
ABSTRACT
Flexible and protease resistant (G4S)n linkers are used extensively in protein engineering to connect various protein domains. Recently, several groups have observed xylose-based O-glycosylation at linker Ser residues that yield unwanted heterogeneity and may affect product quality. Because of this, an engineering effort was implemented to explore different linker sequence constructs. Here, we demonstrate the presence of an unexpected hydroxylation of a prolyl residue in the linker, made possible through the use of high-resolution mass spectrometry (HR-MS) and MSn. The discovery started with the detection of a poorly resolved ∼+17 Da mass addition at the reduced protein chain level of an Fc-fusion construct by liquid chromatography-MS. Upon further investigation at the peptide level using HR-MS, the mass increase was determined to be +15.99 Da and was localized to the linker peptide SLSLSPGGGGGPAR [210–223]. This peptide corresponds to the C-terminus of Fc [210–216], the G4P linker [217–221], and first 2 amino acids of a growth factor [222–223]. The linker peptide was first subjected to MS2 with collision-induced dissociation (CID) activation. The fragmentation profile localized the modification to the GGGPA [218–222] portion of the peptide. Accurate mass measurement indicated that the modification is an addition of an oxygen and cannot be CH4, thus eliminating several possibilities such as Pro→Leu. However, other possibilities cannot be ruled out. Higher-energy collision-induced dissociation (HCD)-MS2 and MS3 using CID/CID were both unable to differentiate between Ala222→ Ser222 or Pro221→ Hyp221. Finally, MS3 using high-resolution CID/HCD confirmed the mass increase to be a Pro221→Hyp221 post-translational modification.
KEYWORDS:
Abbreviations
| HR-MS | = | High-resolution mass spectrometry |
| Hyp | = | Hydroxyproline |
| PTM | = | post-translational modification |
| CID | = | collision-induced dissociation |
| HCD | = | higher-energy collision-induced dissociation |
| scFv | = | single-chain variable fragments |
| LC-MS/MS | = | liquid chromatography tandem mass spectrometry |
Introduction
Linkers are commonly used in protein engineering for three main reasons: 1) to generate recombinant fusion molecules such as bifunctional therapeutics, 2) for half-life extension when joining smaller proteins or peptides with Fc or albumin, and 3) for structural reasons to eliminate steric hindrance or to align protein domains.Citation1,2 Several fusion protein drugs using linkers have been approved by the Food and Drug Administration, including etanercept (Enbrel®) and romiplostim (Nplate®). Recent developments in bispecific or bifunctional antibody engineering has further strengthened the importance of linkers.Citation3 For example, linkers are used to generate single-chain variable fragment (scFv; light and heavy variable domains are covalently linked via linker) fusions, and the scFvs may be further fused to Fc or to a full-length antibody to generate bispecific molecules such as IgG-scFv. AbbVie, formerly Abbott Laboratories, has demonstrated bifunctional DVD-Ig format molecules that contain both an “inner” variable domain (anti-tumor necrosis factor (TNF)), as well as an extra “outer” variable domain against another target.Citation4 These domains are connected using different formats: 1) “natural” linker sequences found in the constant and variable domains of full-length monoclonal antibodies (mAbs), 2) poly-glycine linkers, or 3) hinge sequence from between the CH1 and CH2 region of mAbs, each of which yield different anti-TNF binding affinity for the “inner” variable domain.Citation5
Fc-fusion proteins have been constructed to extend the half-life and increase the efficacy of a potential therapeutic. Romiplostim, which is used for the treatment of thrombocytopenia, is composed of a repeated 14 amino acid thrombopoietin mimetic peptide (TMP) sequence fused to the C-terminus of a human IgG1 Fc dimer using both 5 and 8 amino acid poly-glycine linkers.Citation6 The native TMP peptide was efficacious only with continuous subcutaneous infusion; however half-life was improved following fusion of the TMP peptide with Fc and the aforementioned linkers.Citation7 This increase is a result of 2 factors: added molecular weight beyond the glomerular filtration limit of 50 kDa and utilization of the FcRn recycling mechanism.Citation8,9
Structural considerations were made when constructing the linkers for blinatumomab (BLINCYTO®), a bifunctional, bispecific T-cell engager antibody. Blinatumomab is composed of 2 scFv sequences targeting either CD19 or CD3 that are combined into a single protein chain.Citation10 To properly align the variable sequences, the different domains are connected through (G4S)n; [n = 1–3] linker repeats. The early scFv molecule B6.2 was constructed using the linker sequence GSTSGKSSEGKG… to connect the VL and VH domains.Citation11
In addition to structural considerations, linkers may need to be optimized for activity as well as to overcome any potential manufacturability issues. These issues may include stability, solubility, aggregation, proteolytic resistance, or product homogeneity.Citation1 Several properties of linkers, such as their length, hydrophobicity or hydrophilicity, conformational flexibility or rigidity, and secondary structure, must be taken into account during optimization. As most biotherapeutics are now manufactured in mammalian cells, care needs to be taken to avoid unintended glycosylation and other post-translational modifications (PTMs) that are absent in E. coli expression systems. Currently, the most common linkers are Gly-rich linkers or those with (G4S)n repeats.Citation2 Xylose-containing glycans have recently been observed at Ser residues in (G4S)n and similar linkers.Citation12-14 Following initial placement of xylose on Ser by a xylosyltransferase, rampant extension of the glycan by various other enzymes may occur. More than 20 different species of heterogeneous xylose glycans have been identified, including intermediates of the glycosaminoglycan biosynthetic pathway, and species containing phosphorylation, sulfation, and sialylation.Citation14 Even short non-repeating linkers may contain PTMs, such as the phosphorylated serine residue identified in a G4S linker of a fusion protein by Tsyhchuk.Citation15 This undesirable product heterogeneity prompted us to explore new linker sequences that have better properties.
Linkers containing the amino acid proline are commonly used, especially when conformational rigidity is required, to prevent steric hindrance or when spatial separation of domains is needed. Since domain-to-domain or domain-to-linker interaction/interference has been cited as a cause of poor expression or reduced binding activity of recombinantly expressed fusion proteins, proline-containing rigid linkers are frequently called upon to provide structural independence.Citation16,17 Proline, among other amino acids such as threonine and glutamine, are the most frequently found amino acids in natural linkers such as the proline-rich linker in pyruvate dehydrogenase and cysteine proteinase.Citation18,19 Proline is unique in that its side chain is covalently linked to its backbone main chain forming a cyclic structure. This constrains the backbone dihedral angle phi (ф) to be around −60°, imparting high conformational rigidity.Citation20,21 Further, the lack of amide hydrogen on proline also prevents formation of a hydrogen bond between its amide group and other amino acids located either within the linker or in the domain that are being fused together. Both of these features enable proline-containing linkers to be conformationally constrained and provide structural independence between linked domains.
Considering the abundance of proline in natural linkers, the use of proline along with glycine could be considered as an intermediary solution between highly flexible (e.g., GGGGS) and rigid (e.g., Ala-Pro repeats) linkers. Further, replacing serine with proline would prevent side chain hydrogen bonding and interaction, thus providing limited structural independence for the linker as well as for the domains that are being linked together. The G4P and (G4P)2 [GGGGP and GGGGPGGGGP] linkers are alternative constructs designed to be partially rigid and to eliminate the aforementioned xylose glycans, but they suffer from post-translational modification due to enzymatic action. Here, we demonstrate by HR-MS the presence of unexpected proline hydroxylation in the G4P linker of an Fc-fusion protein. We hope this report will draw attention to the need for careful design and characterization of protein linker sequences to eliminate undesirable attributes in protein therapeutics.
Results
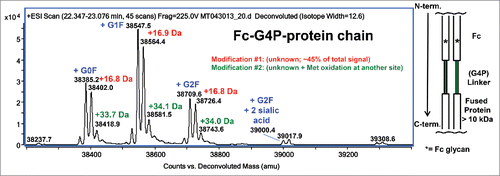
The denatured and reduced protein [an Fc-(G4P)-growth factor fusion] was analyzed by LC-MS as shown in . The observed mass contains heterogeneous Fc glycosylation (G0F, G1F, G2F, G2F + 2 sialic acid), as well as an abundant mass increase of ∼17 Da (unknown modification #1). At the reduced protein chain level, unknown modification #1 occurs at ∼90% of the peak height of the unmodified protein chain of each glycoform and represents about 45% of the total signal. A second lower level mass increase of ∼34 Da (modification #2) was also present in the reduced protein chain data, which was determined to be a combination of unknown modification #1 plus minor Met oxidation at a separate site in the growth factor portion of the protein (data not shown).
Figure 1. Deconvoluted mass spectrum of denatured and reduced Fc-G4P-protein. The Fc is linked to the protein via a G4P linker. Typical Fc glycosylation (G0F, G1F, G2F, etc.) was observed. Modification #1 represents about 45% of the MS signal based on peak height.
Upon reduction, alkylation, tryptic digestion, and analysis using high-resolution tandem mass spectrometry (MS), unknown modification #1 was quickly localized to the peptide containing the G4P linker: SLSLSPGGGGGPAR [210–223] (). Here, residues [210–216] (SLSLSPG) correspond to the C-terminus of Fc, with [217–221] (GGGGP) the G4P linker, and [222–223] (AR) the first 2 amino acids of a growth factor. Using a high-resolution FT-MS scan, the modified peptide with unknown modification #1 (∼+17 Da) has an accurate mass increase of + 15.99 Da, and elutes ∼2 minutes earlier than the unmodified version (T21.8 min vs. T23.8 min). Based on peptide signal intensity, there is a ratio of about 60:40 of unmodified peptide: modified peptide. The initial low-resolution collision-induced dissociation (CID) data of both the doubly charged precursors (m/z 606.82; unmodified and m/z 614.82; modified) could only localize the modification site to the GGGPAR portion of the peptide (). The b13 fragment ion eliminated the terminal Arg residue as a candidate for amino acid substitution or post translational modification.
Figure 2. A high level +15.99 Da mass increase was observed on the G4P linker peptide by LC-MS/MS. Based on peptide signal intensity, there is a ratio of about 60:40 of unmodified peptide: modified peptide. Top panel: XIC (Extracted Ion Chromatogram) of unmodified and modified peptides. Bottom panel: Centroid mass spectrum averaged from T21.5–24.5 min.
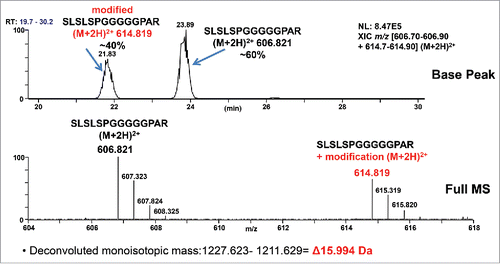
Figure 3. Low-resolution CID mass spectra of unmodified and +15.99 Da modified ([M+2H]2+ = 614.82) linker peptide SLSLSPGGGGGPAR. The modification can be localized to the GGGPAR portion of the sequence by the difference in y6 ions. Different b13 ions eliminate the C-terminal R.
![Figure 3. Low-resolution CID mass spectra of unmodified and +15.99 Da modified ([M+2H]2+ = 614.82) linker peptide SLSLSPGGGGGPAR. The modification can be localized to the GGGPAR portion of the sequence by the difference in y6 ions. Different b13 ions eliminate the C-terminal R.](/cms/asset/9742d882-5a4a-4945-872c-8411846e533f/kmab_a_1325556_f0003_c.gif)
With the remaining Gly, Pro, and Ala residues, there are several plausible amino acid substitutions or PTMs that can yield the observed modified precursor mass, if we only consider the mass with the accuracy of nominal mass. Addition of 16 Da to Gly (57 Da +16 Da = 73 Da) does not yield a recognized amino acid or modification. Addition of 16 Da to Pro (97 Da+ 16 Da = 113 Da) may yield the amino acids Ile or Leu, as well as the post-translationally modified residue hydroxyproline (Hyp). Pro221→Leu/Ile221 would be expected to make the peptide more hydrophobic and to elute later than the wild-type peptide on reverse phase HPLC, which is inconsistent with the actual chromatographic elution of the modified peptide. Addition of 16 Da to Ala (71 Da+ 16 Da = 87 Da) yields Ser. There are also considerations with regard to codon arrangement on the possibilities of the proposed mutations or modifications. Pro221→Ile221 cannot occur with one base change; however, Pro221→Leu221 is possible and would require a single C→T base change in the second codon position (CCT, CCC, CCA, or CCG to CTT, CTC, CTA, or CTG). Ala222→Ser222 can be accomplished by a single G→T base change in the first position of a codon from GCT, GCC, GCA, or GCG to TCT, TCC, TCA, or TCG. Therefore, we are left with 3 possibilities when we consider the nominal mass shift: Pro221→Leu221, Pro221→Hyp221, and Ala222→Ser222.
Based on the accurate doubly charged precursor mass of m/z 614.8187 (or a mass shift of 15.99 Da), Pro221→ Leu221 can be eliminated because the theoretical mass of m/z 614.8364 is ∼29 ppm off the observed mass of m/z 614.8187. This is because the formula of the Pro221→ Leu221 mutation is +CH4, which corresponds to a mass shift of 16.0313 Da, different from the Pro221→Hyp221 modification, which has the formula of +O, with a mass shift of 15.9949 Da. Ala222→ Ser222 has an identical mass shift of 15.9949 Da. shows high-resolution higher-energy collision-induced dissociation (HCD) of m/z 614.82, which yields monoisotopic y3 and y4 fragment ions of 359.204 and 416.225. These observed masses are consistent with either a PSR (Ala222→Ser222) substitution or a *PAR (* = Pro221→Hyp221) PTM whose sequences yield y3 and y4 monoisotopic fragment ions of m/z 359.204 and 416.226, respectively.
Figure 4. High-resolution HCD mass spectrum of +15.99 Da modified ([M+2H]2+ = 614.82) linker peptide SLSLSPGGGGGPAR. Top panel: m/z [100–1250]. Bottom panel: m/z [340–460]. The detected accurate masses of the y3 and y4 fragment ions rule out Pro221→ Leu/Ile221 mutation. The HCD fragmentation cannot discriminate between Pro221→Hyp221 or Ala222→Ser222 as no y2 fragment ion was observed.
![Figure 4. High-resolution HCD mass spectrum of +15.99 Da modified ([M+2H]2+ = 614.82) linker peptide SLSLSPGGGGGPAR. Top panel: m/z [100–1250]. Bottom panel: m/z [340–460]. The detected accurate masses of the y3 and y4 fragment ions rule out Pro221→ Leu/Ile221 mutation. The HCD fragmentation cannot discriminate between Pro221→Hyp221 or Ala222→Ser222 as no y2 fragment ion was observed.](/cms/asset/eea9062e-ebe8-4f1c-ac94-c37e3b969121/kmab_a_1325556_f0004_c.gif)
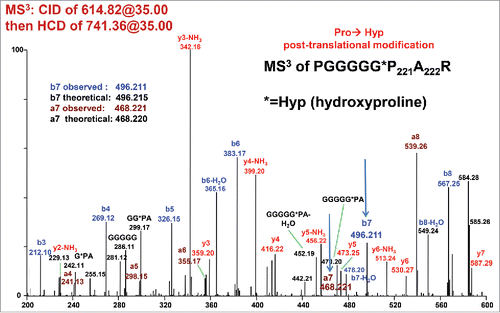
To discriminate between Ala222→Ser222 and Pro221→Hyp221, additional fragmentation experiments were performed. High-resolution CID of the doubly-charged unmodified and modified precursor ions (m/z 606.82 and 614.82) result in abundant y9 fragment ions of m/z 725.37 and 741.36 respectively. These 2 ions were selected for further MS3 fragmentation using targeted CID or HCD. Fragmenting these ions is equivalent to fragmenting unmodified peptide PGGGGGPAR or modified peptides PGGGGG*PAR or PGGGGGPSR. Only 3 fragment ions y2, b7, and a7 may differentiate between PGGGGG*PAR and PGGGGGPSR. PGGGGG*PAR should yield y2 = 246.156, b7 = 496.215, and a7 = 468.220, while PGGGGGPSR should yield y2 = 262.151, b7 = 480.220, and a7 = 452.225. Using high-resolution CID/CID, no discriminating ions were observed (data not shown). By using high-resolution CID/HCD of 614.82/741.36, a Pro221→Hyp221 PTM was confirmed at residue 221 by the presence of a b7 ion at 496.211 and an a7 ion of 468.221 (). Even though the putative a7 ion appears to be at low signal to noise, there is a consistent series of other a- ions including a4, a5, and a6 that suggest this assignment is correct. It should be noted that normalized collision energy (NCE) values of 35 for CID and 27 or 29 for HCD are typically used for LC-MS/MS. Here, a NCE of 35 for CID (MS2) and NCE of 35 for HCD (MS3) were required to see the diagnostic a7 and b7 fragment ions.
Figure 5. CID/HCD MS3 spectrum of m/z 741.36, i.e. y9 ion resulting from CID fragmentation of the +15.99 Da modified peptide (see Bottom Panel of ). HCD fragmentation of the 741.36 ion yields unique a7 and b7 fragment ions that confirm a Pro221→Hyp 221 post-translational modification.
A similar construct using a (G4P)2 repeating linker was also analyzed and yielded similar protein chain level mass data with an ∼17.2 Da mass increase (data not shown) that could be localized to peptide SLSLSPGGGGGPGGGGPAR [210–228] using tryptic peptide mapping and LC-MS/MS. At the peptide level, ∼65% of [210–228] exists unmodified, while ∼34% contains one modification, and ∼1% contains 2 modifications (). With the knowledge of the hydroxyproline PTM observed previously, it is logical to propose that this is the same modification. To localize the major site of modification between Pro215, Pro221, and Pro226, high-resolution CID was performed on the modified doubly charged precursor of m/z 777.39. The presence of the +15.99 Da modification on y3 (m/z 359.20) confirmed that Pro226 was modified to Hyp226 (). There was no evidence of unmodified y3, which would be indicative of unmodified Pro226 and Hyp at either Pro215 or Pro221. This could occur if single Hyp modified peptide species (m/z 777.39) exist, but the modification occurs at different sites with the peptides all co-eluting. The low level species with 2 Hyp residues was also targeted (7.5K res. CID of m/z 785.39). In the CID spectra, the presence of y3 = 359.20 confirms Pro226→Hyp226, while y8 = 700.34 indicates that Pro221→Hyp221 is the second site of modification (data not shown).
Figure 6. XIC of the doubly charged ions for different forms of the (G4P)2 linker peptide, SLSLSPGGGGGPGGGGPAR, expressed in CHO-K1. Peak A: Unmodified peptide, ∼65%, m/z = 769.35–769.45. Peak B: One Hyp, ∼34%, m/z = 777.35–777.45. Peak C: Two Hyp, ∼1%, m/z = 785.35–785.45.
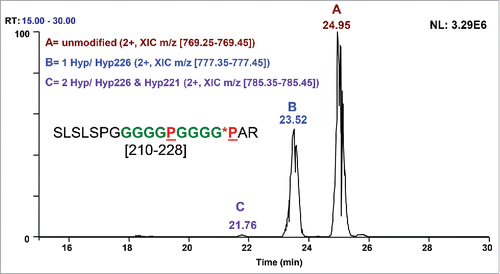
Figure 7. High-resolution CID mass spectrum of the doubly charged ion of (G4P)2 linker peptide SLSLSPGGGGGPGGGGPAR [210–228] with one Hyp. Hyp PTM is present in y3 and higher y ions, confirming Hyp226.
![Figure 7. High-resolution CID mass spectrum of the doubly charged ion of (G4P)2 linker peptide SLSLSPGGGGGPGGGGPAR [210–228] with one Hyp. Hyp PTM is present in y3 and higher y ions, confirming Hyp226.](/cms/asset/ebf76b52-1245-4ef7-808a-80345606525f/kmab_a_1325556_f0007_c.gif)
Discussion
High-resolution MS is one of the most reliable tools for protein characterization and crucial for identifying PTMs and substitutions. At the peptide level, the high mass accuracy of the instrumentation (< 3 ppm RMS with external calibration) allows differentiation of components with the same nominal mass (isobars) with high confidence. This could include sulfation vs. phosphorylation (+79.957 or +79.966 Da), amino acids such as Gln vs. Lys (128.058 or 128.095 Da), and even the addition of oxygen (15.995 Da) vs. certain amino acid substitutions (Asp→Met = +16.014 Da, Met→Phe = +16.028 Da), and Pro→Leu/Ile = +16.031 Da). To localize these changes down to the amino-acid level, effective fragmentation is also required, in addition to the high mass accuracy. As demonstrated in this study, access to various fragmentation techniques, as well as the capability of performing multi-stage fragmentation (MSn) becomes invaluable for identification and localization of modifications.
The identification of xylose-containing glycans in (G4S)n and similar linkers and phosphorylated serine in a G4S linker demonstrate that even commonly used linkers may be susceptible to undesirable post-translational modification.Citation12-15 In screening various new linker candidates, including G4P and (G4P)2, additional PTMs were observed. By combining high-resolution tandem MS with various MSn techniques, an unexpected Pro→Hyp PTM was confidently identified in the G4P linker of our Fc-fusion protein. The high mass accuracy of the Orbitrap Velos Pro eliminated certain possibilities (Pro→Leu/Ile) on the basis of accurate mass of both the precursor and fragment ions. Isobaric modifications such as an Ala→Ser amino acid substitution and a Pro→Hyp PTM could not be so easily reconciled and required multiple stages of fragmentation. Here, MS3 using a combination of CID/HCD detection was needed for final confirmation. It would be very difficult to distinguish between these 2 possibilities without access to an instrument capable of MSn.
Hydroxylation of proline is a PTM in protein synthesis catalyzed by the enzyme prolyl hydroxylase. The resulting Hyp PTM may exist as either 4-Hyp or 3-Hyp; 4-Hyp is very common and known to play a key role in the stability of the collagen structure, while 3-Hyp is rare.Citation22-24 4-Hyp and 3-Hyp can be differentiated by Edman N-terminal sequencing, but they cannot be distinguished by MS-based techniques.Citation23-25 3-Hyp has been observed in vertebrate tendon cartilage at the motif (Gly-Pro-Pro)n, in which the first Pro may become modified to 3-Hyp, while the second Pro becomes 4-Hyp.Citation24 Without confirmatory Edman sequencing data, attempts to classify our identified Hyp residue as 4-Hyp or 3-Hyp would be speculative.
The presence of Hyp in the G4P linker is not particularly surprising when one compares the Gly-Pro-Ala amino acid sequence of the G4P linker/growth factor with certain structural motifs of the protein collagen that result in hydroxyproline at various sites. For collagen to form a right-handed triple helical structure, it requires a motif of Xaa-Yaa-Gly. Every third residue is Gly, while X and Y are frequently Pro and Hyp, respectively.Citation26 The presence of Hyp provides additional stability and rigidity to the backbone via hydrogen bonding of the Hyp side chain OH to the main chain carbonyl. The melting temperature (Tm) of collagen significantly increases due to the presence of Hyp.Citation27 Yang et al. unexpectedly observed Hyp residues in the Xaa-position of Gly-Xaa-Yaa triplets, which resulted in Gly-Hyp-Val and Gly-Hyp-Ala triplets at certain positions in bovine placental collagen.Citation28 There was uncertainty as to the form of the Hyp in the above triplets as 3-Hyp is believed to be exclusive to the Xaa position of the triplet Gly-Pro-Hyp, with 4-Hyp exclusive to the Yaa position.Citation28 Similar results were reported in bovine cartilage by Song, who observed Hyp residues at in the triplets Gly-Hyp-Ala, Gly-Hyp-Val, Gly-Hyp-Gln, and Gly-Hyp-Hyp as well as at other sites.Citation22 Both group's findings agree withthe Hyp findings in our Fc-fusion protein with the G4P linker. Additionally, the Gly-Pro-Ala sequence of the linker/growth factor is favored as a site for Hyp PTM over the Gly-Pro-Gly sequence when using the (G4P)2 linkers as shown in the peptide SLSLSPGGGGGPGGGGPAR. In species with a single Hyp PTM, it is exclusively on Gly-Pro-Ala. Only in the low level species with 2 Hyp PTMs is Hyp seen at Pro in both Gly-Pro-Ala and Gly-Pro-Gly sequences. Because of this, G4P and (G4P)2 linkers may still be of use in the creation of Fc-fusion proteins, but care must be taken with the amino acid residues directly after the linker sequence to avoid creating a Hyp PTM site. The Hyp modification in the two protein constructs discussed here are not isolated incidents. We have since detected Hyp at very high level (2x more abundant than the unmodified) on another fusion protein using a GGGG-(AP)10 linker (data not shown), with the modification site localized to the first Pro of the linker in the sequence GGGGAPAPAP… We hypothesize that the GGGG linker right before the (AP)10 linker was critical as it provided structural flexibility that can be necessary for this modification.
The possibility of hydroxyproline being a critical quality attribute (CQA) has not been extensively discussed in the literature, potentially because of the lack of biotherapeutic agents with this modification selected to move on to late-stage development. On one hand, hydroxyproline is a naturally-occurring non-proteinogenic amino acid, and therefore less likely to be immunogenic. On the other hand, the addition of a hydrogen-bond forming hydroxyl group to the otherwise inert backbone can alter the protein structure. As stated above in the collagen example, Hyp imparts further rigidity to the backbone confirmation due to the additional side-chain-main chain hydrogen bond. This additional rigidity may not be a desired feature in some linkers and could affect the function of the fusion protein. It is also plausible that the OH group of the Hyp side chain could be involved in undesired hydrogen bonding with the fusion protein, thus leading to potential modification of the activity or biophysical properties of the partner protein. It has been suggested through crystal structure analysis that Hyp stabilizes collagen fibrils via mediating direct contact between neighboring molecules.Citation29 Despite the uncertainty of hydroxyproline being a CQA, high levels of hydroxyproline (the fusion molecule we studied serves as a good example) is never the less a significant concern with regard to product homogeneity, especially when it can easily be mistaken for oxidation modifications on residues such as methionine and tryptophan. Therefore, we recommend careful analysis of proteins with proline-containing linkers, and that researchers potentially avoid the use of linkers that demonstrate high levels of hydroxyproline.
Materials and methods
Protein expression and purification
The fusion proteins include linker sequences of either (G4P) or (G4P)2 that attach the C-terminus of IgG1 Fc to the N-terminus of a growth factor protein. The recombinant molecules were produced by the Biologics Optimization group at Amgen. Briefly, the proteins were expressed in CHO-K1 stable cells, then purified using MabSelect SuRe, SP Sepharose HP, and finally Superdex 200 (all from GE Healthcare Life Sciences, Pittsburgh, PA).
LC-MS of reduced protein chains
The Fc-fusion protein was denatured, reduced, and then analyzed by LC-MS using an 1100 HPLC connected to a 6224 ESI-TOF mass spectrometer (all from Agilent Technologies, Santa Clara, CA). The HPLC column, gradient, and running conditions, as well as the ESI-TOF instrumental parameters, are identical to those described previously.Citation14 The resulting Full MS spectra were summed, then deconvoluted to [30,000–50,000 Da] using the MaxEnt algorithm in the Agilent Mass Hunter software. A mass step of 1 Da, a S/N threshold of 30.0, and an average mass % peak height of 50 were used.
Digestion of sample
50 µg of the Fc-fusion protein was dried, resuspended in 25 µL 150 mM Tris, pH 7.5/40 mM hydroxylamine/8 M urea/10 mM dithiothreitol and denatured and reduced for 1 hour at 37°C. The protein was alkylated with 20 mM iodoacetamide for 30 minutes at room temperature in the dark. The sample was diluted to 100 µL with water and 2.5 µg of trypsin, digested overnight at 37°C, acidified, and then analyzed on the LC-MSn system through several separate injections using various methods described below.
LC-MSn
All LC-MSn work was performed on an Orbitrap Velos Pro (Thermo Scientific, Waltham, MA). A NanoAcquity UPLC (Waters Corporation, Milford, MA) equipped with a Waters Symmetry C18 180µm X 20 mm 5µm trapping column and an Agilent Zorbax 300SB-C18 500 µm X 250 mm 5 µm analytical column was used for all peptide separations. The basic instrumental setup is similar to that previously published.Citation14 For data dependent LC-MS/MS, FT-MS was performed over m/z [300–2000] at 30K resolution, followed by low-resolution CID of the top 10 most abundant precursors in the linear ion trap. The instrument used a spray voltage of 3.5 kV, an isolation width of 2.0 Da, a default charge state of 4, an activation time of 10 msec, and an NCE of 35.
LC-MS/MS with high-resolution HCD method consists of FT-MS m/z [600–618] at 30K resolution, followed by HCD fragmentation (7.5K resolution) of the top 3 most abundant precursors. HCD uses a NCE of 35, a default charge state of 4, an isolation width of 3 Da, an activation time of 100 msec, and no dynamic exclusion.
Targeted high-resolution LC-MSn was performed using FT-MS m/z [300–2000] at 30K resolution, followed by CID of m/z 606.82, CID of m/z 614.82, CID/CID of m/z 606.82/725.37, CID/CID of m/z 614.82/741.36, and CID/HCD of m/z 614.82/741.36. HCD used a charge state of 1. Here, all CID and HCD scans were performed at 7.5K resolution and used a NCE of 35, and an isolation width of 2.5 Da. Ions of m/z 725.37 and 741.36 correspond to the proline-directed y9 fragment (peptide PGGGGGPAR) of the unmodified and modified forms of the peptide.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
The authors would like to thank Opas Nuanmanee for producing the proteins, and John Robinson for help in Orbitrap operation and discussions. We also thank Dr. Hsieng Lu and Dr. Greg Flynn for helpful discussions.
References
- Reddy Chichili VP, Kumar V, Sivaraman J. Linkers in the structural biology of protein–protein interactions. Protein Sci 2013; 22:153-67; PMID:23225024; https://doi.org/10.1002/pro.2206
- Chen X, Zaro J, Shen W-C. Fusion protein linkers: Property, design and functionality. Adv Drug Deliv Rev 2013; 65(10):1357-69; PMID:23026637; https://doi.org/10.1016/j.addr.2012.09.039
- Spiess C, Zhai Q, Carter PL. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; https://doi.org/10.1016/j.molimm.2015.01.003
- Wu C, Ying H, Bose S, Miller R, Medina L, Santora L, Ghayur T. Molecular construction and optimization of anti-human IL-1α/β dual variable domain immunoglobulin (DVD-IgTM) molecules. MAbs 2009; 1(4):339-47; PMID:20068402; https://doi.org/10.4161/mabs.1.4.8755
- DiGiammarino EL, Harlan JE, Walter KA, Ladror US, Edalji RP, Hutchins CW, Lake MR, Greischar AJ, Liu J, Ghayur T, et al. Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. MAbs 2011; 3(5):487-94; PMID:21814039; https://doi.org/10.4161/mabs.3.5.16326
- Shimamoto G, Gegg C, Boone T, Quéva C. Peptibodies: A flexible alternative format to antibodies. MAbs 2012; 4(5):586-91; PMID:22820181; https://doi.org/10.4161/mabs.21024
- Molineux G. The development of romiplostim for patients with immune thrombocytopenia. Ann N Y Acad Sci 2011; 1222:55-63; PMID:21434943; https://doi.org/10.1111/j.1749-6632.2011.05975.x
- Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao S-W, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol 2015; 35(2):235-54; PMID:24156398; https://doi.org/10.3109/07388551.2013.834293
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; https://doi.org/10.1038/nri2155
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941-4; PMID:19509221; https://doi.org/10.1158/0008-5472.CAN-09-0547
- Bird RE, Walker BW. Single chain antibody variable regions. Trends Biotechnol 1991; 9:132-7; PMID:1367550; https://doi.org/10.1016/0167-7799(91)90044-I
- Wen D, Foley SF, Hronowski XL, Gu S, Meier W. Discovery and investigation of O-xylosylation in engineered proteins containing a (GGGGS)n linker. Anal Chem 2013; 85:4805-12; PMID:23581628; https://doi.org/10.1021/ac400596g
- Spahr C, Kim JJ, Deng S, Kodama P, Xia Z, Tang J, Zhang R, Siu S, Nuanmanee N, Estes B, et al. Recombinant human lecithin-cholesterol acyltransferase Fc fusion: Analysis of N- and O-linked glycans and identification and elimination of a xylose-based O-linked tetrasaccharide core in the linker region. Protein Sci 2013; 22:1739-53; PMID:24115046; https://doi.org/10.1002/pro.2373
- Spahr C, Shi S D-H, Lu HS. O-glycosylation of glycine-serine linkers in recombinant Fc-fusion proteins: Attachment of glycosaminoglycans and other intermediates with phosphorylation at the xylose sugar subunit. MAbs 2014; 6:904-14; PMID:24927272; https://doi.org/10.4161/mabs.28763
- Tyshchuk O, Völger HR, Ferrara C, Bulau P, Koll H, Mølhøj M. Detection of a phosphorylated glycine-serine linker in an IgG-based fusion protein. MAbs 2017; 9(1):94-103; PMID:27661266; https://doi.org/10.1080/19420862.2016.1236165
- Amet N, Lee HF, Shen WC. Insertion of the designed helical linker led to increased expression of tf-based fusion proteins. Pharm Res 2009; 26:523-8; PMID:19002568; https://doi.org/10.1007/s11095-008-9767-0
- Maeda Y, Ueda H, Kazami J, Kawano G, Suzuki E, Nagamune T. Engineering of functional chimeric protein G-Vargula luciferase. Anal Biochem 1997; 249:147-52; PMID:9212866; https://doi.org/10.1006/abio.1997.2181
- Argos P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J Mol Biol 1990; 211:943-58; PMID:2313701; https://doi.org/10.1016/0022-2836(90)90085-Z
- George R, Heringa J. An analysis of protein domain linkers: Their classification and role in protein folding. Protein Eng 2002; 15:871-9; PMID:12538906; https://doi.org/10.1093/protein/15.11.871
- Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol 1963; 7:95-9; PMID:13990617; https://doi.org/10.1016/S0022-2836(63)80023-6
- MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. J Mol Biol 1991; 218:397-412; PMID:2010917; https://doi.org/10.1016/0022-2836(91)90721-H
- Song E, Mechref Y. LC-MS/MS identification of the O-Glycosylation and hydroxylation of amino acid residues of collagen α-1 (II) chain from bovine cartilage. J Proteome Res 2013; 12(8):3599-609; PMID:23879958; https://doi.org/10.1021/pr400101t
- Hudson DM, Eyre DR. Collagen prolyl 3-hydroxylation: A major role for a minor post-translational modification?. Connect Tissue Res 2013; 54:245-51; PMID:23772978; https://doi.org/10.3109/03008207.2013.800867
- Hudson DM, Werther R, Weis MA, Wu J-J, Eyre DR. Evolutionary origins of C-terminal (GPP)n 3-hydroxyproline formation in vertebrate tendon collagen. PloS One 2014; 9(4):e93467; PMID:24695516; https://doi.org/10.1371/journal.pone.0093467
- Taga Y, Kusubata M, Ogawa-Goto K, Hattori S. Developmental stage-dependent regulation of prolyl 3-hydroxylation in tendon type I collagen. J Biol Chem 2015; 291:837-47; PMID:26567337; https://doi.org/10.1074/jbc.M115.686105
- Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem 2009; 78:929-58; PMID:19344236; https://doi.org/10.1146/annurev.biochem.77.032207.120833
- Sakakibara S, Inouye K, Shudo K, Kishida Y, Kobayashi Y, Prockop DJ. Synthesis of (Pro-Hyp-Gly) n of defined molecular weights. evidence for the stabilization of collagen triple helix by hydroxypyroline. Biochim Biophys Acta 1973; 303(1):198-202; PMID:4702003; https://doi.org/10.1016/0005-2795(73)90164-5
- Yang C, Park AC, Davis NA, Russell JD, Kim B, Brand DD, Lawrence MJ, Ge Y, Westphall MS, Coon JJ, et al. Comprehensive mass spectrometric mapping of the hydroxylated amino acid residues of the α1(V) collagen chain. J Biol Chem 2012; 287:40598-610; PMID:23060441; https://doi.org/10.1074/jbc.M112.406850
- Berisio R, Vitagliano L, Mazzarella L, Zagari A. Crystal structure of a collagen-like polypeptide with repeating sequence Pro–Hyp–Gly at 1.4 Å resolution: Implications for collagen hydration. Biopolymers 2000; 56:8-13; PMID:11582572; https://doi.org/10.1002/1097-0282(2000)56:1%3c8::AID-BIP1037%3e3.0.CO;2-W