Collaborative Access Team (LS CAT) beamline sector 21. Fab crystals diffracted X-rays to 2.0 Å resolution for both crystal forms. Fab crystal structures were solved by molecular replacement in Phaser MRCitation26 using the constant and variable domains of the infliximab Fab from PDB ID 4G3YCitation19 as independent search models. All structures were refined in PhenixCitation27 with manual model building in Coot.Citation28 Structures were validated with MolProbity.Citation29 Data collection and refinement statistics for the I212121 and C2221 crystal forms are shown in .
Fc crystals diffracted X-rays to 1.75 Å. The crystal structures were solved by molecular replacement in Phaser MR using individual immunoglobulin domains derived from PDB ID 4CDHCitation30 as search models (100% sequence identity). The Fc crystal structure was refined in Phenix with automated model building in ARPw/ARP, manual model building in Coot and was validated with MolProbity. Data collection and refinement statistics are shown in . Structure figures were generated using the PyMOL Molecular Graphics System (Schrödinger, LLC).
Coordinates and structure factors have been deposited in the RCSB protein data bank under accession codes 5VH3, 5VH4 and 5VH5.
Size-exclusion chromatography
Analytical size-exclusion chromatography (SEC) was performed using a YMC-Pack Diol-200 column with a mobile phase of 20 mM sodium phosphate, 400 mM sodium chloride, pH 7.2. Intact infliximab, and its Fab and Fc were injected at 0.5, 2.5, and 5 mg/mL to evaluate self-association properties.
Analytical ultracentrifugation – sedimentation velocity
Samples were diluted to 0.1, 0.3, and 0.6 mg/mL with 20 mM sodium phosphate, 400 mM sodium chloride, pH 7.2. Samples were run in triplicate using 1.2 cm centerpieces in a Beckman Coulter Analytical Ultracentrifuge Proteome XL/I at 45,000 RPM at 20°C. One hundred scans were collected at a wavelength of 280 nm. Data were analyzed using Sedfit (ver.14.1)Citation31 to generate c(s) size distribution plots and to determine the relative abundance of the various species. Sednterp (ver. 20130813 BETA) was used to determine partial specific volumes, as well as the solvent viscosity and density. Partial specific volumes of 0.729, 0.725, and 0.729 mL/g were used for the intact infliximab antibody, and its Fab and Fc, respectively, based on their amino acid compositions. A solvent density of 1.017 g/mL and a solvent viscosity of 1.05 cP were used for data analysis.
Analytical ultracentrifugation – sedimentation equilibrium
Infliximab (Lot BGS47015P1) was reconstituted according to manufacturer's instructions to 9.8 mg/mL, and subsequently dialyzed against 5 mM sodium phosphate pH 7.2, 0.0025% polysorbate-80, 2.5% sucrose. Samples were then diluted to 0.07, 0.2, and 0.6 mg/mL in the dialysate buffer. Samples were measured using a Beckman Coulter Analytical Ultracentrifuge Proteome XL/I at 7500, 10000, 12500 and 15000 rpm at 20°C. The protein sedimentation boundary was monitored by repetitive radial scanning at a constant time interval using the XL-I UV absorption optical system. Equilibrium was monitored using the WinMatch program (http://www.biotech.uconn.edu/auf). Equilibrium was considered established when there were no further changes over a 10 hour period by least squares criteria. Data from different rotor speeds and different concentrations were fit simultaneously (i.e., globally) using SedAnal software.
Disclosure of potential conflicts of interest
Thomas F. Lerch, Penelope Sharpe, Hugh D. Conlon, Sharon Polleck, Jason C. Rouse, Yin Luo, and Qin Zou are full-time employees and may be shareholders of Pfizer Inc.
Funding
This study was sponsored by Pfizer Inc.
References
- Rosenberg AS. Effects of protein aggregates: An immunologic perspective. AAPS J 2006; 8:E501-7; PMID:17025268; https://doi.org/10.1208/aapsj080359
- Fang Y, Gao S, Tai D, Middaugh CR, Fang J. Identification of properties important to protein aggregation using feature selection. BMC Bioinformatics 2013; 14:314; PMID:24165390; https://doi.org/10.1186/1471-2105-14-314
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22:1302-6; PMID:15361882; https://doi.org/10.1038/nbt1012
- Tartaglia GG, Cavalli A, Pellarin R, Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci 2005; 14:2723-34; PMID:16195556; https://doi.org/10.1110/ps.051471205
- Fekete S, Beck A, Veuthey JL, Guillarme D. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J Pharm Biomed Anal 2014; 101:161-73; PMID:24816223; https://doi.org/10.1016/j.jpba.2014.04.011
- Krayukhina E, Uchiyama S, Nojima K, Okada Y, Hamaguchi I, Fukui K. Aggregation analysis of pharmaceutical human immunoglobulin preparations using size-exclusion chromatography and analytical ultracentrifugation sedimentation velocity. J Biosci Bioeng 2013; 115:104-10; PMID:22925901; https://doi.org/10.1016/j.jbiosc.2012.07.021
- Oliva A, Farina JB, Llabres M. Pre-study and in-study validation of a size-exclusion chromatography method with different detection modes for the analysis of monoclonal antibody aggregates. J Chromatogr B Analyt Technol Biomed Life Sci 2016; 1022:206-12; PMID:27107247; https://doi.org/10.1016/j.jchromb.2016.04.022
- Courtois F, Schneider CP, Agrawal NJ, Trout BL. Rational design of biobetters with enhanced stability. J Pharm Sci 2015; 104:2433-40; PMID:26096711; https://doi.org/10.1002/jps.24520
- Uchiyama S. Liquid formulation for antibody drugs. Biochim Biophys Acta 2014; 1844:2041-52; PMID:25131621; https://doi.org/10.1016/j.bbapap.2014.07.016
- Kalonia C, Toprani V, Toth R, Wahome N, Gabel I, Middaugh CR, Volkin DB. Effects of protein conformation, apparent solubility, and protein-protein interactions on the rates and mechanisms of aggregation for an IgG1Monoclonal antibody. J Phys Chem B 2016; 120:7062-75; PMID:27380437; https://doi.org/10.1021/acs.jpcb.6b03878
- Plath F, Ringler P, Graff-Meyer A, Stahlberg H, Lauer ME, Rufer AC, Graewert MA, Svergun D, Gellermann G, Finkler C, et al. Characterization of mAb dimers reveals predominant dimer forms common in therapeutic mAbs. mAbs 2016; 8:928-40; PMID:27031922; https://doi.org/10.1080/19420862.2016.1168960
- Skamris T, Tian X, Thorolfsson M, Karkov HS, Rasmussen HB, Langkilde AE, Vestergaard B. Monoclonal antibodies follow distinct aggregation pathways during production-relevant acidic incubation and neutralization. Pharm Res 2016; 33:716-28; PMID:26563206; https://doi.org/10.1007/s11095-015-1821-0
- Liu B, Guo H, Xu J, Qin T, Xu L, Zhang J, Guo Q, Zhang D, Qian W, Li B, et al. Acid-induced aggregation propensity of nivolumab is dependent on the Fc. mAbs 2016; 8:1107-17; PMID:27310175; https://doi.org/10.1080/19420862.2016.1197443
- Tian X, Langkilde AE, Thorolfsson M, Rasmussen HB, Vestergaard B. Small-angle x-ray scattering screening complements conventional biophysical analysis: Comparative structural and biophysical analysis of monoclonal antibodies IgG1, IgG2, and IgG4. J Pharm Sci 2014; 103:1701-10; PMID:24700358; https://doi.org/10.1002/jps.23964
- Zhang H, Cui W, Gross ML. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Lett 2014; 588:308-17; PMID:24291257; https://doi.org/10.1016/j.febslet.2013.11.027
- Gupta AK, Skinner AR. A review of the use of infliximab to manage cutaneous dermatoses. J Cutan Med Surg 2004; 8:77-89; PMID:15685387; https://doi.org/10.1177/120347540400800202
- Knight DM, Trinh H, Le J, Siegel S, Shealy D, McDonough M, Scallon B, Moore MA, Vilcek J, Daddona P, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol 1993; 30:1443-53; PMID:8232330; https://doi.org/10.1016/0161-5890(93)90106-L
- Maini RN, Feldmann M. How does infliximab work in rheumatoid arthritis? Arthritis Res 2002; 4(Suppl 2):S22-8; PMID:12110154; https://doi.org/10.1186/ar549
- Liang S, Dai J, Hou S, Su L, Zhang D, Guo H, Hu S, Wang H, Rao Z, Guo Y, et al. Structural basis for treating tumor necrosis factor alpha (TNFalpha)-associated diseases with the therapeutic antibody infliximab. J Biol Chem 2013; 288:13799-807; PMID:23504311; https://doi.org/10.1074/jbc.M112.433961
- Hu S, Liang S, Guo H, Zhang D, Li H, Wang X, Yang W, Qian W, Hou S, Wang H, et al. Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor alpha-associated diseases from a molecular view. J Biol Chem 2013; 288:27059-67; PMID:23943614; https://doi.org/10.1074/jbc.M113.491530
- Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol 2006; 357:1566-74; PMID:16497332; https://doi.org/10.1016/j.jmb.2006.01.023
- Rupp B. Biomolecular crystallography: Principles, practice, and application to structural biology. New York: Garland Science, 2010; ISBN:9780815340812.
- Brown PH, Balbo A, Schuck P. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Current protocols in immunology. edited by John E Coligan (Ed.) [et al] 2008; Chapter 18:Unit 18 5; PMID:8491296; https://doi.org/10.1002/0471142735.im1815s81
- Howlett GJ, Minton AP, Rivas G. Analytical ultracentrifugation for the study of protein association and assembly. Curr Opin Chem Biol 2006; 10:430-6; PMID:16935549; https://doi.org/10.1016/j.cbpa.2006.08.017
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301-11; PMID:15837620; https://doi.org/10.1016/j.ccr.2005.03.003
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658-74; PMID:19461840; https://doi.org/10.1107/S0021889807021206
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010; 66:213-21; PMID:20124702; https://doi.org/10.1107/S0907444909052925
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486-501; PMID:20383002; https://doi.org/10.1107/S0907444910007493
- Chen VB, Arendall WB, Headd JJ 3rd, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010; 66:12-21; PMID:20057044; https://doi.org/10.1107/S0907444909042073
- Silva-Martin N, Bartual SG, Ramirez-Aportela E, Chacon P, Park CG, Hermoso JA. Structural basis for selective recognition of endogenous and microbial polysaccharides by macrophage receptor SIGN-R1. Structure 2014; 22:1595-606; PMID:25450767; https://doi.org/10.1016/j.str.2014.09.001
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical J 2000; 78:1606-19; PMID:10692345; https://doi.org/10.1016/S0006-3495(00)76713-0