
ABSTRACT
Fcabs (Fc domain with antigen-binding sites) are promising novel therapeutics. By engineering of the C-terminal loops of the CH3 domains, 2 antigen binding sites can be inserted in close proximity. To elucidate the binding mode(s) between homodimeric Fcabs and small homodimeric antigens, the interaction between the Fcabs 448 and CT6 (having the AB, CD and EF loops and the C-termini engineered) with homodimeric VEGF was investigated. The crystal structures of these Fcabs, which form polymers with the antigen VEGF in solution, were determined. However, construction of heterodimeric Fcabs (JanusFcabs: one chain Fc-wt, one chain VEGF-binding) results in formation of distinct JanusFcab–VEGF complexes (2:1), which allowed elucidation of the crystal structure of the JanusCT6–VEGF complex at 2.15 Å resolution. VEGF binding to Janus448 and JanusCT6 is shown to be entropically unfavorable, but enthalpically favorable. Structure-function relationships are discussed with respect to Fcab design and engineering strategies.
Abbreviations
| DSC | = | differential scanning calorimetry |
| Fcab | = | Fc domain with antigen-binding sites |
| Fc-wt | = | Fc wild-type |
| IgG | = | immunoglobulin class G |
| ITC | = | isothermal titration calorimetry |
| Janus448 and JanusCT6 | = | heterodimeric VEGF-binding Fcabs |
| LC-ESI-MS | = | liquid chromatography-electrospray ionization-mass spectrometry |
| MALS | = | multi-angle light scattering |
| SEC | = | size-exclusion chromatography |
| VEGF | = | vascular endothelial growth factor |
| 448 and CT6 | = | VEGF-binding Fcabs |
Introduction
Since the mid-1980s more than 60 therapeutic antibodies or antibody-based products have been granted approval (antibodysociety.org, update: June 2017). Approximately half of them gained market approval in the last 6 years, demonstrating the substantial importance and commercial success of this fast growing class of biopharmaceutical drugs. Beside a considerable number of chimeric, humanized and human antibodies, to date more than 10 Fc-fusion proteins have been approved by the Food and Drug Administration.Citation1 The Fc's ability to induce effector functions, such as antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, and its interaction with the neonatal Fc receptor (FcRn), responsible for the long in vivo half life of IgG,Citation2-4 make the Fc a critical feature for antibody therapeutics that have suitable pharmacokinetics and clinical efficacy.
IgG1-Fc is a homodimeric protein with each chain (T225-K447, Eu numbering systemCitation5) consisting of 2 domains (CH2 and CH3) and one N-linked glycosylation at position 297. The 2 chains are covalently connected via 2 disulfide bridges in the hinge region and non-covalently connected by extensive interactions between the 2 CH3 domains. This IgG1-Fc protein can serve as a starting scaffold for the design of Fc domains with antigen-binding sites (i.e., Fcabs). Each constant domain in IgG1-Fc is composed of 2 antiparallel β-sheets adopting the typical immunoglobulin fold which is known to tolerate variability in sequence and length in its loops connecting the β-strands, similar to the CDR-loops.Citation6 The CH3 domain exhibits 3 C-terminal structural loops that can be engineered for antigen-binding, namely the AB loop (R355-Q362), CD loop (S383-Y391) and EF loop (D413-V422). Incorporation of those artificial binding sites into an IgG1-Fc molecule results in Fcabs, which combine all antibody functions including antigen binding, at a size of only ∼50 kDa. Moreover, by reunion of the Fcab with Fab arms of a different specificity a bispecific antibody (mAbCitation2) can be obtained.Citation7
Proof of concept that it is possible to engineer binding sites in the CH3 domains of IgG1-Fc was demonstrated by generating the human epidermal growth factor receptor 2 (HER2)-binding Fcab H10–03–6. This Fcab was engineered by randomly mutating the AB and EF loops in the CH3 domains, followed by selection for antigen binding using yeast surface display. Remarkable, this Fcab revealed in vivo half life in mice comparable to that of human wild type IgG1-Fc (Fc-wt), in addition to activation of natural killer cells against a HER2-positive cell line and significant retardation of tumor growth in mice xenograft models.Citation8-10 To improve the biophysical properties of this Fcab, a directed evolution approach was developed, resulting in Fcabs with increased conformational stability and retained binding affinity (STAB Fcabs).Citation11 Recently, the X-ray structures of the parental Fcab H10–03–6 and its stabilized version STAB19 were determined, illuminating the effect of loop design on the fold and the stability of Fcabs. The complex structure representing one Fcab binding to 2 HER2 molecules, together with elaborate solution studies, unveiled a negative cooperative binding behavior.Citation12
Moreover, the next generation Fcab FS102 targeting HER2 with high affinity was shown to surpass a combination of the 2 clinically approved antibodies, trastuzumab and pertuzumab, in in vitro and in vivo studies. This Fcab induces internalization and degradation of HER2, leading to tumor cell apoptosis.Citation13 Due to this superior antitumor effect, FS102 entered a clinical Phase 1 study.
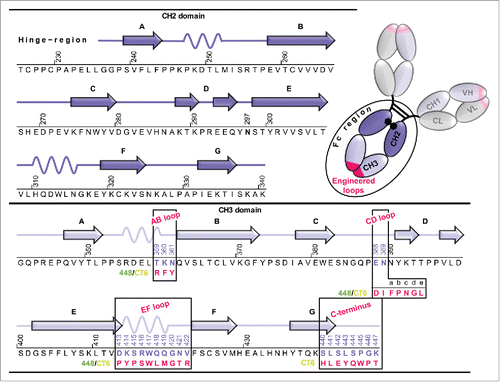
To investigate how a homodimeric Fcab, containing 2 independent binding sites interacts with a soluble homodimeric antigen, vascular endothelial growth factor (VEGF) was chosen as a homodimeric model protein. Depending on the steric constraints in the 2 interaction partners, 1:1, 1:2 or 2:1 complexes, or even the formation of polymers seemed to be possible. Detailed characterization of such an interaction yields valuable information for future engineering strategies using Fcabs or other bivalent binding molecules, such as antibodies. The aim of this study was to investigate the interaction of 2 Fcabs, 448 and CT6, engineered for binding to VEGF-A. Both Fcabs carry the same mutations within the AB, CD and EF loops, with CT6 having additionally the C-terminal region engineered for increased binding affinity (). The biophysical and structural characterization of the 2 Fcabs and the exceptional circumstances that necessitated the generation of a heterodimeric Fcab molecule with only one binding site to finally obtain the 2.15 Å X-ray structure of a complex with VEGF-A are described.
Figure 1. Amino acid sequence of human IgG1-Fc (Fc-wt) and the Fcabs 448 and CT6. Schematic representation of full-length IgG1 consisting of 2 heavy chains (VH, CH1, CH2 and CH3 domains) and 2 light chains (VL and CL domains) with the Fab fragment carrying the antigen-binding sites (pink). The secondary structure elements of one CH2 and CH3 domains (dark and light violet) are shown together with their corresponding amino acid sequence from residue T225 to K447 (Eu numbering systemCitation5). The conserved N-linked glycosylation is located at position 297. The engineered loop regions (AB, CD and EF loops in the CH3 domains) of the Fcabs 448 and CT6 differing in length and sequence and the mutations at the C-terminus of CT6 are depicted in pink.
Results
This study aimed to investigate the interaction and binding stoichiometry between homodimeric Fcabs with their homodimeric antigen VEGF displaying 2 potential epitopes. Structural and thermodynamic studies would give interesting insights into the loop design, the overall structure of the engineered IgG1-Fc molecule, the binding mode and the interface residues that are involved in the interaction.
Homodimeric Fcabs were derived from a mutated IgG1-Fc library displayed on the surface of yeast cells.Citation14 The library was sorted for binding to VEGF in a manner similar to previous studies,Citation8,Citation11 resulting in the 2 Fcabs 448 and CT6. Both binders contain mutations in the AB (positions 359–361), CD (positions 388–389; including 5 insertions at positions 389a-389e) and EF loops (positions 413–422) of the CH3 domains (Eu numbering systemCitation5). Finally, 8 residues at the C-terminal end (position 440–447) of 448 were randomly mutated to improve its affinity, yielding the Fcab CT6. Thus, CT6 represents an affinity-matured version of 448, with the only sequence difference between these 2 Fcabs being 7 C-terminal amino acids ().
Biophysical and structural characterization of VEGF-binding Fcabs
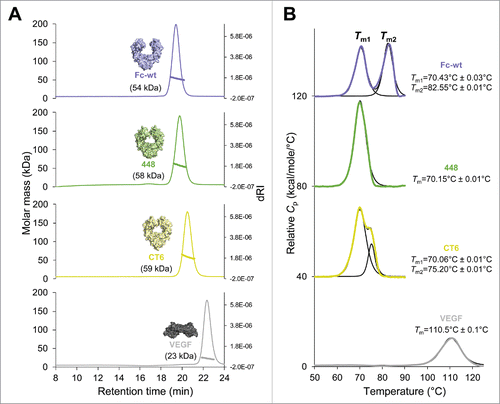
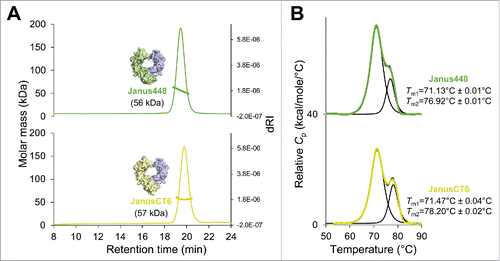
To ensure that the HEK293-expressed Fcabs, as well as E. coli-produced truncated VEGF, were natively folded and stable, they were analyzed by size-exclusion chromatography combined with multi-angle light scattering (SEC-MALS) and differential scanning calorimetry (DSC) (). Evaluation by SEC-MALS revealed no high molecular weight aggregates. Only one symmetric peak was observed for all proteins with molar masses of 54 kDa (Fc-wt), 58 kDa (448), 59 kDa (CT6) and 23 kDa (VEGF) (). Despite the higher molar masses of the 2 Fcabs, their retention times are slightly longer than that of Fc-wt, indicating some nonspecific interaction with the column matrix. Moreover, DSC profiles present the thermal unfolding of the CH2 domains (Tm1) and the CH3 domains (Tm2), which show 2 distinct peaks in Fc-wt with a Tm1 value of 70.4°C and a Tm2 value of 82.6°C, which is consistent with previous studies ().Citation11,Citation12 The Tm1 values of the Fcabs are like wild-type, but, due to the extensive mutations in the CH3 domains of the Fcabs, the Tm2 values are shifted toward lower temperature. The Fcab 448 shows only one main transition with a fitted Tm value of 70.1°C. Interestingly, CT6, having also the C-terminal part mutated, exhibits an increased thermal stability. Its DSC profile clearly shows 2 thermal transitions with an increased Tm2 value of 75.2°C (). The determined Tm value of 110.5°C of recombinant VEGF is in accordance with the published midterm transition value of VEGF measured by DSC.Citation15
Figure 2. Size-exclusion chromatography–multi-angle light scattering (SEC-MALS) and differential scanning calorimetry (DSC) analysis of human IgG1-Fc (Fc-wt), the Fcabs 448 and CT6 and VEGF. (A) A Superdex 200 10/300 GL column (GE Healthcare, USA) pre-equilibrated with PBS plus 200 mM NaCl (pH 7.4) was loaded with 25 µg of Fc-wt (violet), 448 (green), CT6 (yellow) or VEGF (gray), respectively, upstream to MALS analysis. Molar masses were calculated using the ASTRA software. (B) For DSC measurements the Fc proteins and VEGF were diluted to 5 µM and 45 µM in PBS, respectively. The data were fitted to a non-two-state thermal unfolding model using the software Origin 7. The respective Tm values and standard deviations (average of n = 2) are shown next to the graphs.
Structural information on both Fcabs was obtained by crystallization using the sitting drop vapor diffusion method. Crystals of 448 and CT6 had the symmetry of space group P1211 and P22121, respectively (). The highly flexible hinge region of both structures could not be determined, and each chain of the models encompasses residues G236-L443 (448) and G236-W445 (CT6). The electron density is of high quality throughout the structure, besides the loop regions of the CH2 domain of chain A of 448 and chain B of CT6. This is also reflected in higher B-factor values, suggesting higher flexibility of the respective CH2 domain. The CH2 domain of chain B of 448 and chain A of CT6 is stabilized by crystal packing, resulting in lower B-factors and electron density of higher quality. Superimposition of the 2 VEGF-binding Fcabs shows a continuous overlay of the protein backbone and side chain orientation for the CH2 and CH3 domains. Differences can only be seen at the C-terminus, which differs in sequence between these 2 Fcabs, as well as for the N-glycan content at position 297 in the CH2 domain. For each chain, electron density allowed fitting of the typical mammalian-expressed N297-linked pentasaccharide biantennary core (MM) consisting of 2 N-acetylglucosamine and 3 mannose residues in addition to further residues such as β1,2-linked N-acetylglucosamine (GnM) and α1,6-linked fucose (GnMF), depending on the observed electron density. Comparison with the crystal structure of human Fc-wt (Protein Data Bank (PDB) ID: 5JII,Citation12) () revealed differences in the arrangement of the flexible CH2 domains. In contrast to Fc-wt showing a highly open structure of the horseshoe-shaped Fc, the CH2 domains of the Fcabs ( and ) are positioned in a more closed arrangement. This fact could be attributed to quantitative and qualitative variations in the N-glycosylation, as well as altered crystal packing.Citation16-18
Figure 3. Crystal structures of the Fcabs 448 (2.44 Å) and CT6 (2.50 Å) in comparison with human IgG1-Fc (Fc-wt). Cartoon representation of the overall structure of (A) Fc-wt (violet; PDB: 5JII), (B) 448 (green) and (C) CT6 (yellow), each of them rotated by 90° around the vertical axis. (D) Crystal packing of 448 (green) and CT6 (yellow). The 2mFo-DFc density map drawn at RMSD = 1 of the mutated residues in the CD loops of the middle Fcab molecule are depicted as pink mesh. (E) Overlay of the CH3 domains (chain A) of Fc-wt, 448 and CT6. (F) H-bonds (NH-OC and OH-OC, < 3.5 Å) containing mutated regions of CT6 are shown as blue dashed lines. (G) Close-up view of the superimposed CD loop of Fc-wt (violet) and CT6 (yellow) with focus on selected (G) α-helix and (H) β-turns. Blue dashed lines represent important H-bonds (i+3) (I) Network of CT6 residues involved in π-π stacking (< 6.0 Å) is depicted.
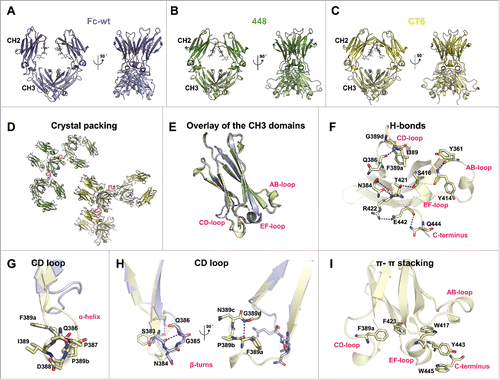
Table 1. Crystallization conditions, data collection and refinement statistics for the Fcabs 448 and CT6 and the JanusCT6–VEGF complex.
After rotating the structures by 90° around the vertical axis ( to ) the elongated CD loops of 448 and CT6 become very conspicuous by protruding to the solvent. Although this loop seems very flexible, enough electron density allows for modeling of all residues in this region. This observation can be attributed to certain crystal packing where the CD loops or parts of the CD loops make crystal contacts with the neighboring molecules (pink mesh in ).
Beside the elongated CD loops, the β-strands of the CH3 domains as well as the backbone of the mutated regions of AB and EF loops of the 2 Fcabs show perfect congruency when superimposed with Fc-wt (PDB: 5JII) (). Due to high structural similarity between the mutated loops of 448 and CT6, the CH3 domain of chain A of the latter was used for illustration of the following findings. Rigidity of the loops was shown to be supported by the formation of novel stabilizing polar bonds (NH-OC and OH-OC, < 3.5 Å) between non-mutated and mutated residues of the AB, EF and CD loops as well as the mutated C-terminus of CT6, namely Y361-Y414, N384-T421, N384-R422, I389-Q386, F389a-G389d, S416-T421, E442-Q444 and E442-R422 (blue dashed lines in ). Moreover, an interesting network of aromatic rings contributing to π-π stacking (< 6.0 Å) that is not present in Fc-wt could be observed. Involved EF and CD loop residues include F389a, F423, W417 and Y443 (present in 448 and CT6), as well as W445 at the mutated C-terminus of CT6 ().
By focusing on the elongated CD loop of 448 and CT6, a right-handed one-turn α-helix stabilized by an H-bond between Q386 and F389a (i+4) can be recognized as indicated by the typical squarish assembly in .Citation19 The non-mutated proline at position P387 was found to be included in the α-helix. Generally, in the rare case where a proline is present within this structural motif, it causes kinks of the α-helical structure.Citation20,Citation21 However, this is not observable in a one-turn helix. Moreover, another proline was selected at position 389b at the end of the short α-helix that is a common feature of this secondary structure motif. This proline is also part of a β-turn (F389a/P389b/N389c/G389d) containing a stabilizing polar bond between F389a and G389d (i+3, yellow structure in ).Citation22 Importantly, this hydrophobic side chain of the phenylalanine at position 389a points toward the hydrophobic core of the protein where it also takes part in π-π stacking as described above. A β-turn is also present in the CD loop of Fc-wt, with the H-bond being located between S383 and Q386 (i+3, violet structure in ).
These structural data suggest that the higher thermal stability of the CH3 domain of CT6 compared with 448 can be assigned to CT6-specific C-terminal residues that exhibit intradomain non-covalent interactions with both CT6-specific residues (E442-Q444, W445-Y443) and the EF-loop (E442-R422). This is also reflected by the fact that, in the CT6 structure (but not in 448), residues 444 and 445 show distinct electron densities.
Formation of Fcab–VEGF polymers
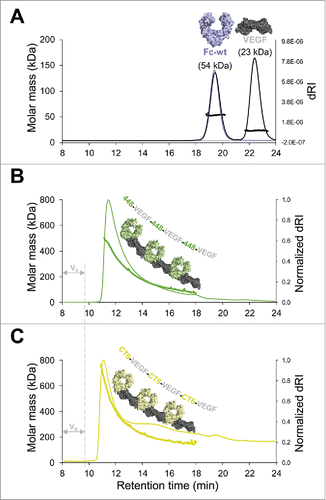
To investigate how homodimeric Fcabs interact with homodimeric VEGF, the 2 proteins were mixed and analyzed by SEC-MALS. As expected, measurements of a 1:3 molar ratio mixture of Fc-wt and VEGF () revealed 2 peaks that were highly similar to the elution profiles of individually analyzed proteins. Also, the molar masses of 54 kDa (Fc-wt) and 23 kDa (VEGF) confirm the absence of binding between Fc-wt and VEGF. In contrast, analysis of 1:1 molar ratio mixtures of Fcabs 448 or CT6 and VEGF revealed no remaining unbound protein, but a tailing peak eluting just after the void volume ( and ). Analysis of molar mass distribution across the eluted fraction shows descending masses of protein complexes starting with ∼500 kDa (448–VEGF, , thick solid line) and ∼760 kDa (CT6–VEGF, , thick solid line), indicating formation of Fcab–VEGF polymers varying in size. In agreement with the MALS signal, the CT6–VEGF polymers also start eluting around 15 sec earlier than 448–VEGF polymers, again suggesting that CT6 is able to form larger complexes (i.e., longer chains) with VEGF compared with 448. Together, these data demonstrate that mixing homodimeric Fcabs with homodimeric VEGF molecules results in the formation of polymeric chains, in which each homodimeric molecule binds to 2 homodimeric interaction partners as schematically represented in and .
Figure 4. Polymerization of Fcab–VEGF complexes. SEC-MALS analysis of Fc protein-VEGF mixtures. (A) Overlay of single measurements of the respective amounts of Fc-wt (violet) and VEGF (gray) that were used for a mixture of Fc-wt and VEGF in a 1:3 molar ratio (black). Calculated molar masses from the mixture run are depicted matching exactly values of the respective single runs. Analysis of 1:1 molar ratio mixtures of (B) 448–VEGF (green) and (C) CT6–VEGF (yellow). Samples elute just after the void volume (V0), referring to high molar masses across the tailing peaks indicating polymerization of the Fcab–VEGF complexes. To facilitate comparison between (B) and (C), the differential refractive index signal (dRI) was normalized. Formation of Fcab–VEGF polymer chains is schematically represented.
Generation and characterization of heterodimeric 2-faced Fcabs (JanusFcabs)
The formation of these Fcab-VEGF polymers prevents isolation of a homogeneous protein fraction, and thus X-ray crystallography studies. To prevent polymerization, the Fcab molecule can be heterodimerized with a second chain encoding the wild-type sequences in the loop regions and at the C-terminus. For generation of a heterodimeric Fc protein, many different strategies resulting in more or less stable proteins have been developed so far. Here, the approach of Spreter Von Kreudenstein et al.Citation23 based on mutations within the CH3/CH3 interface was used for the generation of heterodimeric Fcabs (JanusFcab) composed of one VEGF-binding chain of 448 or CT6 and one Fc-wt chain lacking antigen binding sites.
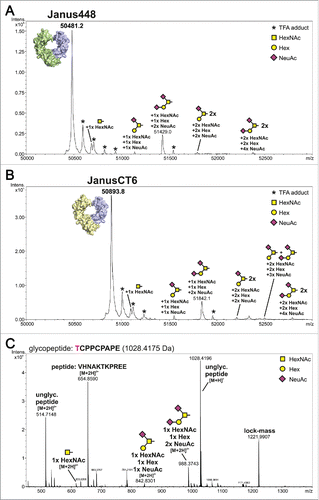
The purity assessment of the heterodimeric samples was done by liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS). For evaluation, the predicted molar masses were determined using the ProtParam tool (http://web.expasy.org/protparam/)Citation24 under consideration of hydrogen release due to disulfide bond formation and PNGaseF digest. Moreover, C-terminal lysine clipping by a carboxypeptidase, which is a common modification in antibody processing, was taken into account.Citation25,Citation26 The theoretical values of 50481.5 Da and 50894.9 Da perfectly match the experimentally determined values of 50481.2 Da (Janus448) and 50893.8 Da (JanusCT6) represented as main peaks in and , respectively. No homodimeric species could be detected, but several small peaks that could be assigned to different O-glycoforms on threonine at position 225 () were observed. Around 10% of this very first amino acid of each Fc chain are occupied with this O-glycosylation. While O-glycosylations are known to be part of antibodies of other classes, IgGs generally only include N-glycans within the CH2 domains. However, Plomp et al.Citation27 reported a partial O-glycosylation in the hinge region of human IgG3 samples derived from various sources. Moreover, O-glycans were also found on Fc constructs of IgG4 and IgG1.Citation16,Citation27 In a full-length IgG1 the accessibility for glycosyltransferases is limited, but the HEK293-produced Fc proteins can be occupied more easily with an O-linked glycan in the N-terminal hinge region. The major O-glycan species found on T225 was a core 1 structure carrying either one or 2 additional N-acetylneuraminic acid residues ().
Figure 5. Intact protein measurement of heterodimeric Fcabs and identification of O-glycosylation site using LC-ESI-MS. The deconvoluted spectra of Janus488 (A) and JanusCT6 (B) after PNGaseF treatment are presented. The theoretical molar mass of the proteins (Janus448: 50481.5 Da; JanusCT6: 50894.9 Da) could be matched to the highest signal of the respective measurement. Several glycoforms of the proteins caused by O-glycosylation were identified and quantified to a relative amount of approximately 10%. The small portion of TFA adducts (marked with an asterisk) derives from the addition of 0.05% of TFA in solvent A. (C) Glycopeptide TCPPCPAPE was identified after endoproteinase Glu-C digestion of Fc-wt. The unglycosylated peptide in 2 different charge states and its observed glycoforms are depicted in the spectrum. The site of O-glycosylation is located on position T225. The signal of m/z = 654.86 could be assigned to a co-eluting peptide. The peak of the lock-mass refers to an internal standard and was used for recalibration.
Additionally, evaluation by SEC-MALS revealed no high molecular weight aggregates, but only one symmetric peak shifted to shorter retention times and slightly smaller molar masses of 56 kDa (Janus448) and 57 kDa (JanusCT6) compared with their respective homodimeric versions (compare ). Analysis of the endothermic transitions showed an increase in thermostability despite the additional heterodimerization mutations that were introduced in the CH3 domains (). In contrast to 448, which showed one endothermic transition, its heterodimeric version Janus448 shows 2 distinct peaks with a significant increase of Tm2 to 76.9°C. In addition, the Tm2 value of JanusCT6 was increased by 3°C compared with its homodimeric version CT6 (compare and ). Surprisingly, for both JanusFcabs, the respective Tm1 values, representing unfolding of the non-mutated CH2 domains, were also slightly increased, exceeding even the Tm1 value of Fc-wt.
Figure 6. SEC-MALS and DSC analysis of the heterodimeric Fcabs Janus448 and JanusCT6. (A) A total amount of 25 µg of Janus448 (green) and JanusCT6 (yellow) was analyzed on a Superdex 200 10/300 GL column (GE Healthcare, USA) pre-equilibrated with PBS plus 200 mM NaCl (pH 7.4) before MALS measurements. Molar masses were calculated using the ASTRA software. (B) For DSC measurements the heterodimeric Fcabs were diluted to 5 µM in PBS. The data were fitted to a non-two-state thermal unfolding model using the software Origin 7. The respective Tm values and standard deviations (average of n = 2) are shown next to the graphs.
JanusFcabs enable biophysical and thermodynamic investigation of the interaction with VEGF
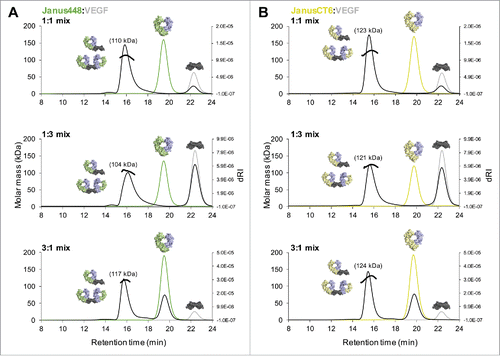
By mixing VEGF with the respective JanusFcab, a binding stoichiometry of 1:2 should be obtained from binding of 2 heterodimeric Fcabs (only containing one VEGF-binding site) to one homodimeric VEGF molecule (containing 2 epitopes). To investigate the molar mass of the complex, SEC-MALS measurements were performed mixing different molar ratios of VEGF and either Janus448 () or JanusCT6 (). No protein fraction was detected right after the void volume, but main peaks with molar masses between 104 kDa and 117 kDa for Janus448–VEGF mixtures and molar masses between 121 kDa and 124 kDa for JanusCT6–VEGF mixtures were observed, suggesting JanusFcab–VEGF complexes revealing mainly a 2:1 (theoretical molar mass of ∼136 kDa) but also 1:1 (theoretical molar mass of ∼80 kDa) binding stoichiometry. The overlay of single measurements of the respective amounts of the interaction partners that were mixed for the complex run fits to the remaining amount of the unbound interaction partner that was added in excess. For example, the complex run of the 1:1 molar ratio mixtures reveals, beside the JanusFcab–VEGF complex peak, no unbound JanusFcab but a small peak corresponding to unbound VEGF, consistent with the formation of 1:2 and 1:1 complexes, resulting in some unoccupied VEGF molecules. Moreover, mixing JanusFcabs and VEGF in molar ratios of 1:1 or 1:3 results in peak tailing, suggesting that, in the absence of a molar excess of JanusFcab, a certain fraction of the formed complexes will show a molar ratio of 1:1. Of note, higher molar masses of the complex were observed with JanusCT6 as interaction partner ( vs. ), suggesting that more complexes are fully saturated (i.e., 1:2) due to stronger binding affinity.
Figure 7. Preventing polymerization by application of heterodimeric Fcabs. SEC-MALS analysis of heterodimeric Fcab-VEGF mixtures of different molar ratios (1:1/1:3/3:1) as indicated in the respective graphs. (A) Overlay of single measurements of the respective amounts of Janus448 (green) and VEGF (gray) that were used for the mixtures of the respective complex run (black). (B) Overlay of single measurements of the respective amounts of JanusCT6 (yellow) and VEGF (gray) that were used for the respective complex run (black). Molar masses of the main peaks of each complex run are depicted.
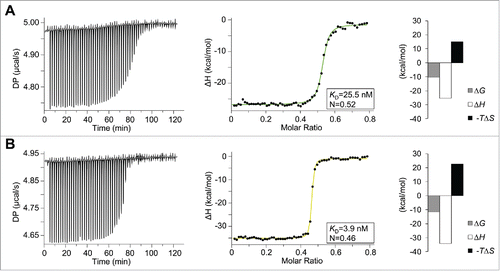
To pursue investigation of binding affinity and mode of interaction, isothermal titration calorimetry (ITC) experiments were performed. By titrating VEGF to the heterodimeric Fcab in the calorimetric cell, an N value of 0.5 clearly indicates a binding stoichiometry of 2:1 for both JanusFcabs–VEGF complexes, confirming the SEC-MALS data (). The steepness of the transition curves obtained by fitting the integrated data to a one-set-of-sites model yields binding affinities for Janus448 (KD = 26 nM, ) and JanusCT6 (KD = 4 nM, ). Both signature plots show a very similar mode of interaction. Binding of both heterodimeric Fcabs to VEGF is enthalpically favorable with ΔH values of −25 kcal mol−1 and −34 kcal mol−1 for Janus448 and JanusCT6, respectively, but entropically unfavorable with -TΔS values of 15 kcal mol−1 (Janus448) and 23 kcal mol−1 (JanusCT6), respectively.
Figure 8. Thermodynamic analyses of the interactions between VEGF and Janus448 or JanusCT6. Calorimetric experiments were performed on a MicroCal PEAQ-ITC instrument at 25°C. The sample cell (V = 200 µL) was either filled with 40 µM Janus448 (A) or JanusCT6 (B), and titrated stepwise with 200 µM VEGF. The left panels show the raw ITC data representing the responses to 59 successive injections of 0.5 µL each. The integrated data were fitted to a one-set-of-sites interaction model (right panels). The insets and the binding signature plots show the fitted and thermodynamic parameters of the respective JanusFcab–VEGF interaction.
Crystal structure of the JanusCT6–VEGF complex at 2.15 Å resolution
For crystallization of the JanusCT6–VEGF complex, the heterodimeric Fcab was mixed with an excess of VEGF and the complex was isolated using preparative SEC. The technique of microseeding was used to gain diffracting crystals with the best having the symmetry of space group P212121 and a resolution of 2.15 Å. One asymmetric unit was composed of 2 JanusCT6 molecules (JanusCT6-A and JanusCT6-B) binding to one homodimeric VEGF molecule (). Due to crystal packing, higher B-factors were observed for the CD loop of the VEGF-binding CT6 chain of JanusCT6-A and the CH2 domain of the non-binding Fc-wt chain of JanusCT6-B.
Figure 9. Crystal structure of the JanusCT6–VEGF complex at 2.15 (Å) resolution (PDB: 5O4E). (A) Surface and cartoon representation of the JanusCT6–VEGF complex. Each of the 2 CT6 chains (yellow) of JanusCT6-A and JanusCT6-B is binding to the opposite poles of the homodimeric VEGF molecule (gray). The non-binding Fc-wt chains are colored in violet. (B) Close-up view on heterodimer mutations in the CH3 domains interface of JanusCT6 (yellow and violet) and the heterodimeric Fc variant (violet, PDB: 4BSW)Citation23 in comparison to the respective residues found at the interface of homodimeric Fc-wt (PDB: 5JII).Citation12
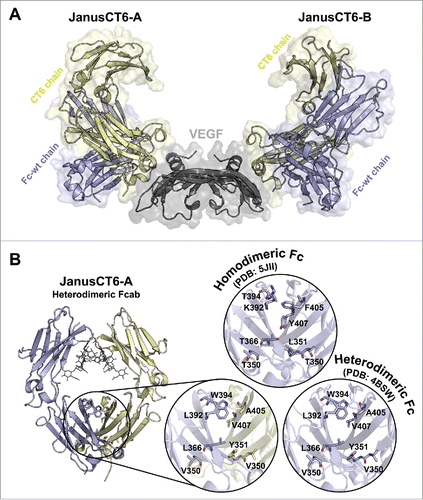
When comparing the 8 heterodimer mutations within the interface of the CH3 domains of JanusCT6 with the original structure of the heterodimeric Fc (PDB: 4BSW), no major differences in side chain orientations can be observed (). Since the heterodimer mutations are located in the interior of the protein, they do not contribute to the crystallization behavior, thus one heterodimeric molecule can be incorporated into the crystal lattice in 2 different orientations (i.e., 0° and 180° rotated around its vertical axis). In short, crystal formation of the heterodimeric Fc (PDB:4BSW) proceeded analogous to the homodimeric Fc-wt protein. As a consequence, both orientations are found to the same extent, and therefore an occupancy of 0.5 is assigned to every amino acid residue. shows the relevant side chains of heterodimeric JanusCT6-A in comparison to one of the 2 orientations of heterodimeric Fc (PDB: 4BSW)Citation23 and to the respective residues found at the interface of homodimeric Fc-wt (PDB: 5JII).Citation12
The CT6 chains of the 2 JanusCT6 molecules bind to opposite parts of the VEGF dimer. The epitope mainly comprises the helix α1 and the connecting loops of β-strands forming the characteristic cysteine knot motif of VEGF.Citation28 Superimposition of homodimeric CT6 with JanusCT6-A and JanusCT6-B revealed no differences in the overall structure beside minor conformational changes in the flexible CD loop ().
Figure 10. Insights into JanusCT6–VEGF interaction. (A) Superimposition of the backbone structure of CT6 (black), JanusCT6-A (yellow and violet) and JanusCT6-B (shades of pink) with close-up view on the CH3 domains of the VEGF-binding CT6 chains. (B, C) Non-covalent interactions between JanusCT6-A (yellow) and VEGF (gray) were identified by help of the RING 2.0 software tool using strict threshold options.Citation29 (B) Van der Waals interactions identified by measuring the distance between the surface of atoms are depicted as black dashed lines. Surface of involved amino acids is represented as spheres. (C) Side chains of aromatic amino acids involved in π-π stacking evaluated below 6.5 Å between the center of mass are shown. H-bonds found between JanusCT6-A and VEGF (yellow and gray) (D) and JanusCT6-B and VEGF (pink and gray) (E) are depicted as blue dashed lines. Bridging water molecules are shown as red spheres.
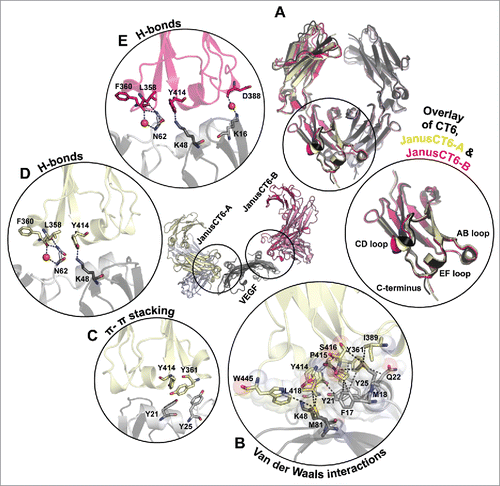
Analysis of residues involved in the binding interface was performed by means of the RING 2.0 web server.Citation29 This tool generates a residue interaction network based on different types of non-covalent interactions. The binding site of JanusCT6-A and JanusCT6-B is stabilized by a network of van der Waals interactions involving residues from all mutated loop regions and the C-terminus of JanusCT6 as well as both chains of VEGF (shown for the JanusCT6-A–VEGF complex, ). Moreover, π-π stacking interaction between Y361 (AB loop of JanusCT6) and Y25 (VEGF), as well as Y414 (EF loop of JanusCT6) and Y21 (VEGF), contribute to the stabilization of the binding interface ().
The resolution of 2.15 Å of the JanusCT6-VEGF complex is high enough to determine several water molecules. Only one could be found to be conserved in both binding sites. This water molecule is responsible for bridging the backbone amide groups of L358 (AB loop) and N62 (VEGF) ( and ). Interestingly, only in the binding site of JanusCT6-B, but not in that of JanusCT6-A, an H-bond between D388 (CD-loop) and K16 (VEGF) is formed via a bridging water molecule (). Since D388 is located in the CD loop, this H-bond might also account for the slightly different structures in the CD loops of JanusCT6-B vs. JanusCT6-A.
Discussion
The Fcab is a promising protein scaffold for therapeutic applications because it combines all antibody functions, including antigen binding, in a protein of only one-third of the mass of a full-length antibody. As with any other engineered protein, immunogenicity is a potential risk. However, the IgG1-Fc itself is a human protein and the non-human sequences are limited to specific loop regions only. In this regard, it is worth mentioning that even approved humanized antibodies contain approximately 15–20% non-human sequences in their variable domains, caused, for example, by grafting of CDR loops from murine antibodies or framework mutations needed for optimized affinity or stability.Citation30
The crystal structures of the HER2-binding Fcabs H10–03–6 and its stabilized version STAB19 both unbound and in complex with the extracellular domain of HER2 were recently described. These complex structures, as well as accompanying biochemical solution studies, revealed a stoichiometry of 1:2, with one Fcab binding to 2 antigen molecules following a negative cooperative binding mode.Citation12 That is, the second antigen molecule binds with lower affinity compared with the first molecule.
Given the close proximity of the 2 binding sites in the Fcab molecule, it was not guaranteed that both binding sites can be occupied simultaneously. To test if this is a more general property of Fcabs, we aimed at investigating the interaction between VEGF and its specific Fcabs 448 and CT6, which differs from the above mentioned HER2-Fcab interaction with regard to 3 properties: 1) in contrast to HER2, VEGF is a small, homodimeric molecule, potentially resulting in the formation of 1:1, 1:2 or 2:1 complexes, or even high molecular weight polymers; 2) the potential binding surface at the C-terminal loops of the CH3 domains was increased by additionally mutating the CD loops (448 and CT6) and also the C-termini (CT6 only); and 3) the VEGF binders in the present study were engineered to contain 5 insertions in the CD loops, whereas the HER2-binding Fcabs contain insertions in the EF loops. In more detail, 3 residues in the AB loop, 7 residues in the CD loop ( 5 of them being insertions) and 10 residues in the EF loop were randomly mutated for selection of VEGF binding. The affinity-matured Fcab CT6 contains additional 8 mutations at the C-terminal end.
The high level of mutations in the CH3 domain that had to be introduced to generate VEGF-binding Fcabs destabilized those domains in these molecules, as shown by DSC. Similar destabilizing effects have previously been observed with particular Fcabs containing a high content of mutations.Citation11,Citation31 Nevertheless, both Fcabs showed monomeric peaks when analyzed by SEC-MALS, demonstrating that they are natively folded and resistant to aggregation at the applied concentration. Only the elution times were slightly extended compared with Fc-wt. This fact could be attributed to the higher flexibility of the elongated CD loop and the significantly higher content of hydrophobic residues in the mutated loop regions, potentially causing non-specific interactions with the column matrix.
In line with the SEC-MALS data, the crystal structures of both VEGF-binding Fcabs clearly show that these engineered molecules largely retain their native structure, as was also seen with the HER2-binding Fcabs H10–03–6 and STAB19,Citation12 again confirming that the IgG1-Fc protein is highly tolerant to mutation in its C-terminal loop regions. Although the AB and EF loops of both Fcabs were mutated, the backbone structures in these loop regions were highly congruent with that of Fc-wt. Only the CD loops show an altered backbone conformation compared with Fc-wt. This observation is not surprising because the CD loops contain an insertion of 5 amino acids. While the CD loops of Fc-wt only comprise one β-turn as a secondary structure element, the elongated CD loops of both VEGF-binding Fcabs contain a newly formed one-turn α-helix, followed by a newly formed β-turn. This is a striking similarity with the previously published structure of the HER2-binding Fcab STAB19, in which a pre-existing α-helix in the EF loops is elongated by the inserted mutations.Citation12 Together, these structural data indicate that, in the case of insertions, the additional amino acids need to be incorporated by forming stable secondary structural motifs to prevent unstructured regions that protrude into the solvent. At least for small insertions in loop regions, an α-helix seems to be an appropriate option. This hypothesis is in line with a previous report, showing that insertions in AB, CD or EF loops strongly reduce the average thermostability of yeast-displayed Fcab libraries.Citation32 Thus, the formation of stable structural elements in the newly inserted regions seems to be one of the driving selection forces in the directed evolution experiment when using yeast surface display, which is known to simultaneously select not only for antigen binding, but also for expressibility and stability.Citation33,Citation34 In other words, parts of the randomly mutated Fcab libraries are probably misfolded, especially in the inserted regions, and the eukaryotic quality control machinery during protein expression in yeast ensures that only mutants with stabilized inserted regions are efficiently displayed on the surface, and are therefore available for selection.
In addition, the interaction between Fcabs 448 and CT6 and their antigen VEGF was examined. SEC-MALS data clearly revealed polymer formation of Fcabs and VEGF, which can be explained by the homodimeric nature of both interaction partners (i.e., Fcab and VEGF). Consistent with the higher affinity of CT6, the molar masses of the CT6-containing polymers were higher compared with the 448-containing polymers. Thus, the higher affinity seems to enable the formation of longer polymer chains. These results again confirm that Fcabs can potentially bind 2 antigen molecules simultaneously, as has already been shown for the HER2-specific Fcabs.Citation12
Since the formation of polymer prevented further investigation of the interaction with VEGF, heterodimeric Fcabs that only contain one binding site were constructed. The design of heterodimeric Fcabs (Janus448 and JanusCT6) consisting of a non-binding Fc-wt chain and a VEGF-binding Fcab chain followed the procedure of heterodimeric Fc protein generation performed by Spreter Von Kreudenstein et al.Citation23 This strategy is based on a combination of negative design (prevention of Fc homodimers) and positive design (enhancement of stability of Fc heterodimers) using an in silico approach and in vitro analysis. While the negative mutations F405A and Y407V (chain A) combined with T366L and T394W (chain B) destroy important interface hotspots for homodimerization and promote heterodimerization, the positive mutations L351Y, T350V (chain A), K392L and T350V (chain B) significantly increase the Tm value of the CH3 domains. The final best performing candidate exhibited a Tm2 value of 81.5°C and heterodimer purity above 95% without detectable homodimeric contaminates, but a small portion of unpaired monomeric chains.Citation23 Consistent with these published results, similar purities of the heterodimeric Fcabs Janus448 and JanusCT6 with only minor monomeric contaminations were observed, which could be separated by SEC. Interestingly, while the heterodimerizing mutations slightly decreased the thermostability of the CH3 domains of Fc-wt by 0.6°C,Citation23 the stabilities of the CH3 domains of the JanusFcabs were markedly increased compared with their corresponding homodimeric versions. This strongly suggests that the 2 different CH3 domains do not denature independently at low and high temperatures, respectively, but together at an intermediate temperature in between the Tm values of Fc-wt and the respective Fcab.
SEC-MALS analysis of JanusFcab-VEGF mixtures confirmed the formation of 2:1 complexes. In general, higher affinities (CT6 vs. 448) and higher excess of JanusFcabs resulted in higher molar masses of the complex peaks, indicating that the complex peaks are composed of 1:1 and 2:1 complexes. ITC analysis of the interaction between both JanusFcabs and VEGF revealed thermodynamic parameters of the interactions in solution. CT6 (KD = 3.9 nM; ΔG = −11.5 kcal mol−1) exhibits a more than 6-times higher affinity compared with 448 (KD = 25.5 nM; ΔG −10.4 kcal mol−1). With both JanusFcabs, binding to VEGF is enthalpically favorable. This effect is more pronounced with JanusCT6, reflecting a higher number of established non-covalent interactions of JanusCT6 or CT6 with VEGF. However, this is accompanied with an increased entropic penalty, suggesting the presence of flexible structural regions in the unbound molecules, which become structurally more restricted in the JanusCT6-VEGF complex.Citation35,Citation36
The increase in binding affinity of JanusCT6 can be attributed to the mutated C-terminal region, which represents the only difference in sequence between these 2 Fcabs. In line with ITC data, the X-ray structure of the JanusCT6–VEGF complex revealed contributions from van der Waals interactions and H-bonding in addition to π-π stacking. More electron density at the C-terminus of CT6 allowed fitting of more residues within this region, suggesting that especially the aromatic amino acids (Y443 and W445) not only contribute to overall stability (π-π stacking), but also to binding affinity. At least W445 was shown to be involved in van der Waals interactions with VEGF. In addition, the intramolecular π-π stacking interaction of W445, which is only present in CT6 but not in 448, might also explain why CT6 is slightly more stable than 448.
Moreover, the complex structure revealed the location of the epitopes positioned at each pole of the homodimeric VEGF molecule. In general, hot spots on VEGF-A for binding to its natural receptor VEGFR-1 (PDB: 5T89)Citation37 and VEGFR-2 (PDB: 3V2A)Citation38 include β-strands connecting loops L1, L2 and L3 and the N-terminal helix α1. A similar but smaller epitope is occupied by 2 clinically approved drugs, the antibody bevacizumab (PDB: 1BJ1)Citation39 and the Fab ranibizumab (PDB: 1CZ8),Citation40 which is an affinity-matured version of the first. JanusCT6 only marginally overlaps with the epitopes of bevacizumab and ranibizumab. All these crystal structures contain a truncated version of VEGF-A encompassing only the important receptor binding domain (∼residues 14–108). However, due to mRNA splicing, 8 isoforms of human VEGF-A are described comprising 121 to 206 amino acids in length.Citation41 Since the epitope of JanusCT6 also covers residues at the C-terminal part of VEGF, binding to different isoforms could indeed lead to changes in affinity due to steric hindrance depending on the amino acid elongation in the respective isoform. The main contributions to VEGF binding derive from mutations within the AB and EF loops. Interestingly, the elongated CD loop is, beside its flexibility as shown when overlaying CT6 with JanusCT6-A and JanusCT6-B, only marginally involved in the interaction with VEGF. This becomes obvious by looking at the cartoon representation of the entire JanusCT6–VEGF complex, where it is located on top of the binding interface, but hardly interacts with the antigen ().
Together, this study demonstrates that homodimeric, bivalent Fcabs can simultaneously interact with 2 antigen molecules. In the case of homodimeric soluble antigens, also comprising 2 epitopes, this potentially leads to the formation of long polymers composed of alternating Fcab and antigen molecules. Since this effect prevented determination of the crystal structure of the Fcab–antigen complex, we established heterodimeric Fcabs that only contain one binding site. This study demonstrates that heterodimeric, monovalent Fcabs enable detailed structural and thermodynamic analysis of their interaction with homodimeric antigens, which is impossible with homodimeric, bivalent Fcabs, nicely demonstrating the great utility of heterodimeric, monovalent Fcabs or even other antibody constructs for structural and biochemical studies with their bivalent antigens.
Material and methods
Recombinant expression and purification of Fc-wt, Fcabs (448, CT6) and heterodimeric Fcabs (Janus448, JanusCT6)
Expression and purification of Fc proteins was conducted as described previously.Citation12 In short, HEK293–6E suspension cells licensed from National Research Council (NRC)Citation42 of Canada were transiently transfected with plasmid preparations of the pTT5 vector (NRC, Canada) containing the coding sequence for either the Fcab clone 448 or CT6 (generous gift of G. Wozniak-Knopp, University for Natural Resources and Life Sciences Vienna, Austria) or Fc-wt. For generation of the heterodimeric Fcabs (Janus448 and JanusCT6) mutations in the CH3 domain [448/CT6 (chain A): T350V/L351Y/F405A/Y407V and Fc-wt (chain B): T350V/T366L/K392L/T394W] were introduced via site-directed mutagenesis using the QuikChange Lightning kit (Agilent, USA).Citation23 For transient transfection, a total of 1 µg of plasmid-DNA and 2.5 µg of linear polyethylenimine (Polysciences, Inc., Germany) per mL culture volume were used. For production of heterodimeric Fcabs, the plasmid-DNA transfection ratio of 5:1 (chain A: chain B) has shown to perform best in terms of high protein yield. Harvesting and purification of the soluble protein was done exactly as described in Lobner et al., including purification steps with a HiTrap Protein A HP column (GE Healthcare, USA) and a HiLoad 16/600 Superdex 200 pg (prep grade) column (GE Healthcare, USA).Citation12,Citation43 The latter purification step was necessary for separation of heterodimeric Fcabs from the small portion of solitary monomeric Fc chains.
Recombinant expression and purification of VEGF
The gene encoding for residues 14–108 of human VEGF-A (UniProtKB: P15692) was synthesized and cloned into pJ414 vector, which carries the inducible T7 promotor (ATUM, USA). The plasmid was transformed into chemically competent E. coli BL21(DE3) cells (Agilent, USA). Expression in inclusion bodies, refolding and purification of the truncated version of VEGF was performed based on the protocol of Heiring and Muller.Citation44,Citation45 Fresh LB medium (8 × 500 mL) supplemented with ampicillin (100 µg mL−1) was inoculated in shake flasks with 5 mL of an overnight culture, respectively. After the culture has reached an optical density of 0.6 at 600 nm at 37°C shaking, overexpression was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and the cells were incubated for another 4 h at 37°C. Cells were harvested by centrifugation (5000 g, 20 min, 4°C), resuspended in 8 × 20 mL of 20 mM TRIS-HCl buffer (pH 7.5) including 5 mM ethylenediaminetetraacetic acid (EDTA), incubated for 10 min and disrupted by ultrasonication with a Vibra-Cell 375 ultrasonic processor (Sonics & Materials, Inc., USA). Pellets obtained after centrifugation (39000 g, 25 min, 4°C) were pooled and washed twice using the same buffer as before. The resulting pellet was subsequently resuspended in 20 mL unfolding buffer (20 mM TRIS-HCl, 5 mM EDTA, 7.5 M urea, 4 mM dithiothreitol (DTT), pH 7.5), stirred for 2 h at room temperature followed by centrifugation (39 000 g, 25 min, 4°C). The supernatant containing the unfolded protein was diluted 10-fold into refolding buffer (20 mM TRIS-HCl, 7 µM CuCl2, pH 8.4) and stirred overnight at room temperature. After dialysis against 20 mM TRIS-HCl (pH 8.0), the protein was loaded onto a 6 mL Resource Q column (GE Healthcare, USA). The protein was eluted by applying a linear gradient from 0 to 500 mM NaCl over 20 column volumes. Fractions containing VEGF were pooled, dialyzed in 50 mM sodium phosphate buffer (pH 7.0) containing 1.2 M (NH4)SO4 and purified using a 1 mL HiTrap Phenyl FF (Low Sub) column (GE Healthcare, USA). The protein was eluted by a linear decrease of the (NH4)2SO4 concentration over 20 column volumes. Before purification with a HiLoad 16/600 Superdex 200 pg (prep grade) column (GE Healthcare, USA), the sample was concentrated and buffer-exchanged into Dulbecco's Phosphate Buffered Saline (PBS, pH 7.4) (Sigma-Aldrich, USA) supplemented with 200 mM NaCl using 10 kDa Amicon Ultra Centrifugal Filter (Merck Millipore, Germany).
Differential scanning calorimetry (DSC)
Analyses of thermal stabilities were performed on a MicroCal VP-Capillary DSC instrument (Malvern Instruments, Ltd., UK). All Fc proteins were diluted to a concentration of 5 µM in PBS buffer (pH 7.4) and heated from 20 to 110°C. The highly thermostable VEGF was diluted to a concentration of 45 µM in PBS buffer (pH 7.4) and heated from 20 to 130°C. All experiments were conducted at a constant heating rate of 1°C min−1. After subtraction of buffer baselines, the data were normalized for protein concentration and fitted with a non-two-state thermal unfolding model using the software Origin 7.
Size-exclusion chromatography-multi-angle light scattering
SEC-MALS was used to determine the molar mass of all proteins and protein complexes under study. Analyses were performed on an LC20 prominence HPLC system equipped with the refractive index detector RID-10A, the photodiode array detector SPD-M20A (all from Shimadzu, Japan) and a MALS Heleos Dawn8+ plus QELS detector (Wyatt Technology, USA). The column (Superdex 200 10/300 GL, GE Healthcare, USA) was equilibrated with PBS plus 200 mM NaCl (pH 7.4) as running buffer. Experiments were performed at a flow rate of 0.75 mL min−1 at 25°C and analyzed using the ASTRA 6 software (Wyatt Technology, USA). Proper performance of molar mass calculation by MALS was verified by the determination of a sample of bovine serum albumin. All proteins were centrifuged (17 000 g, 10 min, 20°C) and filtered (0.1 μm Ultrafree-MC filter, Merck Millipore, Germany) before analysis. A total amount of 25 µg was injected for all single protein measurements. For the complex runs consisting of 1:1 molar ratio mixtures of the respective Fc molecule and VEGF, 25 µg of the antigen were mixed with the appropriate amounts of Fc protein before analysis. For runs at molar ratios of 1:3 and 3:1, 25 µg of the interaction partner present at lower molar concentration was used.
Protein identification using liquid chromatography-electrospray ionization-mass spectrometry
For protein identification and quantification, 30 µg of the respective SEC purified heterodimeric Fcab (Janus448 and JanusCT6) in PBS was treated with 1 U of N-Glycosidase F (Roche, Switzerland) to release N-linked glycans overnight at 37°C. A total of 2.5 µg of the sample was analyzed using a Dionex Ultimate 3000 HPLC-system (Thermo Scientific, USA) directly linked to a QTOF instrument (maXis 4G ETD, Bruker, Germany) equipped with the standard ESI source in the positive ion mode. MS-scans were recorded within a range of 400–3800 m/z. Instrument calibration was performed using ESIcalibration mixture (Agilent, USA). For separation of the proteins, a Thermo ProSwift™ RP-4H Analytical separation column (250 * 0.200 mm) was used. A gradient from 80% solvent A and 20% solvent B (Solvent A: 0.05% trifluoroacetic acid (TFA); B: 80.00% acetonitrile and 20.00% Solvent A) to 62.5% B over 15 min was applied, followed by a 5 min gradient from 62.5% B to 95% B, at a flow rate of 8 µL min−1 and 65°C. The analysis files were deconvoluted using DataAnalysis 4.0 (Bruker, Germany) (Maximum Entropy Method, low mass: 10000, high mass: 100000, instrument resolv. power: 10000) and manually annotated.
O-glycopeptide profiling of T225 using liquid chromatography-electrospray ionization-mass spectrometry
For localization of the O-glycosylation site, the same instruments were used as for protein identification. Therefore, a sample of 20 µg Fc-wt in PBS was S-alkylated with iodoacetamide and further digested with sequencing grade Glu-C (Roche, Switzerland). Measurements were performed as described previously.Citation12 In short, MS-scans were recorded within a range of 150–2200 m/z and the 3 highest peaks were selected for fragmentation in DDA mode. For peptide separation, a Thermo BioBasic C18 separation column (Thermo Scientific, USA) was used. DataAnalysis 4.0 (Bruker, Germany) was used for glycopeptide evaluation. The site of glycosylation (T225) was determined by detection of the glycosylated peptide TCPPCPAPE that only contains a single threonine residue and no serine. Likewise, a tryptic digest with sequencing-grade modified trypsin (Promega, USA) to confirm the findings resulted in the identically glycosylated peptide TCPPCPAPELLGGPSVFLFPPKPK (data not shown).
Isothermal titration calorimetry
ITC measurements were conducted on a MicroCal PEAQ-ITC (Malvern Instruments, Ltd., UK) for determination of binding affinities and thermodynamics of the interaction between the heterodimeric Fcabs and VEGF. Prior to measurements, the samples were prepared in PBS (pH 7.4), centrifuged (17,000 g, 10 min, 20°C) and filtered (0.1 μm Ultrafree-MC filter, Merck Millipore, Germany). The sample cell (V = 200 µL) was washed several times with PBS (pH 7.4) before filling with 40 µM of either Janus448 or JanusCT6. Titrations with 200 µM VEGF were performed at 25°C using an initial injection of 0.1 µL followed by 59 successive injections of 0.5 µL with a 120 sec interval in between. Curve fitting was performed based on a one-set-of-sites binding model using the MicoCal PEAQ-ITC analysis software.
X-ray crystallography and data collection
To provide the best preconditions for X-ray crystallography studies, all proteins were freshly produced and purified with SEC as final polishing step. Fractions in PBS buffer (pH 7.4) containing either 448 or CT6 were pooled and concentrated using 30 kDa Amicon Ultra Centrifugal Filter (Merck Millipore, Germany) to 7.3 mg mL−1and 4.9 mg mL−1, respectively. Crystallization experiments were performed using the sitting drop vapor diffusion method. Crystallization drops were set using a Phoenix HT robot (Art Robins Instruments, USA). The reservoir was filled with 40 µL precipitant solution. Ratios of 150:200 nL, 200:200 nL and 250:200 nL protein to precipitant were dispensed.
For isolation of the JanusCT6–VEGF complex, the heterodimeric Fcab was mixed with an 3-fold molar excess of VEGF and loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare, USA) equilibrated with 20 mM TRIS-HCl buffer (pH 7.5) including 50 mM NaCl. Fractions containing the complex were desalted and concentrated to 5.5 mg mL−1 using 30 kDa Amicon Ultra Centrifugal Filter (Merck Millipore, Germany). Growth of high quality complex crystals was achieved by applying the technique of microseeding, which promotes growth of larger crystals in the metastable zone by providing nucleation seeds. Therefore, a seeding stock solution was prepared from tiny needle-shaped crystals of the JanusCT6–VEGF complex obtained from Morpheus® screen (condition F2) (Molecular Dimensions, UK) using the Seed Bead™ kit (Hampton Research, USA). The crystals were placed into 100 µL crystallization solution, crushed and transferred into a Seed Bead™ tube. The tube was vortexed 3 × 30 s cooling it on ice between the mixing steps. Drops were set using an Oryx8 crystallization robot (Douglas Instruments, Ltd, UK). The reservoir was filled with 40 µL precipitant solution. Ratios of 200:200:100 nL and 300:200:100 nL protein to precipitant to seeding stock were dispensed.
Crystallization plates were stored in a Minstrel DT UV imaging device (Rigaku, USA) at 22°C. Diffracting crystals were grown in a slightly modified version of the Low Ionic Strength screen (Hampton Research, USA) based on Harris et al.Citation46 (448), the Morpheus® screen (CT6) and the JCSG-plus™ screen (JanusCT6–VEGF complex) (both from Molecular Dimensions, UK). Crystals of 448 were soaked with mother liquor supplemented with 20–25% (v/v) glycerol as cryoprotectant. All crystals were harvested using cryo-loops and flash-cooled in liquid nitrogen. Data sets were collected at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) at 100 K on a DECTRIS PILATUS 2M detector ().
Structure determination and refinement of 448, CT6 and the JanusCT6–VEGF complex
Data sets were integrated and scaled with the XDS software-suite.Citation47 The high-resolution cut-off was based on a CC1/2* criterion.Citation48 The structures of 448 and CT6 were determined by molecular replacement with the program Phaser-MRCitation49 using an unpublished Fcab structure as search model for 448 and the structure of 448 as search model for CT6. For determination of the JanusCT6−VEGF complex structure, the phase problem was solved by molecular replacement applying BALBES using Fc-wt as sequence target.Citation50 The initial model was rebuilt using phenix.autobuild providing the sequence of VEGF and JanusCT6.Citation51
The models were improved by iterative cycles of manual model building using COOTCitation52 and maximum likelihood refinement using PHENIX-Refine,Citation53 REFMAC5Citation54 and Rosetta-Refinement.Citation55 Final stages of refinement included Translation Libration Screw (TLS) parameters, isotropic B-factor model, automated correction of N/Q/H errors, automated addition of hydrogens and water molecules and optimization of X-ray/ADP- and X-ray/stereochemistry-weight. Details can be found in .
Validation of the structures
All models were validated with MolProbityCitation56 and PDB_REDO.Citation57,Citation58 Carbohydrates were validated with pdb-careCitation59 and Privateer.Citation60 Figures were prepared with PyMOL Molecular Graphics System (Version 1.3, Schrödinger, LLC). Atomic coordinates have been deposited in the Protein Data Bank under accession codes 5K64 (448), 5K65 (CT6) and 5O4E (JanusCT6–VEGF).
Author contribution
Conceptualization, E.L. and C.O.; Formal Analysis, E.L., A.H., G.M., K.K. and M.K.; Investigation, E.L., A.H., G.M., K.K. and M.K.; Writing – Original Draft, E.L., C.O., M.W.T., G.M. and A.H.; Writing – Review & Editing, E.L., C.O. and M.W.T.; Supervision, C.O. and K.D.-C.; Funding Acquisition, C.O.
Acknowledgments
This work was supported by the Christian Doppler Research Association (Christian Doppler Laboratory for Antibody Engineering), the Austrian Science Foundation (FWF Project W1224 – Doctoral Program on Biomolecular Technology of Proteins – BioToP) as well as the Federal Ministry of Economy, Family and Youth through the initiative “Laura Bassi Centers of Expertise,” funding the Center of Optimized Structural Studies, No. 253275. We thank Dr. Gordana Wozniak-Knopp (University for Natural Resources and Life Sciences Vienna, Austria) for providing plasmid DNA of the Fcabs 448 and CT6. We thank Dr. Mike R. Williams from Malvern Instruments Ltd. (Malvern, UK) for assisting in ITC measurements. We acknowledge the European Synchrotron Radiation Facility (Grenoble, France) for provision of synchrotron radiation facilities and the Local Contacts for providing assistance in using beamline ID30A-3.
Related Research Data
References
- Jafari R, Zolbanin NM, Rafatpanah H, Majidi J, Kazemi T. Fc-fusion proteins in therapy: an updated view. Curr Med Chem. 2017;24:1228-37. doi:10.2174/0929867324666170113112759. PMID:28088904
- Kubota T, Niwa R, Satoh M, Akinaga S, Shitara K, Hanai N. Engineered therapeutic antibodies with improved effector functions. Cancer Sci. 2009;100:1566-72. doi:10.1111/j.1349-7006.2009.01222.x. PMID:19538497
- Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs. 2011;3:415-6. doi:10.4161/mabs.3.5.17334. PMID:21785279
- Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol. 2015;35:235-54. doi:10.3109/07388551.2013.834293. PMID:24156398
- Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci U S A. 1969;63:78-85. doi:10.1073/pnas.63.1.78. PMID:5257969
- Halaby DM, Poupon A, Mornon J. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng. 1999;12:563-71. doi:10.1093/protein/12.7.563. PMID:10436082
- Lobner E, Traxlmayr MW, Obinger C, Hasenhindl C. Engineered IgG1-Fc-one fragment to bind them all. Immunol Rev. 2016;270:113-31. doi:10.1111/imr.12385. PMID:26864108
- Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschlager M, Antes B, Ettl K, Kainer M, Weberhofer G, Wiederkum S, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel. 2010;23:289-97. doi:10.1093/protein/gzq005. PMID:20150180
- Jez J, Antes B, Castilho A, Kainer M, Wiederkum S, Grass J, Rüker F, Woisetschläger M, Steinkellner H. Significant impact of single N-glycan residues on the biological activity of Fc-based antibody-like fragments. J Biol Chem. 2012;287:24313-9. doi:10.1074/jbc.M112.360701. PMID:22589538
- Woisetschlager M, Antes B, Borrowdale R, Wiederkum S, Kainer M, Steinkellner H, Wozniak-Knopp G, Moulder K, Rüker F, Mudde GC. In vivo and in vitro activity of an immunoglobulin Fc fragment (Fcab) with engineered Her-2/neu binding sites. Biotechnol J. 2014;9:844-51. doi:10.1002/biot.201300387. PMID:24806546
- Traxlmayr MW, Lobner E, Antes B, Kainer M, Wiederkum S, Hasenhindl C, Stadlmayr G, Rüker F, Woisetschläger M, Moulder K, et al. Directed evolution of Her2/neu-binding IgG1-Fc for improved stability and resistance to aggregation by using yeast surface display. Protein Eng Des Sel. 2013;26:255-65. doi:10.1093/protein/gzs102. PMID:23267121
- Lobner E, Humm AS, Goritzer K, Mlynek G, Puchinger MG, Hasenhindl C, Rüker F, Traxlmayr MW, Djinović-Carugo K, Obinger C. Fcab-HER2 interaction: a menage a trois. Lessons from X-Ray and solution studies. Structure. 2017;25:878–89 e5. doi:10.1016/j.str.2017.04.014
- Leung KM, Batey S, Rowlands R, Isaac SJ, Jones P, Drewett V, Carvalho J, Gaspar M, Weller S, Medcalf M, et al. A HER2-specific modified Fc Fragment (Fcab) induces antitumor effects through degradation of HER2 and apoptosis. Mol Ther. 2015;23:1722-33. doi:10.1038/mt.2015.127. PMID:26234505
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553-7. doi:10.1038/nbt0697-553. PMID:9181578
- Muller YA, Heiring C, Misselwitz R, Welfle K, Welfle H. The cystine knot promotes folding and not thermodynamic stability in vascular endothelial growth factor. J Biol Chem. 2002;277:43410-6. doi:10.1074/jbc.M206438200. PMID:12207021
- Ahmed AA, Giddens J, Pincetic A, Lomino JV, Ravetch JV, Wang LX, Bjorkman PJ. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol. 2014;426:3166-79. doi:10.1016/j.jmb.2014.07.006. PMID:25036289
- Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol. 2012;7:1596-602. doi:10.1021/cb300130k. PMID:22747430
- Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol Immunol. 2013;56:131-9. doi:10.1016/j.molimm.2013.03.018. PMID:23628091
- Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W. H. Freeman; 1999; 10-12.
- Barlow DJ, Thornton JM. Helix geometry in proteins. J Mol Biol. 1988;201:601-19. doi:10.1016/0022-2836(88)90641-9. PMID:3418712
- Hardy JA, Nelson HC. Proline in alpha-helical kink is required for folding kinetics but not for kinked structure, function, or stability of heat shock transcription factor. Protein Sci. 2000;9:2128-41. doi:10.1110/ps.9.11.2128. PMID:11305238
- Fu H, Grimsley GR, Razvi A, Scholtz JM, Pace CN. Increasing protein stability by improving beta-turns. Proteins. 2009;77:491-8. doi:10.1002/prot.22509. PMID:19626709
- Spreter Von Kreudenstein T, Escobar-Carbrera E, Lario PI, D'Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. MAbs. 2013;5:646-54. doi:10.4161/mabs.25632. PMID:23924797
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: John M. Walker, editor. The Proteomics Protocols Handbook. Humana Press; 2005. p. 571-607.
- Harris RJ. Processing of C-terminal lysine and arginine residues of proteins isolated from mammalian cell culture. J Chromatogr A. 1995;705:129-34. doi:10.1016/0021-9673(94)01255-D. PMID:7620566
- van den Bremer ET, Beurskens FJ, Voorhorst M, Engelberts PJ, de Jong RN, van der Boom BG, Cook EM, Lindorfer MA, Taylor RP, van Berkel PH, et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. MAbs. 2015;7:672-80. doi:10.1080/19420862.2015.1046665. PMID:26037225
- Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CA, Kammeijer GS, Jansen BC, Rispens T, Hensbergen PJ, Vidarsson G, et al. Hinge-region o-glycosylation of human immunoglobulin G3 (IgG3). Mol Cell Proteomics. 2015;14:1373-84. doi:10.1074/mcp.M114.047381. PMID:25759508
- Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci U S A. 1997;94:7192-7. doi:10.1073/pnas.94.14.7192. PMID:9207067
- Piovesan D, Minervini G, Tosatto SC. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res 2016;44:W367-74. doi:10.1093/nar/gkw315. PMID:27198219
- Jones TD, Carter PJ, Pluckthun A, Vasquez M, Holgate RG, Hotzel I, Popplewell AG, Parren PW, Enzelberger M, Rademaker HJ, et al. The INNs and outs of antibody nonproprietary names. MAbs. 2016;8:1-9. doi:10.1080/19420862.2015.1114320. PMID:26716992
- Traxlmayr MW, Lobner E, Hasenhindl C, Stadlmayr G, Oostenbrink C, Ruker F, Obinger C. Construction of pH-sensitive Her2-binding IgG1-Fc by directed evolution. Biotechnol J. 2014;9:1013-22. doi:10.1002/biot.201300483. PMID:24964247
- Hasenhindl C, Traxlmayr MW, Wozniak-Knopp G, Jones PC, Stadlmayr G, Ruker F, Obinger C. Stability assessment on a library scale: a rapid method for the evaluation of the commutability and insertion of residues in C-terminal loops of the CH3 domains of IgG1-Fc. Protein Eng Des Sel. 2013;26:675-82. doi:10.1093/protein/gzt041. PMID:24006374
- Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol. 2000;18:754-9. doi:10.1038/77325. PMID:10888844
- Traxlmayr MW, Obinger C. Directed evolution of proteins for increased stability and expression using yeast display. Arch Biochem Biophys. 2012;526:174-80. doi:10.1016/j.abb.2012.04.022. PMID:22575387
- Garbett NC, Chaires JB. Thermodynamic studies for drug design and screening. Expert Opin Drug Discov. 2012;7:299-314. doi:10.1517/17460441.2012.666235. PMID:22458502
- Chodera JD, Mobley DL. Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu Rev Biophys. 2013;42:121-42. doi:10.1146/annurev-biophys-083012-130318. PMID:23654303
- Markovic-Mueller S, Stuttfeld E, Asthana M, Weinert T, Bliven S, Goldie KN, Kisko K, Capitani G, Ballmer-Hofer K. Structure of the Full-length VEGFR-1 extracellular domain in complex with VEGF-A. Structure. 2017;25:341-52. doi:10.1016/j.str.2016.12.012. PMID:28111021
- Brozzo MS, Bjelic S, Kisko K, Schleier T, Leppanen VM, Alitalo K, Winkler FK, Ballmer-Hofer K. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood. 2012;119:1781-8. doi:10.1182/blood-2011-11-390922. PMID:22207738
- Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998;6:1153-67. doi:10.1016/S0969-2126(98)00116-6. PMID:9753694
- Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB. Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J Mol Biol. 1999;293:865-81. doi:10.1006/jmbi.1999.3192. PMID:10543973
- Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8:880-7. doi:10.1038/nrc2505. PMID:18923433
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi:10.1093/nar/30.2.e9. PMID:11788735
- Traxlmayr MW, Faissner M, Stadlmayr G, Hasenhindl C, Antes B, Ruker F, Obinger C. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta Prot Proteomics. 2012;1824:542-9. doi:10.1016/j.bbapap.2012.01.006
- Heiring C, Muller YA. Folding screening assayed by proteolysis: application to various cystine deletion mutants of vascular endothelial growth factor. Protein Eng. 2001;14:183-8. doi:10.1093/protein/14.3.183. PMID:11342715
- Christinger HW, Muller YA, Berleau LT, Keyt BA, Cunningham BC, Ferrara N, de Vos AM. Crystallization of the receptor binding domain of vascular endothelial growth factor. Proteins. 1996;26:353-7. doi:10.1002/(SICI)1097-0134(199611)26:3%3c353::AID-PROT9%3e3.0.CO;2-E. PMID:8953654
- Harris LJ, Skaletsky E, McPherson A. Crystallization of intact monoclonal antibodies. Proteins. 1995;23:285-9. doi:10.1002/prot.340230218. PMID:8592710
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125-32. doi:10.1107/S0907444909047337. PMID:20124692
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030-3. doi:10.1126/science.1218231. PMID:22628654
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658-74. doi:10.1107/S0021889807021206. PMID:19461840
- Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125-32. doi:10.1107/S0907444907050172. PMID:18094476
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61-9. doi:10.1107/S090744490705024X. PMID:18094468
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486-501. doi:10.1107/S0907444910007493. PMID:20383002
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213-21. doi:10.1107/S0907444909052925. PMID:20124702
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240-55. doi:10.1107/S0907444996012255. PMID:15299926
- DiMaio F, Echols N, Headd JJ, Terwilliger TC, Adams PD, Baker D. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat Methods. 2013;10:1102-4. doi:10.1038/nmeth.2648. PMID:24076763
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12-21. doi:10.1107/S0907444909042073. PMID:20057044
- Joosten RP, Joosten K, Murshudov GN, Perrakis A. PDB_REDO: constructive validation, more than just looking for errors. Acta Crystallogr D Biol Crystallogr. 2012;68:484-96. doi:10.1107/S0907444911054515. PMID:22505269
- Joosten RP, Long F, Murshudov GN, Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213-20. doi:10.1107/S2052252514009324. PMID:25075342
- Lutteke T, von der Lieth CW. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics. 2004;5:69. doi:10.1186/1471-2105-5-69. PMID:15180909
- Agirre J, Iglesias-Fernandez J, Rovira C, Davies GJ, Wilson KS, Cowtan KD. Privateer: software for the conformational validation of carbohydrate structures. Nat Struct Mol Biol. 2015;22:833-4. doi:10.1038/nsmb.3115. PMID:26581513