
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The intrinsic complexity and heterogeneity of therapeutic monoclonal antibodies is built into the biosimilarity paradigm where critical quality attributes are controlled in exhaustive comparability studies with the reference medicinal product. The long-term success of biosimilars will depend on reassuring healthcare professionals and patients of consistent product quality, safety and efficacy. With this aim, the World Health Organization has endorsed the need for public bioactivity standards for therapeutic monoclonal antibodies in support of current controls. We have developed a candidate international potency standard for rituximab that was evaluated in a multi-center collaborative study using participants' own qualified Fc-effector function and cell-based binding bioassays. Dose-response curve model parameters were shown to reflect similar behavior amongst rituximab preparations, albeit with some differences in potency. In the absence of a common reference standard, potency estimates were in poor agreement amongst laboratories, but the use of the candidate preparation significantly reduced this variability. Our results suggest that the candidate rituximab standard can support bioassay performance and improve data harmonization, which when implemented will promote consistency of rituximab products over their life-cycles. This data provides the first scientific evidence that a classical standardization exercise allowing traceability of bioassay data to an international standard is also applicable to rituximab. However, we submit that this new type of international standard needs to be used appropriately and its role not to be mistaken with that of the reference medicinal product.
| Abbreviations | ||
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| CDC | = | complement-dependent cytotoxicity |
| CPP | = | critical process parameters |
| CQA | = | critical quality attributes |
| FcγR | = | Fcɣ receptors |
| GCV | = | geometric coefficient of variation |
| GM | = | geometric mean |
| HSA | = | human serum albumin |
| IgG | = | immunoglobulin G |
| IS | = | International Standard |
| IU | = | International Units |
| MAA | = | marketing authorization application |
| mAb | = | monoclonal antibody |
| NFAT-RE | = | Nuclear factor of activated T cells response element |
| NK | = | natural killer cells |
| PBMCs | = | peripheral blood mononuclear cells |
| RMP | = | reference medicinal product |
| WHO | = | World Health Organization |
Introduction
Biotherapeutic monoclonal antibodies (mAbs) have had a major impact on healthcare, primarily in the fields of oncology and inflammation.Citation1,2 Their success and the patent expiry of some of the earliest products has stimulated the interest of biosimilar drug developers, and to date six biosimilar mAbs are already approved in Europe. Remsima® and Flixabi® are approved biosimilar products to the infliximab innovator product Remicade®; Amgevita® and Imraldi® are biosimilars to the adalimumab innovator product Humira®; and Truxima® and Rixathon® are approved rituximab biosimilars for Mabthera®/Rituxan®. In addition, a number of applications are currently under assessment,Citation3 and there are reports of at least 140 biosimilar mAb development programs of which greater than 40 are of rituximab.Citation4,5 With many of these at the later stages of development, it is expected that the increased availability of biosimilars will further increase market competition and translate into improved accessibility of these medicines to patients.
Biosimilarity is demonstrated through extensive comparability studies that form part of both the non-clinical and clinical modules of the marketing authorization application (MAA) dossier. The biosimilar should conform to the reference medicinal product (RMP) quality target profile, which defines the tolerance level associated with its proven safe and efficacious clinical record.Citation6 Therefore, understanding the significance of any product micro-heterogeneity is critical to the biosimilarity concept.Citation7 This micro-heterogeneity is scrutinized during the process using orthogonal methods and contributes to what is known from the regulatory review as the “totality of evidence” regarding biosimilarity.Citation8 Once the biosimilar is approved, it becomes an independent product with its own life-cycle. Post-marketing changes in the manufacturing process for both innovator and biosimilar products are common place and are approved by the regulatory authorities.Citation9,10 Careful control of the product critical quality attributes (CQA) and critical process parameters (CPP) are key, but potential drift, evolution and divergence over a product's life is acknowledged as a consequence of the complexity of mAbs and their production in biological systems.Citation11,12 So far, the extent and effects of product drift within the growing number of approved biosimilars remains a concern that as yet cannot be quantified.
The assessment of the biological properties of a mAb is an essential component of the comparability exercise, and proprietary “in house” reference standards are used by manufacturers throughout the product's life. MAb products are typically marketed and dosed in mass units, with their biological activity defined relative to the “in house” proprietary reference standard. To date they are approved in the absence of public standards; however, this represents a challenge for biosimilar manufacturers because no reagents are available to calibrate bioassays and manufacturer's standard preparations. While the biosimilar regulatory pathway is rigorous,Citation13–17 the success of the biosimilarity regulatory process is dependent on establishing and sustaining confidence in the quality of biosimilar products.
The World Health Organization (WHO) has recognized the need for global standardization of biotechnology products, including mAbs, to ensure their safety, quality and efficacy.Citation18–20 Therapeutic mAbs are bioengineered products with no naturally occurring equivalent, so crucially they represent a new class of WHO international standard (IS) with as yet no precedence. WHO IS for mAbs would be publically available stable lyophilized preparations with the sole application in the control and calibration of potency bioassays and “in house” reference standards by different stakeholders. They would define international units (IU) of bioactivity, and hence facilitate the assessment and management of mAbs across different manufacturers and jurisdictions. In doing so, these ISs would support both innovator and biosimilar manufacturers at various stages of product development, as well as post-approval (i.e., upon manufacture process change, product surveillance) when bioassays play a pivotal part in ensuring the consistency, safety and efficacy of mAbs products. The use of a common bioactivity IS to which manufacturers can calibrate their “in house” reference standards presents a unique opportunity to understand any potential drift in bioactivity in a complex multi-product marketplace.
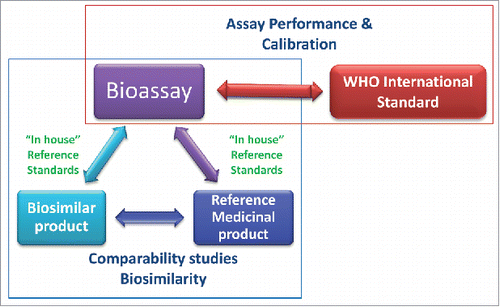
The existence of these ISs, however, would not infer changes in the current dosage in mass units, and neither would these ISs be a substitute for the regulatory role of the RMP in defining biosimilarity or specific product activity (IU/mg), which are the explicit role of the RMP and the remit of the competent authorities.Citation14,17 Specifically, the IS should not bear any additional burden on the current requirement for an extensive comparability exercise with the RMP, but provide the necessary additional reassurance in bioassay and local standards performance by allowing traceability to IU overtime. In other words, the quality characteristics of the WHO IS preparation, such as purity or specific activity, would not be used to define the acceptable specific activity or purity of a biosimilar product. Nevertheless, if adopted, bioactivity measurements expressed in units traceable to the IS would assist in harmonizing global product data, which could in principle support regulatory decision-making. The distinct characteristics and roles of the RMP and the IS are summarized in and .
Table 1. A comparison of the characteristics and roles of the reference medicinal product with that of a WHO international standard for mAb biological activity.
Figure 1. The roles of the WHO international standards (IS) and the reference medicinal product (RMP) in relation to the bioassay. The WHO IS supports bioassay performance and calibration. Bioassays are used as part of the comparability studies to demonstrate biosimilarity between the biosimilar product and the RMP.
Rituximab is on the WHO list of essential medicines for a basic healthcare system.Citation21 It is a chimeric mAb that is used as a monotherapy or in combination with chemotherapy regimens for the treatment of CD20+ B cell lymphoproliferative malignancies, transplant rejection and autoimmune disorders.Citation22-24 Current therapeutic indications include non-Hodgkin's lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis and microscopic polyangiitis,Citation25,26 and there is growing interest in its use for the treatment of autoimmune diseases.Citation27 With a molecular weight of approximately 144,544 Da, rituximab is composed of 1328 amino acids and consists of a human kappa constant region, a human immunoglobulin G1 (IgG1) Fc portion and murine variable regions. It has a conserved N-glycosylation site at Asn297 of both heavy chains to which biantennary glycan structures are attached.Citation28 The exact mechanism of action in vivo remains unclear, but it is assumed that it exerts its effects by binding to CD20+ B lymphocytes with the concomitant induction of apoptosis, either directly or by engagement of the Fc region with immune effectors. Rituximab promotes B cell lysis through complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) upon binding to Fcγ receptors (FcγR) on effector cells that include natural killer cells (NK), granulocyte and macrophages.Citation29-31
Following WHO endorsement,Citation32 we developed a candidate IS for the biological activities of rituximab, and, for the first time, data is available illustrating the potential benefits of using these new class of reagents. Here, we report results of an international collaborative study designed to assess the suitability of a lyophilized candidate antibody preparation to serve as an IS for rituximab bioactivity with associated arbitrary IU. The results are discussed and highlight the value of using ISs for the bioactivity of mAbs. Using rituximab as a case study, we argue that this new class of biological standard could assist in managing product consistency over time and consequently promote market assurance.
Results
International collaborative study data analysis
A multi-center international collaborative study was organized to evaluate the suitability of a lyophilized candidate preparation as an IS for the bioactivities of rituximab. Additionally, the study also provides valuable information on other rituximab preparations, as well as bioassay systems currently used to test rituximab products. Sixteen laboratories performed bioassays for this study using their own “in house” qualified methods and reference standards when available. The CDC assay is the current batch release assay for rituximab, but the study was extended to include other relevant bioactivities that are typically assessed during product development. Of the participants, 16 performed CDC assays, 11 laboratories performed ADCC assays, 5 laboratories performed cell-based binding assays and 1 participant performed a direct apoptosis assay. A summary of the bioassays that contributed to the study is shown in . The complete data set was reviewed and included in an independent statistical analysis performed for the purposes of this study. In the first instance, we addressed whether different rituximab preparations were comparable in the various bioassays prior to calculating the relative potency estimates of the study preparations.
Table 2. Summary of the bioassays that contributed to the study.
Qualitative comparison between rituximab preparations bioassay responses
Rituximab products have inherent micro-heterogeneities and the different laboratories in the study used different methodologies to characterize the various biological activities of rituximab. Thus, it is important to evaluate whether different rituximab preparations have a comparable biological behavior across this setting, and therefore whether a standardization exercise for rituximab is plausible. Typically, this is the case for other biological medicines of similar complexity to mAbs, such as the tumor necrosis factor receptor II Fc fusion protein etanercept,Citation33 or coagulation factors such as Factor VIII,Citation34 where product bioactivities are traceable to the established WHO IS. Three rituximab study preparations, including a coded duplicate of the candidate (samples A and B) and a lyophilized Mabthera® batch (sample C), as well as the participants' “in house” reference standard preparations when available, were compared. “In house” reference standards were typically innovator batches or manufacturers' biosimilar preparations. No single defined statistical method to compare bioassay dose-response curve data is available in the current European and United States Pharmacopoeias. To assess comparability between the rituximab preparations, we used an “equivalence testing” approach where curve similarity was demonstrated using pre-defined acceptable ranges for the differences in model parameters. For this, equivalence bounds were based on the data for the coded duplicate samples tested in each assay, as model parameters (upper asymptote, asymptote difference and slope factor) are expected to be equivalent for these samples. The equivalence bounds were calculated using the complete data set, and they were intended for use only in the analysis of the data from this study. The choice of equivalence bounds and the effect on the qualitative similarity of the preparations (what defines and is also referred to here as assay validity) for each independent assay type is shown in the supplementary data (Figures S1, S2, S3 and Tables S1-S5).
Non-parallelism of the dose-response curves as per defined equivalence criteria was found in 0 to 78% of the bioassays when individual laboratory data was assessed and the preparations compared to the candidate (sample A). When the study preparations were compared to the “in house” reference standards, non-parallelism was found in 0 to 100% of the bioassays as per individual laboratory. For the CDC assay, laboratories 2, 10 and 12 showed a high percentage of invalid assays; however, these were observed across various sample comparisons, including coded duplicates, and therefore likely attributable to individual laboratory assay performance. For these laboratories, individual assay data was variable, which contributed to the differences in model parameters that were outside the pre-defined acceptable range. Although limited data is available to stratify the ADCC assay by type (5 reporter assays, 3 NK cell line killing assays and 3 primary cell-based killing assays) and assay validity is also dependent on individual laboratory performance, we noted that, in general, the NK cell line killing assays seemed to show greater comparability between preparations (lower number of invalid assays). The reporter assays trended towards showing more pronounced differences between the candidate preparation and the “in house” references. Extreme values (defined as the maximum of the upper 90% confidence limit and reciprocal of the lower 90% confidence limit) for the ratio of parameter estimates between the sample preparations for each individual assay, laboratory and bioassay type can be found as supplementary data (Figures S4-S6). All of the laboratories in the study, with the exception of one laboratory that did not return any valid CDC assay data when comparing sample C to their “in house” reference standard, produced assays where preparations were qualitatively comparable, and thus acceptable under the set criteria. These assays were used to calculate the sample relative potency estimates (supplementary data Table S5).
Preliminary assays were performed by each laboratory to establish suitable dilution ranges for the study preparations using their qualified bioassays. Laboratory 11 reported failed system suitability criteria for their peripheral blood mononuclear cells (PBMC)-killing ADCC assay when their “in house” rituximab reference standard was used at the concentration optimized for the study preparations, and only returned data for samples A, B and C. It was noted that single gene reporter ADCC assays showed a decrease in bioactivity at the higher antibody concentration(s) and one or two doses were excluded in the analysis (laboratories 1, 7, 8, 10 and 12). Furthermore, laboratory 15 also reported reduced activity for the study preparations at the highest concentration using their validated ADCC reporter assay, whilst this effect was not observed when using their “in house” reference standard. However the laboratory successfully performed an end-point killing assay using PBMC with both the study preparations and their “in house” reference standard, and returned data that was included in the analysis contributing to the potency results.
The data showed that, in general, all of the rituximab preparations had a comparable behavior other than some differences in potency. Invalid assays were mostly related to poor relative laboratory bioassay performance and were subsequently removed for the calculation of relative potencies. This data demonstrates that the candidate reference standard closely resembles other rituximab preparations in the bioassay systems used to test them. Therefore, the results reflect that the applicability of the bioassay IS concept can be extended to rituximab mAb products.
Estimates of relative potency to “in house” reference standards
Manufacturers develop “in house” reference standards to characterize their products during development, and to calibrate their bioassay (system suitability standards). From preliminary standards to primary and working standards, these reagents adapt to the different stages of the product life accommodating different applications, such as managing process changes, scale up of production and product stability. Reference standards used in late development and in analytical assessment of biosimilarity are validated and undergo extensive characterization. In addition, strategies for periodical re-evaluation and replacement of primary and working standards are essential in product regulatory submission dossiers. Potency estimates for the study preparations (samples A, B and C) were calculated for each laboratory and bioactivity using “in house” reference standards when available (). “In house” reference standards used in the study are assumed to be those routinely supporting product development, system suitability or non-clinical/clinical studies by the participating manufacturers. Where an “in house” reference preparation was unavailable, some laboratories used a clinical batch of the innovator product.
Table 3. Individual laboratory relative potency estimates (geometric mean) for CDC assays.
The CDC activity potency estimates (geometric mean (GM) value) for the individual laboratories for samples A and C relative to the “in house” reference standard ranged from 0.95 to 1.48 and 0.93 to 1.47, respectively (). The intra-laboratory variability of these estimates ranged from 3% to 13%. For ADCC activity, potency estimates ranged from 0.44 to 2.17 and 0.64 to 2.42 for sample A and C, respectively, with intra-laboratory variability (geometric coefficient of variation (GCV)) ranging from 7% to 36% (). The GM potency values for cell-binding range between 0.57 and 1.09 and from 0.57 to 1.04 for samples A and C, respectively with GCV between 7% and 17% (). The apoptosis assay was performed by only one laboratory that estimated a relative potency of 1.09 for sample A and 1.01 for sample C with a GCV of 13% and 11%, respectively ().
Table 4. Individual laboratory relative potency estimates (geometric mean) for ADCC assays.
Table 5. Individual laboratory relative potency estimates (geometric mean) for cell-binding assays.
Table 6. Individual laboratory relative potency estimates (geometric mean) for apoptosis assays.
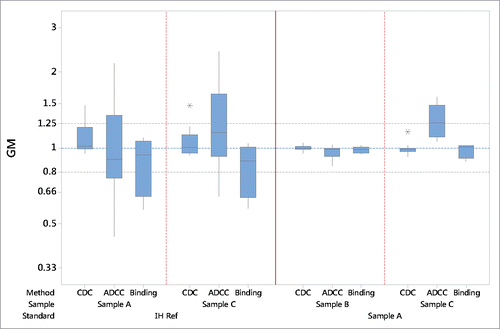
The combined potency estimates for each bioactivity, associated GCV and confidence limits are shown in . The high GCV values represent a measure of the large inter-laboratory variability reflecting the wide distribution of bioactivity potency values found for the individual laboratories. For CDC bioactivity, the GCV values of the combined GM potency estimates for samples A and C were 16% and 14%, respectively. For ADCC activity, the GCVs were 62% and 51%, and for cell binding assays, the GCVs were 33% and 29% for samples A and C, respectively. In all cases the high variability associated with the combined estimate potency values demonstrated poor agreement between laboratories when potency was calculated relative to “in house” reference standards ().
Table 7. Potency summary data relative to “in house” reference standards (IH Ref).
Figure 2. Summary bioassay data of geometric mean (GM) relative potency estimates using “in house” reference (IH Ref) or the candidate (sample A) as a standard. Boxes represent the interquartile range and the line shows the median. The bars represent the range and * shows outliers defined as 1.5 times the interquartile range.
Estimates of relative potency to the candidate preparation
A rituximab potency IS will be used to define IU of bioactivity for rituximab, and it will become the highest order public standard for rituximab bioassays. The participants used their “in house” qualified methodologies to assess the bioactivity of two rituximab preparations, sample B (coded duplicate) and sample C, using the candidate preparation as reference standard. Potency estimates for the individual laboratories and bioassays are summarized in .
The GM potency estimates for CDC activity were in good agreement between the 16 laboratories ranging from 0.95 to 1.05 and 0.92 to 1.16 for sample B and sample C, respectively (). Intra-laboratory GCV values ranged from 2% to 19%, similarly to the GCV associated with the potency estimates when expressed relative to “in house” references as shown before. For ADCC assays, intra-laboratory variability ranged from 7% to 35% for the individual laboratories, also comparable to the GCV values for potency estimates calculated relative to “in house” references (). The GM potency estimates for ADCC ranged from 0.86 to 1.03 and from 1.06 to 1.60 for samples B and C, respectively, values that were much less dispersed than those calculated relative to “in house” reference standards. The potency estimates for sample C skewed towards values above 1.0, which suggest a higher ADCC bioactivity relative to the candidate preparation. The calculated GM relative potencies for cell-binding assays ranged from 0.95 to 1.02 and 0.88 to 1.02 for sample B and C, respectively, with an intra-laboratory variability (GCV values between 1% and 19%) also comparable to that calculated with “in house” reference standards (). The apoptosis assay performed by one laboratory showed a GM potency of 1.00 and 0.89 for samples B and C, respectively and an intra-laboratory GCV of 12% for both estimates ().
The combined potency data relative to the candidate for each bioactivity, associated GCV and confidence limits are shown in . In general, the inter-laboratory variability was low with close agreement between GM relative potency values. For CDC, the GCV was 3% and 5% and the combined potency estimates were 1.00 and 0.99 for sample B and C, respectively. These GCVs were considerably lower than those calculated relative to “in house” reference standards. The inter-laboratory GCVs for the combined ADCC potency data was 6% and 16% for samples B and C, respectively, values that were also significantly lower than those found when the “in house” reference was used as a standard. The combined GM value of 1.28 for sample C suggests a higher ADCC bioactivity relative to the candidate. Similarly, a reduced inter-laboratory variability for cell-binding assays with GCVs of 3% and 7% for sample B and C, respectively, was observed. The use of the candidate rituximab preparation as reference standard clearly showed increased harmonization of the bioactivity data amongst the participant laboratories ().
Table 8. Potency summary data relative to the candidate preparation.
Accelerated degradation studies
Accelerated degradation studies were performed to predict the long-term stability of the candidate preparation and calculate expected annual bioactivity loss. The relative bioactivities of accelerated thermal degradation samples can be used to fit an Arrhenius equation relating degradation rate to absolute temperature assuming first-order decay,Citation35 and hence predict the degradation rates when stored at −20 °C. At the time we analyzed the data, only one data point at 10 months was available, and no detectable loss of CDC or ADCC bioactivity was found even at the higher temperatures (). Stability over an extended period of time is a pre-requisite for an IS. Both stability prediction and monitoring studies are currently ongoing.
Table 9. Accelerated degradation studies: Potency estimates for the candidate preparation stored at elevated temperatures for 10 months relative to ampoules stored at baseline temperature of −70 °C. The 95% upper and lower confidence limits are also shown.
Proposed assignment of international units of bioactivity for the candidate preparation
Prior to our work, no rituximab IS or reference method to test the bioactivity of the rituximab candidate existed. Thus, typically a variety of methods have been used to assign an arbitrary value to the IS, and the definition of an IU is therefore not dependent on a specific method of determination.Citation36 In line with its role to support bioassay performance, calibration and validation, and the various biological activities of rituximab, arbitrary IU for the 4 independent bioactivities assessed are proposed. Each ampoule of the candidate rituximab preparation (National Institute for Biological Standards and Control (NIBSC) code 14/210) will be proposed for assignment of 1,000 IU of CDC activity, 1,000 IU of ADCC activity, 1,000 IU of cell-binding activity and 1,000 IU of apoptotic activity. It is important to emphasize that the content of rituximab per ampoule is nominal and no declared mass content for the proposed preparation will be given. As such, the proposed unitage will not define a specific activity for the preparation (that is U/mg) and will not be intended for any use that derives or infers specific activity for regulatory purposes ().
Discussion
In this study, the criteria describing comparable bioassay behavior between rituximab preparations, hence assay validity, was based on data from coded duplicate preparations where differences can only be attributed to assay performance. In general, only a low and a similar percentage of assays in the study revealed differences in the qualitative behavior of any two given preparations regardless of the rituximab samples compared (i.e., coded duplicate, sample C or “in house” reference). This data suggests that the rituximab preparations were comparable in the individual bioassay types, despite the differences in potency observed. Between 70–85% of the bioassays were valid, and all of the 16 laboratories reported data that contributed to the overall potency estimates. It is important to note that an invalidity rate of at least 10% is to be expected due to the statistical methods applied to determine equivalence criteria. This data provides the first scientific evidence to suggest that the applicability of a classical standardization exercise that allows traceability of bioassay potency data to the IS is also possible for rituximab. Therefore, this work presents the first candidate standard for the biological activities of a mAb with no natural equivalent, representing the introduction of a new class of IS. In general, data for the CDC, ADCC and cell-based binding activities showed very poor agreement between laboratories when the potency estimates of the study preparations (sample A, B and C) were expressed relative to the “in house” reference standards. These results presumably reflect the differences between the “in house” reference standards and highlight a general lack of harmonization. The greatest variability between laboratories was observed for the ADCC assay (GCV of 62% and 51% for sample A and C, respectively). This may be explained by the intrinsic complexity of the ADCC assay, the different assay platforms used or the differences in the individual laboratory's assay sensitivity. The later may relate to potential variations in some quality attributes of the “in house” reference standards that may be significant for ADCC, but may not have such an effect on other rituximab bioactivities, i.e., different glycosylation profile may affect ADCC activity as described before,Citation37,38 but may be less likely to affect CDC activity or target cell binding.Citation39,40 The higher intra-laboratory variability (7–36%) for ADCC compared to CDC, cell-based binding or apoptosis assays (3–17%) may also reflect specific challenges of the ADCC bioassay. However, no association with individual ADCC assay platforms or intra-laboratory assay variability was found. Neither could the inter-laboratory variability in ADCC activity be linked to the assay platform suggesting that the reported differences mainly reflect the differences between the “in house” reference standards. Moreover, when calculating relative bioactivity ratios for the “in house” references (i.e., CDC/ADCC), different values are observed, further suggesting product heterogeneity that translates into different relative functional effects.
The inhibition of ADCC bioactivity in the reporter assays at the highest concentrations of the study preparations is an interesting observation. Direct effects on the surrogate effector cells and direct cytotoxicity or steric hindrance as the total protein concentration increases could be a contributing factor. In addition, the lyophilized preparations contain 1% human serum albumin (HSA, with ≥ 95% albumin) for stabilization purposes during the freeze-drying process, which represents approximately 100 times higher HSA protein content in mass than rituximab itself. Thus, we speculate that potential traces of IgG in the HSA at high concentrations may interfere with the reporter assay by competing for the FcR on the surrogate effector cells or affecting the nuclear factor of activated T-cells response element (NFAT-RE) signaling pathway, thus reducing the reporter signal. Excess endogenous human IgG in serum has previously been shown to inhibit therapeutic antibody ADCC activity.Citation41,42 The use of low concentration (i.e., 4% v/v) of low IgG-containing serum is typically recommended in ADCC reporter assays to avoid IgG interference (Promega, ADCC reporter bioassay “instructions for use”). Although inhibitory effects due to competition for CD16 binding could also be expected on ADCC killing assays, media is often supplemented with higher sera concentrations (i.e., 10% v/v fetal bovine serum).Citation43 Also, for primary cell-based killing assays, the higher expression of CD16 receptors compared to reporter cells has been shown to have a positive impact on rituximab ADCC activity.Citation44 In addition, for PBMC assays, other cell types such as monocytes may also mediate ADCC activity in the co-culture environment.Citation43 This could provide an explanation for laboratory 15 finding their ADCC reporter assay unsuitable at high concentrations, whilst successfully performing a PBMC end-point killing assay. No inhibitory effects were reported for one laboratory performing a dual reporter assay, suggesting that the potential effects on effector cells may be normalized or the dual reporter cell line used was more resilient to the effects of high HSA concentrations. However, it should be mentioned that, although some of these may be contributing factors, the antibody concentration range used by each laboratory is different, and therefore these effects may depend on the method or may have remained undetected for some labs at the concentration range tested.
The most relevant finding of this study was the excellent agreement in rituximab potency estimates when the candidate preparation was used as a standard despite different methodologies being used by participating laboratories. The results from the study substantiate the conclusion for CDC and ADCC activities, and, although the data is limited, cell-binding results further support these findings. The results suggest that the candidate preparation 14/210 is able to harmonize potency assay data between laboratories, which is in line with its intended role. We also note that the CDC potency for sample C was similar to that of the candidate standard, but the ADCC potency estimate of sample C was significantly greater (GM 1.28) and had a higher associated variability (GCV 16%). This variability was also greater than that observed for the coded duplicate (sample B) (GCV 6%) and was unrelated to the ADCC assay platform used. We hypothesize that this reflects differences between the two rituximab preparations (samples A and C) that were identified by individual labs in their ADCC assays to different extents. Differences between the ADCC potency and efficacy of trastuzumabCitation45 and an anti-CD20 mAbCitation43 were reported when different effector cells, primary PBMC or primary NK cells, engineered NK cell lines or engineered reporter cell lines, were used. These differences were attributed to differences in the biological properties and expression profiles of activating or inhibitory receptors in the effector cells used, i.e., number of CD16 (FcɣIII receptors), receptor polymorphisms, molecular interactions between the Fc domain of the mAb and the FcɣIII receptor, the involvement of specific FcɣIII receptor subunits and transduction signals. Additionally, ADCC activity using primary cells may be affected by cryopreservation and overnight resting of PBMCs prior to performing the assay, which can have an effect on CD16 expression and NK activation.Citation46 Method-dependent differences in ADCC activity have also been reported between an infliximab biosimilar and the innovator product.Citation7,47 These results draw attention to the complexity of in vitro ADCC assays and suggest that a deeper understanding of these critical in vitro biological assays and correlation with clinical studies may be needed.
The ‘similar-but-not-identical’ concept applies to the RMP itself as much as to the biosimilar and is central to the regulatory process.Citation7,48 A well-documented example is the approved change in the glycosylation profile of Rituxan®/Mabthera® batches. This change was associated with a manufacturing process change which correlated with increased ADCC activity,Citation37 but with no apparent clinical effects to date. Recently, drifts in ADCC-related quality attributes of Herceptin® have also been described.Citation40 Although product changes are recognized,Citation11,12 in a global and rapidly developing biosimilars landscape,Citation49 there is no clear understanding of what the effects, if any, might be on the safety and efficacy of a given mAb over time.Citation50 For illustration, a hypothetical situation where 4 approved mAb products may drift in different directions overtime has been simulated in . The figure shows how the IS would allow traceability of products through their life-time to IU of bioactivity, aligning potency data from different manufacturers and therefore allowing the detection and better understanding of potential changes in products' quality attributes. We argue that the availability of bioassay data calibrated to an IS would have a positive impact on the ability of regulators to interpret product heterogeneity of both marketed products and biosimilar products under development. This is of no relevance in a single innovator product situation, but as the RMP undergoes changes with time and the number of approved and investigational biosimilar products increase, the need for ISs becomes increasingly evident. WHO ISs have supported potency harmonization of biological medicines across the globe, both for natural and recombinant products, for more than a century.Citation51 However, their long history and success has not paved the way for IS for mAbs as their role is received with skepticism across some sectors. Primary amongst these concerns are fears of retrospective effects for marketed products imposing new regulatory demands or of the IS being inappropriately used.Citation52 This opposition may have been due to the lack of data to support their role so that stakeholders can appreciate the benefits in assuring confidence in biosimilar products pre- and post- marketing. Based on the results of the multi-center collaborative study presented here, we propose the suitability of the rituximab candidate preparation NIBSC Code 14/210 as an IS for the in vitro biological activities of rituximab. The assignment of 1,000 IU of bioactivity per ampoule for each of the 4 independent bioactivities assessed (CDC, ADCC, cell-binding and apoptosis) will be proposed with the proviso that this unitage infers no regulatory role in defining biosimilarity, labelling or dosing requirements. These regulatory roles are fulfilled exclusively by the RMP under the scope of the competent authorities. Caveats of this work are: 1) only a limited amount of data were gathered for cell-binding, and 2) only one laboratory performed apoptosis assays. However, the assignment of 4 independent bioactivities would be needed as different rituximab preparations might differ in their relative ratio of individual bioactivities. The data suggests that the use of IS for rituximab may assist stakeholders in the calibration and validation of bioactivity assays and help with harmonizing potency values between laboratories. Using this study as an example, it is anticipated that IS for mAbs may assure confidence in a rapidly expanding biosimilar market. Furthermore, improving the understanding of mAb structure-function relationship is critical, and ISs for mAbs may become a vital tool in the identification of bioactivity changes during the life-cycle of both innovator and biosimilar products. As an independent reference standard, the IS could also assist in any investigation of adverse events by Official Medicine Control Laboratories (OMCL), as well as support Centrally Authorised Product (CAP) testing.
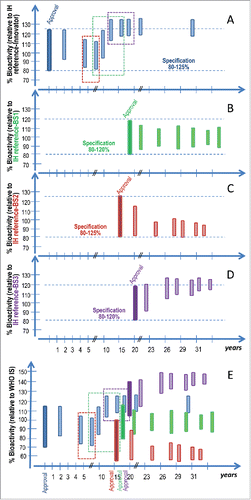
Figure 3. Simulation of the potential impact of product drifting on mAb bioactivity over time. The Figures Show a hypothetical situation for 4 approved mAb products namely the innovator, a biosimilar 1 (BS-1), a biosimilar 2 (BS-2) and a biosimilar 3 (BS-3), in blue, green, red and purple respectively, with various post-approval changes and effects on mAb bioactivity. The x-axis represents time in years relative to the approval of the innovator product. The colored boxes represent the bioactivity range for the products for a given biological activity at the time of approval (darker shade) and post various manufacturing process changes (lighter shaded bars). Product specification at the time of approval is set in relation to the target product profile and is also noted in figures A-D and indicated by the dotted lines. The colored dotted rectangles in figures A and E indicate the biological activity range of the innovator product batches available and used as RMP during the comparability exercise for the three color-matching biosimilars. Figures A, B, C and D show biological activity relative to the proprietary “in house” reference standards (IH reference innovator, IH reference BS-1, IH reference BS-2 and IH reference BS-3) and figure E shows the bioactivity relative to the WHO IS. The innovator product batches used at the time of the biosimilarity assessment for different biosimilars have an impact on the target product profile and thus the characteristics of the approved biosimilar. Further, product drifting may occur for the 4 approved independent products upon post-marketing process changes. Currently in the absence of public higher order standards, those changes can only be characterized based on the comparison of a small number of available product batches post-change, available pre-change product batches, the proprietary “in house” reference standard and defined bioactivity specifications at the time of approval. Relative bioactivity changes between different approved products cannot be characterized in the absence of a common reference standard. The use of a WHO IS for the bioactivity of mAbs allows data harmonization and therefore a better understanding of potential product drift and evolution.
In conclusion, we developed a lyophilized candidate WHO IS for bioassays relevant to the mechanisms of action of rituximab and demonstrated its suitability through an international collaborative study. In view of the ability of the candidate preparation to harmonize potency data between laboratories, NIBSC 14/210 will be proposed to serve as IS for rituximab bioactivities with an independent assignment of 1,000 IU per individual bioactivity (CDC, ADCC, cell-binding and apoptosis). In addition, the study revealed differences in the potency of rituximab “in house” reference standards, substantiating the need for a publically available common reference standard that supports bioassay performance, facilitating calibration of local standards that are traceable to IU. This role is without exception distinct from the regulatory role of the RMP, which has the exclusive purpose of defining biosimilarity. This study reports the first candidate standard for the biological activities of a mAb for which there is no naturally occurring protein of equivalent bioactivity, and therefore represents the introduction of a new class of IS. Upon adoption, the WHO IS is expected to have a significant positive impact on new and existing rituximab products throughout their life-cycle, and in doing so will enhance confidence in the use of biosimilars so they can realize their full potential benefit for patients.
Materials and methods
Materials, study preparations processing and characterization
A preparation of recombinant chimeric rituximab was kindly donated to WHO for the purpose of this study (see acknowledgements). A suitable certificate of analysis and safety data sheet was also provided. The material was formulated, freeze-dried, and the CDC activity of trial fills was compared with that of the bulk material. A clinical batch of Mabthera® (Roche) was purchased from ADAllen Pharma Ltd, formulated and filled as per conditions optimized for the candidate. Details of the preparations used in the collaborative study are shown in . The rituximab standard candidate production fill was prepared and lyophilized under controlled conditions following standardized procedures for the preparation of International Biological Standards.Citation36 Buffers and excipients were prepared using water for irrigation (Baxter, Switzerland) and filtered using sterile non-pyrogenic 0.22 μm filters (Nalgene®, Nalge Nunc International). Briefly, 1 mL of rituximab solution containing approximately 100 μg rituximab protein in formulation buffer (1% (v/v) clinical grade HSA (BPL Zenalb 20, Bio Products Laboratory Limited), 25 mM tri-sodium citrate dehydrate, 150 mM sodium chloride, pH 6.5) was filled using an automated filling line (Bausch & Stroebel, Ilshofen, Germany) with constant stirring of the material at 2–8 °C. The filled ampoules were freeze-dried using a Serail CS100 freeze-dryer (Le Coudray St Germer, France). The process was initiated on the day of filling and the product was frozen over 120 minutes to −50 °C and held for 6 hours before applying vacuum. Primary drying was at −30 °C shelf temperature and 100 μbar vacuum for 40 hours, followed by a 15 hour ramp to +30 °C and a secondary drying at the same temperature and 30 µbar vacuum for a further 20 hours. The glass ampoules were sealed under dry nitrogen by heat fusion, coded as 14/210 and stored at −20 °C in the dark at NIBSC until shipment. The nominal content of rituximab per ampoule was calculated from the dilution of the bulk material and assumed protein mass content as per manufacturer's data. The characteristics of the lyophilized preparations comply with approved specifications suitable for WHO reference materials and are described in . Residual moisture was measured by the coulometric Karl-Fischer method (Mitsubishi CA100) and the headspace oxygen content was determined by frequency modulated spectroscopy using a Lighthouse FMS-760 Instrument (Lighthouse Instruments, LLC). No evidence of microbial contamination was found by total viable count method. For the collaborative study, coded duplicates from the candidate preparation were labelled as samples A and B and the lyophilized and re-formulated Mabthera® preparation was labelled as sample C.
Table 10. Preparations used in the collaborative study.
Table 11. Fill production details of the preparations used in the collaborative study.
Participants in the collaborative study, study design and bioassay methods
A total of sixteen participants from nine different countries kindly contributed to the bioassay data used in the study (). Amongst the participants 9 were manufacturers, 5 were control laboratories, 1 was a Pharmacopoeia and 1 was a contract research organization. The laboratories are identified with a number, from 1 to 16, that has no relation to the order in the listing. The bioassays performed by each laboratory are summarized in . The participants received a collaborative study protocol that included bioassay methods and layouts as examples only, instructions for use, template sheets to record data and methodologies. The preparations were shipped at room temperature and participants were instructed to store the samples at −20 °C upon arrival. The ampoules were reconstituted with 1 mL of sterile distilled/deionized water on the day of the assay as per instructions provided. The laboratories were encouraged to use their qualified bioassay methods, including routine controls and qualified “in house” reference standards where possible. Data was requested from three independent assay runs performed on three different days using fresh preparations. Enough ampoules were provided to perform the three assay runs, conduct preliminary assays to establish a suitable working dilution range for the test materials and in case of accidental loss. Typically participants returned data from a total of 9 assays accommodating the three study preparations, the “in house” reference when available and two independent dilution series per sample with some randomization. summarizes the bioassays that contributed to the study.
Table 12. List of participants in the collaborative study.
Statistical analysis
An independent statistical analysis of all bioassay data was performed at NIBSC. Analysis of dose-response curve data was performed using a four-parameter logistic model:where y denotes the assay response, x is the concentration, α is the upper asymptote, δ is the difference between upper and lower asymptotes, β is the slope factor and γ is the EC50 (50% effective concentration). Models were fitted using the R package ‘drc’. Parallelism (similarity) for a pair of dose-response curves was concluded by demonstrating equivalence of the parameters α, β and δ independently for each of the 3 bioassays (CDC, ADCC and cell binding). For this approach, ratios of these parameters for the two samples under consideration were calculated and log transformed. Approximate 90% confidence limits for these values were determined using the delta method. Extreme values (defined as the maximum of the upper 90% confidence limit and the reciprocal of the lower 90% confidence limit) were calculated and equivalence (assay validity) concluded in cases where these were below the pre-defined upper equivalence bounds for all three model parameters. Upper equivalence bounds were set as the 90th percentile of observed extreme values for the candidate preparation and its coded duplicate (samples A & B). The calculated upper equivalence bound values and how the percentage of invalid assays changes for each sample with different bounds depicting the rationale for determining them is shown as supplementary data (Table S1 and Figures S1-S3). The validity of the apoptosis assays, performed by one laboratory only, was concluded when no significant non-parallelism was found by analysis of variance. Relative potency estimates were calculated as the ratio of EC50 estimates in assays where acceptable parallelism was concluded. All relative potency estimates were combined to generate unweighted geometric mean (GM) potencies for each laboratory and these laboratory means were used to calculate overall unweighted geometric mean potencies. Variability between assays and laboratories was expressed using geometric coefficients of variation (GCV = {10s-1} × 100% where s is the standard deviation of the log10 transformed potencies).
Stability studies
Ampoules of the lyophilized candidate preparation (NIBSC 14/210) were stored at elevated temperatures, namely 56 °C, 45 °C, 37 °C, 20 °C and 4 °C and tested at indicated time points together with ampoules stored at the recommended temperature of −20 °C and −70 °C as baseline reference temperature. To date, tests for CDC and ADCC bioactivities have been performed after 10 months of storage. Accelerated thermal degradation and “real time” stability studies for stability prediction and monitoring are ongoing.
Disclosure of interest
The authors report no potential conflicts of interest.
SUPPL_MAT.zip
Download Zip (3.5 MB)Acknowledgments
We are grateful to Sandoz GmbH (Austria) for kindly donating the material to develop the candidate preparation and to the participating laboratories that performed the bioassays and provided helpful discussions and comments. We also thank Paul Matejtschuk and Kiran Malik for their assistance in the pilot fills and process optimization, and staff in Standards Processing Division for the preparation of the candidate fill and dispatching the study preparations. Finally we would like to thank Meenu Wadhwa, Susan Thorpe and Chris Burns for their helpful discussions and Christian Schneider for the critical review of this manuscript. This work was funded, in part, by a grant from UK Department of Health's Policy Research Programme, Grant Number 044/0069.
References
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7(1):9–14. doi:10.4161/19420862.2015.989042. PMID:25529996
- Reichert JM. Metrics for antibody therapeutics development. MAbs. 2010;2(6):695–700. doi:10.4161/mabs.2.6.13603. PMID:20930555
- European Medicines Agency. Medicines under evaluation by the Committee for Medical Products for Human Use (CHMP). 2017; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/document_listing/document_listing_000349.jsp&mid=WC0b01ac05805083eb#section1 (Last accessed 13 June 2017).
- Udpa N, Million RP. Monoclonal antibody biosimilars. Nat Rev Drug Discov. 2016;15(1):13–14 doi:10.1038/nrd.2015.12. PMID:26678619
- Patent expiry dates for best-selling biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):178–9. doi:10.5639/gabij.2015.0404.040
- McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91(3):405–417. doi:10.1038/clpt.2011.343. PMID:22318617
- Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196. doi:10.1182/blood-2014-06-583617. PMID:25298038
- Markus R, Liu J, Ramchandani M, Landa D, Born T, Kaur P. Developing the Totality of Evidence for Biosimilars: Regulatory Considerations and Building Confidence for the Healthcare Community. BioDrugs 2017;31(3):175–187. doi:10.1007/s40259-017-0218-5. PMID:28439817
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013:72(3):315–318. doi:10.1136/annrheumdis-2012-202941. PMID:23390018
- Vezer B, Buzas Z, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016:32(5):829–834. doi:10.1185/03007995.2016.1145579. PMID:26808864
- Grampp G, Ramanan S. The Diversity of Biosimilar Design and Development: Implications for Policies and Stakeholders. BioDrugs. 2015;29(6):365–372. doi:10.1007/s40259-015-0147-0. PMID:26581551
- Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363–372. PMID:24567263
- Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013:5(5):621–623. PMID:23924791
- Nikolov NP, Shapiro MA. An FDA perspective on the assessment of proposed biosimilar therapeutic proteins in rheumatology. Nat Rev Rheumatol 2017;13(2):123–128. PMID:28053335
- European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2012; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf.
- US Food and Drug Administration. Guidance to Industry: Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product. 2015; Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
- World Health Organization, Expert Committee on Biological Standardization. Fixty seventh report. Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs). 2017; WHO Technical Report series 1004:93-127. Available from: http://apps.who.int/iris/bitstream/10665/255657/1/9789241210133-eng.pdf
- World Health Organization, Expert Committee on Biological Standardization. Fixty sixth report. Global needs in standardization of products derived by biotechnology. 2016; WHO Technical Report series. 999:13–15. Available from: http://www.who.int/biologicals/expert_committee/WHO_TRS_999_FINAL.pdf?ua=1
- Wadhwa M, Kang HN, Knezevic I, Thorpe R, Griffiths E. WHO/KFDA joint workshop on implementing WHO guidelines on evaluating similar biotherapeutic products, Seoul, Republic of Korea 24-26 August, 2010. Biologicals. 2011;39(5):349–357.
- Thorpe R, Wadhwa M. Intended use of reference products & WHO International Standards/Reference Reagents in the development of similar biological products (biosimilars). Biologicals. 2011;39(5):262–265. PMID:21880508
- World Health Organization, Expert Committee. The 19th WHO Model list of Essential Medicine. 2015; WHO Technical Report series 994:417–470. Available from: http://apps.who.int/iris/bitstream/10665/189763/1/9789241209946_eng.pdf.
- Forstpointner R, Unterhalt M, Dreyling M, Bock HP, Repp R, Wandt H, Pott C, Seymour JF, Metzner B, Hanel A, et al. Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: Results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood. 2006;108(13):4003–4008. PMID:16946304
- Hiddemann W, Kneba M, Dreyling M, Schmitz N, Lengfelder E, Schmits R, Reiser M, Metzner B, Harder H, Hegewisch-Becker S, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106(12):3725–3732. doi:10.1182/blood-2005-01-0016. PMID:16123223
- European Medicines Agency. MabThera: European Public Assessment report (EPAR)—Scientific Discussion. European Medicines Agency. 2005; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000165/WC500025817.pdf.
- European Medicines Agency. MabThera: European Public Assessment report (EPAR)—Product information. 2016; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000165/WC500025821.pdf.
- Dotan E, Aggarwal C, Smith MR. Impact of Rituximab (Rituxan) on the Treatment of B-Cell Non-Hodgkin's Lymphoma. P T. 2010:35(3):148–157. PMID:20442809
- Sanz I. Indications of rituximab in autoimmune diseases. Drug Discov Today Ther Strateg. 2009;6(1):13–19. doi:10.1016/j.ddstr.2009.10.001. PMID:20379381
- Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. 2009;30(7):356–362. doi:10.1016/j.tips.2009.04.007. PMID:19552968
- Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–3837. doi:10.1016/j.molimm.2007.06.151. PMID:17768100
- Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22(47):7359–7368. doi:10.1038/sj.onc.1206939. PMID:14576843
- Jaglowski SM, Alinari L, Lapalombella R, Muthusamy N, Byrd JC. The clinical application of monoclonal antibodies in chronic lymphocytic leukemia. Blood. 2010;116(19):3705–3714. doi:10.1182/blood-2010-04-001230. PMID:20610811
- World Health Organization, Expert Committee on Biological Standardization. Sixty fifth report. Proposed First WHO reference reagent for Rituximab for use in Complement dependent cytotoxicity assays. 2015; WHO Technical Report Series. 993:36–37. Available from: http://apps.who.int/iris/bitstream/10665/173739/1/9789240694095_eng.pdf
- Wadhwa M, Bird C, Dilger P, Rigsby P, Jia H, Gross MEB, participants of the s. Establishment of the first WHO International Standard for etanercept, a TNF receptor II Fc fusion protein: Report of an international collaborative study. J Immunol Methods. 2017;447:14–22. doi:10.1016/j.jim.2017.03.007. PMID:28288790
- Hubbard AR. Potency labeling of novel factor VIII and factor IX concentrates: past experience and current strategy. Semin Thromb Hemost. 2015;41(8):849–854. doi:10.1055/s-0034-1395353. PMID:25703515
- Kirkwood TB. Predicting the stability of biological standards and products. Biometrics. 1977;33(4):736–742. doi:10.2307/2529472. PMID:588659
- World Health Organization, Expert Committee on Biological Standardization.Fifty fifth report. Recommendations for the preparation, characterization and establishment of international and other biological reference standards. 2006; WHO Technical Report Series. 932:73–130. Available from: http://apps.who.int/iris/bitstream/10665/43278/1/WHO_TRS_932_eng.pdf.
- Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312. doi:10.1038/nbt.1839. PMID:21478841
- Cheng ZJ, Garvin D, Paguio A, Moravec R, Engel L, Fan F, Surowy T. Development of a robust reporter-based ADCC assay with frozen, thaw-and-use cells to measure Fc effector function of therapeutic antibodies. J Immunol Methods. 2014;414:69–81. doi:10.1016/j.jim.2014.07.010. PMID:25086226
- Kellner C, Derer S, Valerius T, Peipp M. Boosting ADCC and CDC activity by Fc engineering and evaluation of antibody effector functions. Methods. 2014:65(1):105–113. doi:10.1016/j.ymeth.2013.06.036. PMID:23851282
- Kim S, Song J, Park S, Ham S, Paek K, Kang M, Chae Y, Seo H, Kim HC, Flores M. Drifts in ADCC-related quality attributes of Herceptin(R): Impact on development of a trastuzumab biosimilar. MAbs. 2017;9(4):704–714. doi:10.1080/19420862.2017.1305530. PMID:28296619
- Iida S, Misaka H, Inoue M, Shibata M, Nakano R, Yamane-Ohnuki N, Wakitani M, Yano K, Shitara K, Satoh M. Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin Cancer Res. 2006;12(9):2879–2887. doi:10.1158/1078-0432.CCR-05-2619. PMID:16675584
- Preithner S, Elm S, Lippold S, Locher M, Wolf A, da Silva AJ, Baeuerle PA, Prang NS. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol Immunol. 2006:43(8):1183–1193. doi:10.1016/j.molimm.2005.07.010. PMID:16102830
- Chung S, Lin YL, Reed C, Ng C, Cheng ZJ, Malavasi F, Yang J, Quarmby V, Song A. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies - impact of effector cells. J Immunol Methods. 2014;407:63–75 doi:10.1016/j.jim.2014.03.021. PMID:24704820
- Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007:110(7):2561–2564. doi:10.1182/blood-2007-01-070656. PMID:17475906
- Hsieh YT, Aggarwal P, Cirelli D, Gu L, Surowy T, Mozier NM. Characterization of FcgammaRIIIA effector cells used in in vitro ADCC bioassay: Comparison of primary NK cells with engineered NK-92 and Jurkat T cells. J Immunol Methods. 2017;441:56–66. PMID:27939300
- Mata MM, Mahmood F, Sowell RT, Baum LL. Effects of cryopreservation on effector cells for antibody dependent cell-mediated cytotoxicity (ADCC) and natural killer (NK) cell activity in (51)Cr-release and CD107a assays. J Immunol Methods. 2014:406:1–9. doi:10.1016/j.jim.2014.01.017. PMID:24561308
- European Medicines Agency. Committee for Medical Products for Human Use (CHMP). Assessment report, Remsima. 2013; EMA/CHMP/589317/2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002576/WC500151486.pdf
- Schiestl M, Li J, Abas A, Vallin A, Millband J, Gao K, Joung J, Pluschkell S, Go T, Kang HN. The role of the quality assessment in the determination of overall biosimilarity: a simulated case study exercise. Biologicals. 2014;42(2):128–132. doi:10.1016/j.biologicals.2013.11.009. PMID:24373974
- Biosimilars of Rituximab. Generics and Biosimilars Initiative Journal (GaBI Online). Mol, Belgium: Pro Pharma Communications International; [cited 2017 Apr 14]. Available from: http://www.gabionline.net/Biosimilars/General/Biosimilars-of-rituximab.
- Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, Thirstrup S. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30(12):1179–1185. doi:10.1038/nbt.2447. PMID:23222783
- Bristow AF, Barrowcliffe T, Bangham DR. Standardization of biological medicines: the first hundred years, 1900-2000. Notes Rec R Soc Lond. 2006;60(3):271–289. doi:10.1098/rsnr.2006.0153. PMID:17212227
- World Health Organization, Expert Committee on Biological Standardization. Sixty sixth report. Report of a WHO informal consultation on international standards for biotherapeutic products. 2016; WHO Technical Report series 999:13–15. Available from: http://apps.who.int/iris/bitstream/10665/208900/1/9789240695634-eng.pdf